

Abstract
A mutation (Cam7) to the single endogenous calmodulin gene of Drosophila generates a mutant protein with valine 91 changed to glycine (V91G D-CaM). This mutation produces a unique pupal lethal phenotype distinct from that of a null mutation. Genetic studies indicate that the phenotype reflects deregulation of calcium fluxes within the larval muscles, leading to hypercontraction followed by muscle failure. We investigated the biochemical properties of V91G D-CaM. The effects of the mutation on free CaM are minor: Calcium binding, and overall secondary and tertiary structure are indistinguishable from those of wild type. A slight destabilization of the C-terminal domain is detectable in the calcium-free (apo-) form, and the calcium-bound (holo-) form has a somewhat lower surface hydrophobicity. These findings reinforce the indications from the in vivo work that interaction with a specific CaM target(s) underlies the mutant defects. In particular, defective regulation of ryanodine receptor (RyR) channels was indicated by genetic interaction analysis. Studies described here establish that the putative CaM binding region of the Drosophila RyR (D-RyR) binds wild-type D-CaM comparably to the equivalent CaM-RyR interactions seen for the mammalian skeletal muscle RyR channel isoform (RYR1). The V91G mutation weakens the interaction of both apo- and holo-D-CaM with this binding region, and decreases the enhancement of the calcium-binding affinity of CaM that is detectable in the presence of the RyR target peptide. The predicted functional consequences of these changes are consonant with the in vivo phenotype, and indicate that D-RyR is one, if not the major, target affected by the V91G mutation in CaM.
Keywords: calmodulin mutant phenotype, calcium signaling in Drosophila, ryanodine receptor, calcium channels
The control of intracellular processes by calcium ions is complex, with regulatory information encoded not only in the calcium ion concentration per se but also in the frequency, duration, and location of calcium fluxes within the cell (Clapham 1995; Putney Jr. 1998). The ubiquitous calcium-binding protein calmodulin (CaM) plays a role in converting these aspects of calcium activity into cellular responses (Cohen and Klee 1988; Van Eldik and Watterson 1998). Calcium-binding events at the four “EF hand” sites of CaM induce conformational changes in CaM that alter its activity toward its various intracellular targets. CaM regulates a diverse array of targets by a variety of mechanisms, presumably reflecting the need for widespread and varied responses to fluctuations in calcium levels. The best studied class of CaM targets, a group of protein kinases, only bind strongly to the holo- (calcium-bound) form of CaM, which engulfs a short amphipathic helical region in the kinase. The resulting conformational change in the protein relieves auto-inhibition at the active site (for review, see Schulman and Braun 1999). CaM regulation of several calcium-regulated ion channel types is proving to be significantly different. The calcium-free (apo-) form of CaM binds constitutively to the channel and upon calcium binding, channel activation or inhibition is produced (for review, see Saimi and Kung 2002).
Given the large array of potential CaM targets identified by in vitro work, a key issue in understanding CaM function in intact cells and organisms is identifying the target regulation in operation under particular physiological conditions or at different developmental stages. A genetic approach to this problem has proved useful in Saccharomyces cerevisiae, where individual point mutations to the single CaM gene produce four different phenotypes, suggesting that different regions of CaM are required in different intracellular processes (for review, see Cyert 2001). Nelson et al. (1997) applied a similar approach in Drosophila melanogaster. Random chemical mutagenesis was used to produce point mutations to the single Cam gene of this organism, and associated phenotypes that differed from that of a null mutation of the Cam gene were identified.
One mutation, Cam7, which generates a valine-to-glycine change at position 91 of CaM (V91G D-CaM), produced a completely novel phenotype. Mutant animals form shortened, indented pupal cases with a “Michelin Man” appearance. Although adult bodies are formed in the pupal cases, none ever emerge as live adults, and head defects are common, including “inside-out” heads resulting from failed eversion of the head capsule. Further study of the phenotype (Wang et al. 2003) established that the lethal pupal defects all arise in the musculature, and that the indented pupal cases reflect hypercontraction of the muscles at pupariation. Additional evidence demonstrated that calcium fluxes in the body wall muscles are severely disrupted, and suggested that the function of the major calcium-release channel of the musculature, the ryanodine receptor (RyR, a CaM-regulated channel) could be affected. Thus, (1) a mutant allele of the single D-RyR channel gene suppresses the pupal hypercontraction and allows survival through to adulthood; and (2) in another Dipteran fly species, injection of ryanodine into larvae immediately before pupariation produces pupal cases strikingly similar to those produced by Cam7 (Zdarek and Fraenkel 1972).
We present here an analysis of the biochemical properties of the V91G D-CaM encoded by Cam7, with particular emphasis on examination of its interaction with D-RyR channels. The mutation produces only small changes in the properties of the free protein, but interactions with the CaM-binding region of D-RyR are weakened in both the apo- and holo-states.
Results
The effect of the V91G mutation on the conformation and calcium-binding properties of apo- and holo-D-CaM
Apo- and holo-CaM adopt different conformations and can differ dramatically in the way they interact with targets (Cohen and Klee 1988; Van Eldik and Watterson 1998). It was important therefore to determine whether the V91G mutation affects the properties of CaM in either the presence or absence of calcium. The UV absorption spectrum, the near- and far-UV CD spectra, and the tyrosine fluorescence emission spectrum of V91G D-CaM, recorded at 10°C in the presence or absence of 1 mM Ca2+, all proved effectively identical to those of wild-type (WT) D-CaM under the same conditions (data not shown). Thus, at relatively low temperatures, in either the apo- or holo-form, the V91G mutation does not affect the secondary structure of D-CaM, or the local tertiary structural environment of the single tyrosine at position 138 in the C-terminal domain. However, at 20°C, the far-UV CD spectra indicated a small reduction in the stability of the C-terminal domain of V91G D-CaM. This effect was investigated further by examining both urea and heat-induced unfolding of WT and V91G D-CaM in the presence and absence of calcium.
In the presence of urea, previous unfolding studies have shown that WT CaM undergoes two overlapping transitions due to unfolding of the two domains, which exhibit similar overall thermodynamic stability (Masino et al. 2000). In the apo-state, unfolding of the C-domain precedes that of the N-domain. However, as a result of the higher affinity of the C-domain for calcium, this unfolding order is reversed for the holo-form. By using far-UV CD to monitor unfolding (Fig. 1A ▶), we found that, in the absence of calcium, V91G D-CaM unfolds somewhat before WT D-CaM in the first phase of the transition, indicating that the C-terminal domain is slightly less stable in the mutant protein. In the presence of 0.3 mM Ca2+ however, the urea unfolding curves are closely similar (Fig. 1A ▶). These findings suggest that the V91G mutation has little effect on the calcium-binding properties of the protein (see also Masino et al. 2000).
Figure 1.

Urea and thermal unfolding of WT and V91G D-CaM. (A) Urea-induced unfolding of apo-V91G D-CaM (circles) and WT D-CaM (squares) in the presence (closed symbols) and absence (open symbols) of 0.3 mM CaCl2 monitored using far-UV circular dichroism. (B) Urea-induced unfolding of apo-V91G D-CaM (circles) and WT D-CaM (squares) in the presence (closed symbols) and absence (open symbols) of 0.3 mM CaCl2 monitored using Tyr-138 fluorescence. (C) Thermal unfolding of apo-V91G D-CaM (circles) and WT D-CaM (squares) in the presence (closed symbols) and absence (open symbols) of 5 mM MgCl2 monitored using far-UV circular dichroism. (D) Thermal unfolding of apo-V91G D-CaM (circles) and WT D-CaM (squares) monitored using Tyr-138 fluorescence.
Monitoring of the unfolding of the C-terminal domain by the fluorescence of Tyr-138 (Fig. 1B ▶) allowed a more quantitative analysis of the effect of V91G on C-terminal stability. For the apo-proteins at 10°C, analysis of the unfolding curves as a function of [Urea] was performed by the linear extrapolation method, that is, (ΔG(obs) = RT ln Keq(obs); and ΔG(obs) = ΔG° − m[U] (see Masino et al. 2000). This gave for V91G, ΔG°apo = 1.89 ± 0.05 kcal/mol, m = 0.893 ± 0.013 kcal/mol.M (and [U]1/2 = ΔG°/m = 2.12 M), and for WT D-CaM, ΔG°apo = 2.17 ± 0.04 kcal/mol, m = 0.908 ± 0.015 kcal/mol.M (and [U]1/2 = 2.39 M). The close similarity of the m-values indicates that a similar total surface area is exposed on unfolding of the C-domain of either protein. For the holo-proteins at 20°C in 0.3 mM Ca2+, analysis gave for V91G, ΔG°holo,0.3mM = 6.09 ± 0.15 kcal/mol, m = 0.865 ± 0.017 kcal/mol.M (and [U]1/2 = 7.04 M) and for WT D-CaM, ΔG°holo,0.3mM = 6.53 ± 0.17 kcal/mol, m = 0.918 ± 0.014 kcal/mol.M (and [U]1/2 = 7.11 M).
The attribution of the destabilizing effects of the V91G mutation to the C-domain was confirmed by analysis of thermally induced unfolding. As with urea, thermal unfolding of apo-D-CaM produces a biphasic curve with the C-terminal domain unfolding at lower temperature (Masino et al. 2000). Thermal unfolding of apo-WT and V91G D-CaM, as monitored by far-UV CD, is shown in Figure 1C ▶. Destabilization of the C-terminal domain by the V91G mutation with no effect on the N-domain is clearly indicated by the curves. Magnesium is known to bind preferentially to the N-domain of CaM (Masino et al. 2000) and thus preferentially stabilizes this domain against thermally induced unfolding. The thermal unfolding curves for the apo-forms of WT and V91G D-CaM in the presence of magnesium (Fig. 1C ▶) show similar parallel shifts to higher temperature, indicating that they have similar magnesium-binding properties, and further confirming that the effects of V91G on stability are in the C-domain.
Further quantitative information on the effects of the V91G mutation on the stability of the apo-form was obtained by monitoring unfolding by the fluorescence from Tyr-138 (Fig. 1D ▶). The following thermodynamic parameters for C-domain unfolding were derived from these curves (Masino et al. 2000): for apo-V91G, ΔHm = 32.8 kcal/mol and Tm = 35.1°C; for apo-WT D-CaM, ΔHm = 35.6 kcal/mol and Tm = 40.9°C. If we assume that ΔCp, the heat capacity change per residue = 0.8 ± 0.1 kcal/mol.deg (Masino et al. 2000), then ΔG°(10°C) = 1.83 ± 0.1 kcal/mol and ΔG° (20°C) = 1.31 ± 0.1 kcal/mol for the C-domain in apo-V91G, compared with ΔG°(10°C) = 2.25 ± 0.1 kcal/mol and ΔG°(20°C) = 1.80 ± 0.1 kcal/mol for the C-domain of apo-WT D-CaM. Thus the mutation destabilizes the C-domain by ~0.5 kcal/mol in the range of 10°–20°C. The values of ΔG° (T) for both proteins are in good agreement with those from urea unfolding. The very similar values of ΔΔG° (20°C) (=ΔG°holo,0.3mM − ΔG°Apo) for the two proteins (4.7 ± 0.2 kcal/mol for V91G and 4.8 ± 0.21 for WT D-CaM) are also consistent with the C-terminal domains of the two proteins binding calcium with similar affinity.
Decreased thermal stability of the V91G C-terminal domain in its apo-form may underlie a difference in electrophoretic mobility detected between apo-V91G and apo-WT D-CaM in nondenaturing gels. As shown in Figure 2A ▶, although at high calcium (200 μM), the electrophoretic mobility of the two proteins at room temperature is identical; at very low calcium (< 10 nM) V91G D-CaM migrates detectably slower than WT. This difference is sufficient to permit resolution of the two proteins as separate bands when they are co-electrophoresed as one sample in these gels (Fig. 2C ▶).
Figure 2.

Nondenaturing gel electrophoresis of WT and V91G D-CaM. (A) Relative mobilities of holo- and apo-forms of WT and V91G D-CaMs. At high [Ca2+] (200 μM), there is no difference in the mobility of WT and V91G D-CaM; at low [Ca2+] (<10 nM), V91G D-CaM migrates more slowly than WT. (B) Ca2+-induced mobility shifts for WT and V91G D-CaM. Both WT and V91G D-CaM show a strong Ca2+-induced shift in mobility, although for V91G the shift is smaller because the apo-form migrates more slowly. Diffusion of EGTA leads to sequestering of Ca2+ and thus to a zone of protein migrating at intermediate mobilities between the high and low [Ca2+] samples. (C) Co-electrophoresis of apo-WT and apo-V91G D-CaM. The two proteins migrate as clearly separable bands in the absence of calcium.
A technique was recently developed that allows the intrinsic calcium-binding affinities of the N- and C-domains of CaM to be investigated independently within the intact protein. Tyrosine (Tyr)138, within site IV of CaM, reports on calcium binding to the C-domain sites. Although both the N- and C-domains of CaM contain phenylalanine residues, steady-state and time-resolved fluorescence studies of full-length CaM and its domain fragments have shown that, in the absence of a target peptide, Tyr138 quenches the phenylalanines within the C-domain of mammalian CaM (VanScyoc et al. 2002). Thus the decrease in phenylalanine (Phe) fluorescence detected during calcium titration of free CaM is entirely due to calcium binding to the N-terminal sites, whereas the increase in Tyr fluorescence reflects calcium binding to the C-terminal sites. This result was confirmed for Drosophila CaM, in the absence of a target peptide, by comparing the calcium binding properties of sites I and II in (1) full-length D-CaM, and (2) an N-domain fragment of D-CaM. The resolved free energies from Phe fluorescence were identical (data not shown). Thus, Tyr and Phe fluorescence can be used to monitor domain-specific calcium binding in free D-CaM.
This technique was therefore used to compare the calcium binding properties of WT and V91G D-CaM. The fluorescence of Phe and Tyr for WT and V91G D-CaM during calcium titrations proved very similar (Fig. 3 ▶). As shown from the resolved dissociation constants in Table 1, the V91G mutation had no significant effect on the calcium-binding affinity of either domain. The differences in calcium binding to WT and V91G D-CaM were extremely small and within the standard deviation of replicate determinations. The ratio of V91G and WT D-CaM Kd values was 0.89 for calcium binding to sites I and II and 1.07 for calcium binding to sites III and IV (see Table 1), representing ~7%–10% relative differences.
Figure 3.

Calcium binding to WT and V91G D-CaM as monitored by Phe and Tyr fluorescence. (A) Representative calcium titration of WT (filled circles) and V91G (open circles) D-CaM N-domain sites I and II monitoring a calcium-dependent decrease in phenylalanine fluorescence (λex of 250 nm, λem of 280 nm). Solid and dashed lines are simulated curve fits from parameters resolved in the nonlinear least squares analysis of the V91G and WT D-CaM Phe data, respectively, using an Adair function. (B) Calcium titration of WT (filled squares;) and V91G (open squares) D-CaM C-domain sites III and IV monitoring a calcium-dependent increase in tyrosine fluorescence (λex of 277 nm, λem of 320 nm). Dashed lines represent the simulated curve fit of WT D-CaM Tyr, providing a comparison of V91G D-CaM calcium-binding affinity (solid line) to that of WT D-CaM.
Table 1.
Effest of D-RyR-peptide on calcium binding of WT and V91G D-CaM
| D-CaM | WT Kd (μM)a | V91G Kd (μM)a | Ratio (mutant)b |
| Phenylalanine fluorescence | |||
| Alone | 15.88 ± 0.72 | 14.07 ± 1.18 | 0.89 |
| +D-RyR-Peptide | 0.25 ± 0.01 | 0.69 ± 0.14 | 2.71 |
| Ratio (peptide)c | 62.35 | 20.37 | |
| Tyrosine fluorescence | |||
| Alone | 2.31 ± 0.09 | 2.47 ± 0.31 | 1.07 |
| +D-RyR-Peptide | 0.28 ± 0.01 | 0.60 ± 0.12 | 2.16 |
| Ratio (peptide)c | 8.35 | 4.13 | |
a Average dissociation constants determined by macroscopic equilibrium constants from Adair fits (see Eq. 1, Materials and Methods). Reported averages and standard deviations are representative of at least three trials. The var. for individual trials ranged from 0.009 to 0.094, with an average of 0.029.
b Ratio (mutant) = Kd (V91G)/Kd (WT).
c Ratio (peptide) = Kd (Alone)/Kd (+D-RyR-peptide).
Calcium titrations of both WT and V91G D-CaM in the presence of the fluorescent reporter 9-anthroylcholine (9AC) were used to compare the surface hydrophobicity of the proteins. Fluorescence from 9AC is strongly enhanced upon binding to solvent-accessible hydrophobic regions, and thus 9AC has been used previously to demonstrate (1) the appearance of hydrophobic patches on the surface of WT CaM in response to calcium binding (Laporte et al. 1980), and (2) the diminished surface hydrophobicity developed upon calcium titration of mutants with defective calcium binding sites (Beckingham 1991; Zhu et al. 1998). Figure 4A ▶ compares the 9AC fluorescence calcium titration curves for WT and V91G D-CaM. For comparison, the 9AC curves for two previously studied calcium-binding site mutants of D-CaM, B1Q and B3Q (Maune et al. 1992a) are presented. In B1Q, binding site 1 is incapacitated by mutation of the bidentate glutamate at the -Z position, to a glutamine; in B3Q, the equivalent mutation is used to incapacitate site 3. As can be seen, the calcium-induced conformational change is drastically altered in B3Q, with a very limited increase in 9AC fluorescence occurring at a higher overall calcium concentration than for WT CaM (Fig. 4 ▶). The paired sites in each domain of WT CaM are known to show strong cooperativity, with the C-terminal sites showing a 10–40-fold greater affinity for calcium than the N- terminal sites. Thus the low amplitude of the 9AC titration curve for B3Q reflects the limited calcium-induced conformational change due to calcium binding at the weaker N-terminal sites. For B1Q, a greater enhancement of 9AC fluorescence is seen than for B3Q, although the change is still considerably lower than that seen for the wild-type protein. The characteristics of this B1Q curve reflect the larger conformational change associated with calcium binding to the higher-affinity C-terminal sites of CaM.
Figure 4.
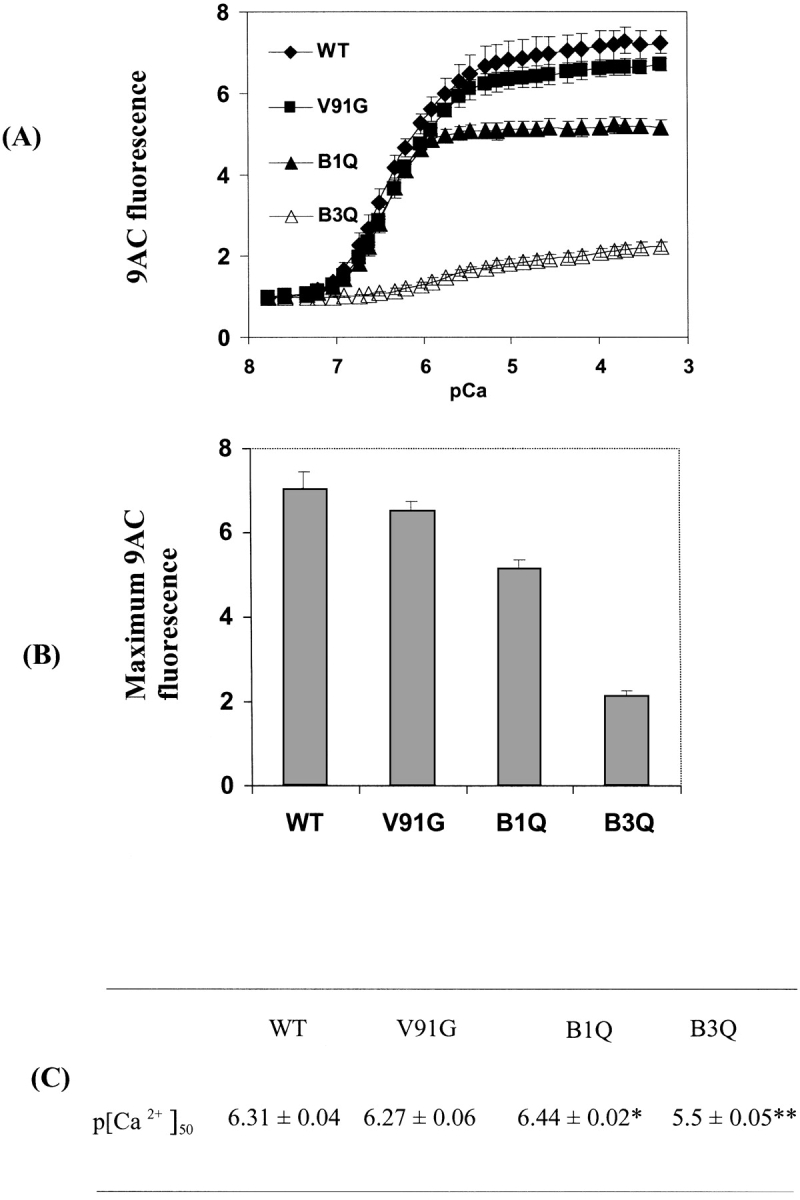
9AC fluorescence enhancement during calcium titration of WT and mutant D-CaMs. (A) Fluorescence enhancement curves. 9AC fluorescence enhancement during calcium titration is shown for WT D-CaM, V91G D-CaM, and for mutants with no calcium binding at site 3 (B3Q) or site 1 (B1Q). (B,C). Maximum fluorescence enhancement (Max) (B) and p[Ca2+]50 (C) for all CaMs studied. Max and p[Ca2+]50 were calculated by fitting the data to sigmoidal dose-response curves with variable slopes using Prism software (see Materials and Methods). Data for WT and V91G are from five repetitions; for B3Q and B1Q, from three repetitions. Error bars = std. dev. Significance of differences from WT CaM are as follows: *, P < 0.01, **, P < 0.0001.
In contrast to these two calcium-binding site mutants, the midpoint for the 9AC fluorescence titration curve of V91G D-CaM is indistinguishable from that of WT CaM (Fig. 4A,C ▶), thus confirming that the V91G mutation does not alter the calcium affinity of CaM. However, the maximum fluorescence enhancement level of the V91G 9AC titration curve is slightly, but reproducibly, lower, than that of WT CaM (Fig. 4C ▶), suggesting that the V91 residue may contribute to the final level of surface hydrophobicity detectable in holo-CaM.
Effects of the V91G mutation on interactions between D-CaM and the putative D-RyR CaM binding region
Prior genetic studies demonstrated that the Cam7 pupal phenotype is caused by muscle defects, and suggested that RyR function is involved (Wang et al. 2003). The ryanodine receptor is the major channel involved in regulating Ca2+ mobilization during muscle excitation-contraction (EC) coupling. In mammals, three isoforms, RYR1, 2, and 3, are present. For RYR1, the isoform of skeletal muscle, both the apo- and holo-forms of CaM regulate function. Apo-CaM potentiates channel opening whereas holo-CaM inhibits opening, thus providing feedback regulation of channel activity (Tripathy et al. 1995). A CaM-binding region spanning residues 3614–3643 has been identified in RYR1, and a synthetic peptide derived from this region exhibits high affinity for both apo- and holo-CaMs (Rodney et al. 2001a). Apo-CaM binds to the C-terminal region of this peptide, and calcium binding is proposed to produce a shift in binding to a more N-terminal region of the peptide (Rodney et al. 2001a).
Drosophila possesses a single gene encoding RyR, and the encoded protein shares 45% identity with mammalian RYR1 (Takeshima et al. 1994). The homologous CaM-binding region (3723–3752) of Drosophila RyR (D-RyR) shares a 76% similarity (46% identity) with the 3614–3643 region of RYR1 (M-RyR) (Fig. 5A ▶). No prior studies of CaM interactions with the Drosophila RyR have been performed. In order to determine whether the putative CaM-binding region of D-RyR functions similarly to that in mammalian RYR1 and if so, to address the effect of the V91G mutation on this interaction, we synthesized peptides (M-RyR-Peptide and D-RyR-Peptide) corresponding to the CaM-binding region of RYR1 (3614–3643) and D-RyR (3723–3752). Interactions between CaM and these peptides were initially probed using native gel mobility shift assays. As noted previously (Rodney et al. 2001a), at low [Ca2+], CaM-RyR peptide complexes do not enter the gels, and the extent of complex formation is indicated by the quantity of unbound CaM still migrating at the position of the free CaM band (Fig. 5B ▶). At high [Ca2+], a band corresponding to the CaM-peptide complex is detected in the gels, thus providing another marker for complex formation (Fig. 5C ▶).
Figure 5.

D-CaM binding to the CaM-binding region of RYR1 and the homologous Drosophila RyR peptide. (A) Comparison of RyR CaM-binding region peptide sequences. MRyR = sequence of peptide representing mammalian skeletal muscle RYR1 CaM-binding region (residues 3614–3643 of protein). DRyR = sequence of peptide representing the homologous region of Drosophila RyR (residues 3723–3752 of protein). (B) CaM-peptide binding at low [Ca2+] (<10 nM). Free peptide and the CaM-peptide complex do not enter the gel under these conditions. (C) CaM-peptide binding at high [Ca2+] (200 μM). The CaM-peptide complexes enter the gels under these conditions. M-RyR-Peptide contains more positively charged residues than D-RyR-Peptide (see A), and thus retards the migration of D-CaM more than D-RyR-Peptide.
As can be seen from Figure 5, B and C ▶, D-RyR-Peptide forms complexes with D-CaM at both low (<10 nM) and high (200 μM) [Ca2+], indicating conservation of function in this region of D-RyR. D-CaM differs by only three conservative amino acid changes from mammalian CaM, and was shown to interact with M-RyR-Peptide comparably to mammalian CaM (Rodney et al. 2000). The affinity of WT D-CaM for the two peptides is fairly comparable, with the gels indicating that Drosophila CaM shows somewhat stronger binding to M-RyR-Peptide than to D-RyR-Peptide at low [Ca2+] and somewhat stronger binding to D-RyR-Peptide than to M-RyR-Peptide at high [Ca2+] (Fig. 5B,C ▶). In the mammalian system, apo-CaM binds to a more C-terminal position within the RYR1 target region than holo-CaM. Specifically, a peptide representing residues 3614–3627 of M-RyR-Peptide shows binding to holo-CaM but no detectable binding to apo-CaM (Rodney et al. 2001a). Given that the C-terminal regions of M-RyR-Peptide and D-RyR-Peptide show strong conservation and the N-terminal region significantly less so (Fig. 5A ▶), it seems likely that differences in the two CaMs themselves contribute to the differences in binding between M-RyR-Peptide and D-RyR-Peptide under low [Ca2+] conditions, whereas at high [Ca2+], differences in the peptide sequences may play a greater role.
We used D-RyR-Peptide to examine the effect of the V91G mutation on the D-CaM/D-RyR interaction. Solution binding assays as described (Martin and Bayley 2002) were used to compare the affinity of WT and V91G D-CaM for D-RyR-Peptide at high and low [Ca2+]. In the presence of 1 mM calcium, 100 mM KCl, V91G D-CaM shows a somewhat weaker affinity for the peptide than the WT protein (130 nM Kd as opposed to 80 nM Kd for WT) (Table 2). In the presence of EGTA, the affinity of both proteins for the peptide was considerably weaker and could only be measured in the absence of salt. In 1 mM EGTA, the Kd values for WT and V91G D-CaM were 470 nM and 1250 nM, respectively. Thus the V91G mutation weakens the interaction between D-CaM and the D-RyR CaM binding region by ~two–threefold, both in the presence and absence of calcium.
Table 2.
Affinity of WT and V91G D-CaM for the CaM binding region of Drosophila RyR (D-RyR-peptide)
| Peptide [Ca2+] | Conditions | Method | Kd (nM) WT | Kd (nM) V91G |
| D-RyR-Peptide + high | 100 mM KCl | Displacement | ~73 | ~125 |
| Ca2+ | 1 mM CaCl2 | |||
| D-RyR-Peptide + high | 100 mM KCl | Competition | 85 (13) | 132 (18) |
| Ca2+ | 1 mM CaCl2 | |||
| D-RyR-Peptide + low | 100 mM KCl | Competition | Not measurable | Not measurable |
| Ca2+ (<1 nM) | 1 mM EGTA | |||
| D-RyR-Peptide + low | 0 mM KCl | Competition | 470 (60) | 1250 (175) |
| Ca2+ (<1 nM) | 1 mM EGTA |
Affinities were determined by fluorescence titration methods as detailed in Materials and Methods.
The affinity of CaM for calcium is strongly influenced by target interactions (Keller et al. 1982; Bayley et al. 1996; Johnson et al. 1996; Martin et al. 1996). Recent studies established that binding of mammalian CaM to the M-RyR-Peptide increases the affinity of the C-terminal lobe of CaM for calcium, with lesser effects on N-terminal calcium binding (Xiong et al. 2002). In these prior studies, calcium binding was monitored via fluorescence changes for versions of CaM with a tryptophan substitution at position 19 (N-domain) or 91 (C-domain) of the protein. Here we used the Phe and Tyr fluorescence of D-CaM (as described above) to address the effect of the V91G mutation on calcium affinity in the presence of D-RyR-Peptide. Marked shifts of both the Phe and Tyr fluorescence curves (Fig. 6 ▶, Table 1) to lower calcium concentrations indicate that calcium binding to WT D-CaM is enhanced significantly by the presence of the D-RyR-Peptide. The V91G mutation weakens this enhancement effect: The calcium binding affinity is two–threefold less for V91G D-CaM than for the wild-type protein in the presence of the peptide. Thus, the Kd as monitored by Phe fluorescence is 2.7 times higher, and 2.2 times higher as monitored by Tyr fluorescence (Table 1). A change of this magnitude is commensurate with a decrease in affinity of V91G D-CaM for D-RyR-Peptide of two–threefold (see above).
Figure 6.

Effects of D-RyR-Peptide on calcium affinity of WT and V91G D-CaM. Calcium titrations of D-CaM in the presence of D-RyR-Peptide as monitored by changes in (A) phenylalanine fluorescence (λex, 250 nm; λem, 280 nm), WT (filled circles), and V91G (open circles) D-CaM, and (B) tyrosine fluorescence (λex, 277 nm; λem, 320 nm), WT (filled squares), and V91G (open squares) D-CaM. Dashed curves represent the titration of WT D-CaM in the absence of D-RyR-Peptide.
Comparison of panels A and B of Figure 6 ▶ shows that, in the presence of peptide, the change in Phe fluorescence as a function of [Ca2+] is effectively coincident with that for Tyr fluorescence, which is reporting C-domain calcium binding. If, as previously (see above), the Phe titration is interpreted as monitoring Ca2+ affinity in the N-domain, this finding would indicate that the N- and C-domain sites now have an identical affinity for Ca2+, with the affinity of the N-domain sites increased ~60-fold, and that of the C- domain sites increased ~eightfold relative to the affinities in the absence of peptide (see Table 2). However, as noted above, the studies with the mammalian system indicate that calcium-binding affinity in the N-domain does not show such an enhancement (Xiong et al. 2002).
The affinity of the N-domain sites in the presence of the peptide was therefore further investigated by a stoichiometric calcium-binding experiment, performed at higher protein concentration, in which calcium binding to the C-domain sites was monitored using Tyr-138 fluorescence. The occupancy of the C-domain was determined as a function of [Ca2+] with/without the D-RyR-Peptide. If, in the presence of the peptide, the affinity of the N-domain is comparable to that of the C-domain, occupancy of the C-domain sites should be decreased at low calcium levels, with complete saturation only occurring in the presence of four equivalents of calcium. However, as shown in Figure 7 ▶, the Tyr-138 signal saturates at two molar equivalents of calcium both in the presence and absence of peptide. Thus, under these conditions, the N-domain calcium sites are not able to compete effectively with the C-domain sites in the presence of D-Ryr-Peptide.
Figure 7.

Stoichiometric binding of calcium to WT D-CaM ± D-RyR-Peptide. Calcium titrations of D-CaM (45 μM) in the presence and absence of D-RyR peptide (65 μM) as monitored by tyrosine fluorescence emission, λex = 280 nm and λem = 306 nm. The contribution from the tyrosine in D-RyR-Peptide has been subtracted, and the curves have been offset in order to facilitate comparison.
Given these findings, alternative explanations for the behavior of the Phe fluorescence in the presence of D-RyR-Peptide were considered. If C-domain calcium binding alone is sufficient for D-CaM interaction with the peptide, this binding could bring a quenching Tyr residue from either the CaM C terminus (Tyr-138) or possibly the D-RyR-Peptide (Tyr-7; see Fig. 5A ▶), sufficiently close to the N terminus such that, in this altered configuration, the observed decrease in Phe fluorescence reflects C-domain, rather than N-domain, calcium binding. Further research is required to investigate these possibilities. Nevertheless, in terms of the central issues under investigation here, two conclusions can be drawn from these studies: (1) the interaction of D-CaM with its target binding region in D-RyR is qualitatively similar to that of mammalian CaM with RYR1; and (2) in the presence of D-RyR-Peptide, the V91G mutation reduces the calcium affinity of CaM by two–threefold compared to the wild-type protein.
The effect of V91G mutation on the interaction between D-CaM and intact mammalian RYR1 channels
Although target peptide binding assays are informative, ultimately it is the effects of the V91G mutation on the interaction of D-CaM with intact RyR channels that is of interest. No functional preparations of RyR channels have yet been generated from Drosophila muscle, and so we addressed this question using purified mammalian skeletal sarcoplasmic reticulum (SR) membrane preparations. Skeletal muscle SR is very rich in RYR1, and RYR1 is the only detectable CaM binding protein in these SR preparations (Rodney et al. 2000). The competitive binding assay developed by Rodney et al. (2000, 2001b) was used for this analysis (see Materials and Methods). That is, the ability of nonradioactive WT or V91G D-CaM to inhibit binding of radiolabeled mammalian CaM to the SR membranes at increasing concentrations was examined. As shown in Figure 8 ▶, at low [Ca2+], V91G D-CaM is impaired in its interaction with the channel, whereas at high [Ca2+], V91G D-CaM binding is very similar to WT D-CaM binding.
Figure 8.

Affinity of WT and V91G D-CaM for mammalian skeletal muscle sarcoplasmic reticulum (SR) membranes. SR membranes were incubated with 5 nM [35S]mammalian CaM and increasing concentrations (0.24 nM – 5 mM) of WT or V91G D-CaM at (A) <10 nM free [Ca2+] or (B) 200 μM free [Ca2+]. The solid lines are derived from fitting the data as described (VanScyoc et al. 2002). B/Bo = amount of radioligand bound normalized to the amount bound in the absence of competing unlabeled CaM.
Discussion
We present here in vitro studies aimed at determining the effects of the V91G mutation on the properties of D-CaM, with a view to understanding the molecular basis of the in vivo mutant phenotype. In vivo, the mutation primarily affects larval body wall muscle function, deregulating calcium fluxes and ultimately producing hypercontraction at pupariation. Further, genetic data suggest that defective regulation of D-RyR channels underlies these defects (Wang et al. 2003).
Analysis of V91G D-CaM by several spectroscopic techniques indicates that the overall secondary and tertiary structure of the protein is identical to that of the WT protein. Further, the calcium-binding properties of the mutant protein, as judged by two methods—calcium titration of (1) endogenous fluorescence from Phe and Tyr residues and (2) exogenous fluorescence from 9AC—are also unchanged. However, three significant differences in the properties of V91G mutant D-CaM from WT were detected: (1) a slight decrease in the stability of the C-terminal domain in the apo-form at temperatures above 20°C, (2) slower migration of the apo-form upon native gel electrophoresis, and (3) somewhat lower surface hydrophobicity of the holo-form.
In the atomic structures for both the apo- and holo-forms of CaM (Taylor et al. 1991; Finn et al. 1993; Kuboniwa et al. 1995; Zhang et al. 1995), V91 is located at the end of the short α-helix entering the third calcium binding loop in the C-terminal domain. Glycine has a negative effect on helix formation, and thus the destabilization of the C-terminal domain by the V91G mutation may reflect an increased flexibility of this helix in the apo-form. The altered electrophoretic mobility of apo-, but not holo-, V91G D-CaM could have similar origins. Binding of calcium to CaM increases the rigidity of its structural elements (Komeiji et al. 2002), and thus in the presence of calcium, the effects of the glycine mutation on this helix may be less detectable at low temperatures (10°–20°C).
Calcium binding causes a major, concerted movement of two helices in each terminal domain of CaM such that a hydrophobic target interaction pocket is opened up in each globular domain. Residue V91 is part of the C-terminal hydrophobic pocket exposed upon calcium binding. The slight loss of hydrophobicity on the surface of V91G D-CaM could simply reflect loss of the contribution of V91 to the overall surface properties of the protein. Alternatively, the V91G mutation may allow closer side-chain packing in this region such that the “openness” and hence, accessibility, of the hydrophobic target interaction pocket in the C-domain is decreased.
These findings on the effects of the V91G mutation help explain some aspects of its in vivo phenotype. Any mutation that substantially altered the calcium-binding properties of free, unbound CaM would produce widespread effects on holo-CaM-regulated activities throughout the organism. Thus the unchanged calcium-binding properties of V91G D-CaM are almost a prerequisite for this mutant to produce limited, tissue-specific effects in vivo. The fact that the mutation has an effect on the apo-conformation of CaM demonstrates its potential to specifically affect targets that bind apo-CaM, many of which are ion channels. However, the lower surface hydrophobicity in the presence of calcium also allows for altered target interactions in the holo-form. In this context we used CSU analysis (Sobolev et al. 1999) to characterize contacts between V91 of CaM and target residues in 14 structures available through the protein database. In all but two of these structures, V91 is in close structural contact with residues of the target protein.
Given the prior genetic findings (Wang et al. 2003), we specifically investigated the effects of the V91G mutation on interaction with the D-RyR receptor. We provide the first evidence that residues 3723–3752 of D-RyR function similarly to the well characterized equivalent CaM-binding region of mammalian RYR1 (residues 3614–3643) in that both apo-CaM and holo-CaM bind this region with high affinity. However, the overall affinity of the interaction appears to be lower in Drosophila, particularly in the apo-state (Table 2; Rodney et al. 2001a).
The V91G mutation has detectable effects on this interaction with the D-RyR CaM-binding region: At both high and low calcium, the interaction of D-CaM with the peptide is weakened, and the enhanced affinity of CaM for calcium produced by peptide binding is decreased by about two- to threefold. In addition, the mutation weakens the affinity of CaM for intact mammalian RYR1 channels at low calcium levels. This latter finding must be interpreted cautiously, given that it involves a cross-species interaction; nevertheless, on balance, it reinforces the conclusion that the V91G mutation weakens interactions with RyR channels.
How might the changed properties of the D-CaM/D-RyR peptide interaction produced by V91G influence RyR channel function? The apo-version of mammalian CaM is a weak activator of RYR1, and so the decreased V91G D-CaM binding to D-RyR at low calcium could have a small effect on channel activation upon membrane depolarization. Holo-mammalian CaM is a strong inhibitor of RYR1 activity, and thus the combined effects of (1) decreased affinity of V91G D-CaM for the D-RyR CaM-binding region and (2) the decreased calcium affinity of V91G D-CaM when bound to this region, could be expected to decrease the effectiveness of channel closure and require that higher levels of calcium are needed to effect channel inhibition.
On balance then, a failure of prompt channel closure might be the predominant effect of the mutation. This prediction is consonant with the genetic findings: The effects of the V91G (Cam7) mutation in muscle were alleviated by reducing RyR channel activity with one copy of a defective Ryr allele. Further, the relatively small change (two–threefold) in the interaction properties of V91G D-CaM with the D-RyR peptide also fits the in vivo phenotype. Thus, whereas a complete loss of RyR channel regulation by CaM would almost certainly lead to early, cataclysmic, organismal death, a small change in RyR regulation could produce the progressive deterioration seen for Cam7 animals (Wang et al. 2003)—ultimately culminating in lethal hypercontraction at pupariation.
The involvement of other CaM targets in the (Cam7) phenotype remains an open question, however. In particular, defective regulation of the dihydropyridine receptor (DHPR) may play a small role in the Cam7 defects. These L-type voltage-sensitive calcium channels are activated upon muscle membrane depolarization and activate RyR channels on the SR through conformational coupling. L-type channels share many similarities with RyR channels in terms of CaM regulation. Apo-CaM binds constitutively to L-type channels, and holo-CaM causes channel inactivation, with the C-terminal calcium-binding sites playing a determinative role in this inactivation (Peterson et al. 1999; Zuhlke et al. 1999). Thus, the V91G mutation could diminish the inhibitory effects of the holo-C terminus on DHPR function. Indeed, we have shown that the presence of one copy of a mutated allele of the relevant DHPR gene (Ca- 1DX7) produces a very mild rescue effect of the Cam7 phenotype; however, this effect is much smaller than that detected with a mutation in the Ryr gene (Wang et al. 2003).
Could other muscle targets be affected by the V91G mutation and contribute to the in vivo phenotype? Although in mammals CaM regulates several kinases with roles in muscle physiology, these CaM targets are unlikely to play a role in the Cam7 phenotype. Failed activation of myosin light chain kinase would produce a phenotype opposite to that of V91G CaM, that is, decreased as opposed to increased contraction. Further, the isoform of the single MLCK gene of Drosophila (Kojima et al. 1996) predominantly expressed in larval muscles lacks the CaM-binding region (Tohtong et al. 1995), suggesting that MLCK-induced phosphorylation has no role in these muscles. In mammals, phosphorylation of the Ca2+-ATPase of cardiac SR by CaM-kinase II (CaMKII) stimulates calcium re-uptake into the SR and is indicated to facilitate cardiac muscle relaxation (Xu et al. 1993; Xu and Narayanan 1999). Thus, a mechanism whereby defective regulation of this kinase could increase sarcoplasmic calcium levels, and hence contraction, is indicated. Further, previous studies have shown that holo-V91G D-CaM has a reduced ability to activate various enzyme isoforms from the single Drosophila CaMKII gene (GuptaRoy et al. 2000). However, the Drosophila Ca2+-ATPase lacks the phosphorylation site for CaMKII, suggesting lack of regulation by CaMKII (Magyar and Varadi 1990; Shi et al. 1998). Phosphorylase kinase (PhK), a prominent muscle CaM-regulated kinase, also seems unlikely as a source of defects in Cam7 animals, because lowered CaM activation would lead to decreased muscle ATP production and muscle failure.
Nevertheless, given that V91G D-CaM has been shown in vitro to affect a CaM target (CaMKII) with presumed roles in many tissues (GuptaRoy et al. 2000), it is relevant to ask why, in vivo, the effects of the mutation are so tissue-specific. Presumably the key issues here are (1) the extent to which the mutation affects a particular target, and (2) how critical that target is to the functioning of a given tissue. The limitation of the lethal effects of V91G D-CaM to muscle tissue must indicate (1) that the V91G mutation affects one or more targets that are essential to muscle function but that have negligible, or nonexistent, roles in other tissues, and (2) all other targets are minimally affected and/or are not critical to the functioning of any tissue.
Materials and methods
Protein expression and purification
Both WT and V91G D-CaM were cloned into the pET15b vector (Novagen), and cultures were prepared and induced as described (Mukherjea et al. 1996). CaM was purified by two rounds of affinity chromatography on phenyl Sepharose essentially as described (Maune et al. 1992a), except that initial cell lysis was performed using an air pressure disruptor (Emulsiflex-C5 by Avestin) at a pressure of 7000–10,000 psi. CaM protein concentrations were determined using the extinction coefficients previously determined for D-CaM (Maune et al. 1992a). Mutant Drosophila CaMs B1Q and B3Q were purified and quantitated as described (Maune et al. 1992a).
Peptide synthesis
Peptides were synthesized in the core facility at Baylor College of Medicine (Houston, TX) under the direction of Dr. Richard Cook. Peptides were characterized by HPLC and amino acid analysis.
Spectroscopic studies
UV-absorption spectra, near- and far-UV CD spectra, and tyrosine emission spectra for both WT and V91G D-CaM were generated and analyzed as described (Maune et al. 1992a,b; Masino et al. 2000). Tryptophan fluorescence emission spectra of M-RyR-Peptide were recorded in 25 mM Tris (pH 8.0), 100 mM KCl (plus 1 mM CaCl2 or 1 mM EGTA) using a SPEX FluoroMax fluorimeter with λex = 290 nm (bandwidth 1.7 nm) and emission scanned from 300 to 450 nm (bandwidth 5 nm). The stoichiometry for the interaction of M-RyR-Peptide with D-CaM was demonstrated to be 1:1 by fluorescence titrations performed at high concentration using solutions of CaM and peptide whose concentrations were established using known or calculated extinction coefficients. The dissociation constants were then determined by direct fluorometric titrations performed at lower concentrations using standard analysis methods (Martin and Bayley 2002), giving a value for Kd (M-RyR-Peptide) of 2.8 ± 0.3 nM (1 mM Ca2+, 100 mM KCl); 520 ± 70 nM (−Ca2+, 100 mM KCl), and 15.5 ± 2.1 nM (−Ca2+, 0 KCl). Dissociation constants for the Drosophila peptide were determined using competition titrations in which the tryptophan-containing M-RyR-Peptide (1.5 μM) was displaced from its complex with D-CaM (1.2 μM) by addition of D-RyR-Peptide, (Table 2, line 1): Alternatively, a competition titration of both M-RyR-peptide (0.3 μM) and D-RyR-Peptide (different concentrations in the range 2–10 μM) was performed with D-CaM (Table 2, lines 2–4).
Unfolding studies
Thermal unfolding was performed in 20 mM HEPES, 100 mM KCl (or 85 mM KCl, 5 mM MgCl2) at pH 8.0, as described (Masino et al. 2000). Urea-induced unfolding was performed in 25 mM Tris, 100 mM KCl (pH 8.0) at 10°C and in 25 mM Tris, 100 mM KCl, 0.3 mM CaCl2 (pH 8.0) at 20°C. Molar ellipticity (λɛM) at 220 nm was used to monitor loss of secondary structure during unfolding. Unfolding of the C-terminal domain was measured by monitoring changes in the environment of the single tyrosine of D-CaM (residue 138) using fluorescence at 306 nm after excitation at 280 nm, as above.
9-AC fluorescence titrations
Purified D-CaMs were dialyzed extensively against titration buffer (0.5 mM CaCl2, 100 mM KCl, and 10 mM MOPS [pH 7.0]) and 2-mL aliquots of titration buffer containing 5 μM D-CaM and 20 μM 9-AC were used for fluorescence measurements. Free [Ca2+] was progressively reduced by additions of 20 mM, 100 mM, 200 mM, or 1 M EGTA. Fluorescence was measured in an SLM 8100 fluorimeter, with excitation at 363 nm and emission measured at 480 nm and above. Free [Ca2+] was calculated using a program kindly donated by Dr. Susan Hamilton. For statistical analysis, data were fit to an empirical sigmoidal dose-response curve with variable slopes [Equation: Y = Min + (Max − Min)/1+10^((Log[Ca2+]50 − X) * Hill Slope)] using Prism software (GraphPad). Y = normalized fluorescence level at a given [Ca2+]free; X = log[Ca2+]free; Min = constant set to 1. Max (maximum fluorescence enhancement) and [Ca2+]50 (free [Ca2+] at 50% fluorescence change) were calculated by the program. p[Ca2+]50 and Max were determined for each individual experiment and then analyzed by unpaired t-tests using Statview software (version 4.51; Abacus Concepts).
Nondenaturing gel mobility shift assay
D-CaM binding to RyR-derived peptides was analyzed by a modification of the technique used by Rodney et al. (2001a). CaM and peptide were incubated together at room temperature for 30–60 min before loading.
Incubation solutions: (1) High [Ca2+] buffer: 50 mM Tris (pH 7.0), 12 μM CaM, 1 mM DTT, 200 μM CaCl2. Peptide:CaM ratios as described in figure legends. (2) Low [Ca2+] buffer: 50 mM Tris (pH 7.0), 7 μM CaM, <10 nM CaCl2, with peptide:CaM ratios as described in figure legends.
Sample loading buffer: 50 mM Tris (pH 7.0), 10% glycerol, 200 μM CaCl2 or 1 μM EGTA, 0.003% Bromophenol Blue.
Gel electrophoresis
Polyacrylamide gels contained 373 mM Tris (pH 8.5), 200 μM CaCl2 or 1 mM EGTA, 1% ammonium persulfate, and 16% acrylamide/bis (29:1). To induce polymerization, 0.05% (high [Ca2+] gels) or 0.1% TEMED was added. Tris-glycine electrophoresis running buffer contained 25 mM Tris (pH 8.3), 250 mM glycine, and 200 μM CaCl2 or 1 mM EGTA. All electrophoresis was at room temperature. Gels were run at a constant current of 11–12 mA for 2 h. Gels were fixed in 50% methanol, 7% acetic acid for 1 h, washed in distilled water for 1 h, stained with GelCode Blue (Pierce) for 2 h, washed in distilled water for 2 h, and stored in 20% glycerol at 4°C.
Equilibrium calcium titrations monitored by fluorescence
Calcium binding to D-CaM was monitored using calcium-induced changes in phenylalanine (Phe) and tyrosine (Tyr) fluorescence as described (VanScyoc et al. 2002). Phe residues were selectively monitored using λex of 250 nm, λem of 280 nm; the single Tyr residue (Y138) in the C-domain was monitored by λex of 277 nm, λem of 320 nm. Samples containing 6 μM D-CaM ± 7.5 μM D-RyR-Peptide in 50 mM HEPES, 100 mM KCl, 50 μM EGTA, 5 mM NTA (pH 7.4), 22°C, were titrated with concentrated CaCl2 solutions containing the same buffer components. The concentration of free calcium at each titration point was determined by monitoring the degree of calcium saturation of 0.1 μM Oregon Green 488 BAPTA-5N (λex of 494 nm, λem of 521 nm) or 1 μM difluoro-BAPTA (λex of 257 nm, λem of 369 nm) as described (VanScyoc and Shea 2001), using Kd values of 29.60 μM and 2.48 μM for calcium binding to Oregon Green and difluoro-BAPTA, respectively. At least three replicate titrations of WT and V91G D-CaM in the absence and presence of D-RyR-Peptide were performed, using an SLM 4800 Fluorimeter with a xenon arc lamp and 8-nm band passes. Representative titrations of each set are shown in Figures 3 ▶ and 6 ▶.
Analysis of calcium binding to CaM
Macroscopic equilibrium constants of calcium binding were determined by fitting the titration data to a model-independent two-site Adair function as shown in the equation below (VanScyoc et al. 2001).
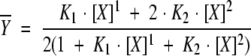 |
K1 is the equilibrium association constant for binding a single calcium ion to a domain. K2 is the equilibrium association constant for saturating both calcium-binding sites within a domain. Values for the average dissociation constants for calcium binding to wild-type and V91G D-CaM in the absence and presence of D-RyR-Peptide are shown in Table 1.
Equilibrium [35S] calmodulin binding to mammalian RyR channels
Procedures were largely based on published methods (Rodney et al. 2000). Sarcoplasmic reticulum (SR) membrane proteins (10 μg) were incubated with 5 nM [35S] mammalian CaM and increasing concentrations of WT or mutant D-CaM (0, 4.9, 9.8, 19.5, 39, 78, 156, 312, 625, 1250, and 2500 nM) for 2 h at room temperature in binding buffer (low [Ca2+] or high [Ca2+]). Unbound radioactive CaM was washed off by filtering through Whatman GF/F filters (presoaked in 0.3 mg/mL BSA in binding buffer) and washing with matched binding buffer. Bound [35S] CaM was quantitated by scintillation counting.
Binding buffer: (1) Low [Ca2+]: 300 mM NaCl, 50 mM MOPS (pH 7.4), 1 mM EGTA, 100 μg/mL BSA, 0.1% CHAPS; (2) High [Ca2+]: 300 mM NaCl, 1.2 mM CaCl2, 50 mM MOPS (pH 7.4), 1 mM EGTA, 100 μg/mL BSA, 0.1% CHAPS.
Washing buffer: As binding buffer without CHAPS.
Acknowledgments
We thank G.G. Rodney for helpful discussions and technical advice. This work was supported by the following fellowships and grants: a graduate fellowship to R.A.N. from the University of Iowa Center for Biocatalysis and Bioprocessing (NIH T32 GM08365-13); graduate support to B.W. from a NASA Specialized Center of Research and Training (NSCORT) in Gravitational Biology at Rice University; grant R01 GM57001 to M.A.S. from the NIH; grant C-1119 to K.B. from the Robert A. Welch Foundation; grant AR41802 to S.L.H. from the NIAMS; and funding to K.B. and P.M.B. from the Texas-UK Collaborative Initiative.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.04928204.
References
- Bayley, P.M., Findlay, W.A., and Martin, S.R. 1996. Target recognition by calmodulin: Dissecting the kinetics and affinity of interaction using short peptide sequences. Protein Sci. 5 1215–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckingham, K. 1991. Use of site-directed mutations in the individual Ca2(+)-binding sites of calmodulin to examine Ca2(+)-induced conformational changes. J. Biol. Chem. 266 6027–6030. [PubMed] [Google Scholar]
- Clapham, D.E. 1995. Calcium signaling. Cell 80 259–268. [DOI] [PubMed] [Google Scholar]
- Cohen, P. and Klee, C.B. 1988. Calmodulin: Molecular aspects of cellular regulation. Elsevier Press, New York.
- Cyert, M.S. 2001. Genetic analysis of calmodulin and its targets in Saccharomyces cerevisiae. Annu. Rev. Genet. 35 647–672. [DOI] [PubMed] [Google Scholar]
- Finn, B.E., Drakenberg, T., and Forsen, S. 1993. The structure of apo-calmodulin. A 1H NMR examination of the carboxy-terminal domain. FEBS Lett. 336 368–374. [DOI] [PubMed] [Google Scholar]
- GuptaRoy, B., Marwaha, N., Pla, M., Wang, Z., Nelson, H.B., Beckingham, K., and Griffith, L.C. 2000. Alternative splicing of Drosophila calcium/calmodulin-dependent protein kinase II regulates substrate specificity and activation. Brain Res. Mol. Brain Res. 80 26–34. [DOI] [PubMed] [Google Scholar]
- Johnson, J.D., Snyder, C., Walsh, M., and Flynn, M. 1996. Effects of myosin light chain kinase and peptides on Ca2+ exchange with the N- and C-terminal Ca2+ binding sites of calmodulin. J. Biol. Chem. 271 761–767. [DOI] [PubMed] [Google Scholar]
- Keller, C.H., Olwin, B.B., LaPorte, D.C., and Storm, D.R. 1982. Determination of the free-energy coupling for binding of calcium ions and troponin I to calmodulin. Biochemistry 21 156–162. [DOI] [PubMed] [Google Scholar]
- Kojima, S., Mishima, M., Mabuchi, I., and Hotta, Y. 1996. A single Drosophila melanogaster myosin light chain kinase gene produces multiple isoforms whose activities are differently regulated. Genes Cells 1 855–871. [DOI] [PubMed] [Google Scholar]
- Komeiji, Y., Ueno, Y., and Uebayasi, M. 2002. Molecular dynamics simulations revealed Ca(2+)-dependent conformational change of calmodulin. FEBS Lett. 521 133–139. [DOI] [PubMed] [Google Scholar]
- Kuboniwa, H., Tjandra, N., Grzesiek, S., Ren, H., Klee, C.B., and Bax, A. 1995. Solution structure of calcium-free calmodulin. Nat. Struct. Biol. 2 768–776. [DOI] [PubMed] [Google Scholar]
- Laporte, D.C., Wierman, B.M., and Storm, D.R. 1980. Calcium-induced exposure of a hydrophobic surface on calmodulin. Biochemistry 19 3814–3819. [DOI] [PubMed] [Google Scholar]
- Magyar, A. and Varadi, A. 1990. Molecular cloning and chromosomal localization of a sarco/endoplasmic reticulum-type Ca2(+)-ATPase of Drosophila melanogaster. Biochem. Biophy. Res. Commun. 173 872–877. [DOI] [PubMed] [Google Scholar]
- Martin, S.R. and Bayley, P.M. 2002. Regulatory implications of a novel mode of interaction of calmodulin with a double IQ-motif target sequence from murine dilute myosin, V. Protein Sci. 11 2909–2923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin, S.R., Bayley, P.M., Brown, S.E., Porumb, T., Zhang, M., and Ikura, M. 1996. Spectroscopic characterization of a high-affinity calmodulin-target peptide hybrid molecule. Biochemistry 35 3508–3517. [DOI] [PubMed] [Google Scholar]
- Masino, L., Martin, S.R., and Bayley, P.M. 2000. Ligand binding and thermodynamic stability of a multidomain protein, calmodulin. Protein Sci. 9 1519–1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maune, J.F., Klee, C.B., and Beckingham, K. 1992a. Ca2+ binding and conformational change in two series of point mutations to the individual Ca(2+)-binding sites of calmodulin. J. Biol. Chem. 267 5286–5295. [PubMed] [Google Scholar]
- Maune, J.F., Beckingham, K., Martin, S.R., and Bayley, P.M. 1992b. Circular dichroism studies on calcium binding to two series of Ca2+ binding site mutants of Drosophila melanogaster calmodulin. Biochemistry 31 7779–7786. [DOI] [PubMed] [Google Scholar]
- Mukherjea, P., Maune, J.F., and Beckingham, K. 1996. Interlobe communication in multiple calcium-binding site mutants of Drosophila calmodulin. Protein Sci. 5 468–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson, H.B., Heiman, R.G., Bolduc, C., Kovalick, G.E., Whitley, P., Stern, M., and Beckingham, K. 1997. Calmodulin point mutations affect Drosophila development and behavior. Genetics 147 1783–1798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson, B.Z., DeMaria, C.D., Adelman, J.P., and Yue, D.T. 1999. Calmodulin is the Ca2+ sensor for Ca2+-dependent inactivation of L-type calcium channels. Neuron 22 549–558. [DOI] [PubMed] [Google Scholar]
- Putney Jr., J.W. 1998. Calcium signaling: Up, down, up, down, what’s the point? Science 279 191–192. [DOI] [PubMed] [Google Scholar]
- Rodney, G.G., Williams, B.Y., Strasburg, G.M., Beckingham, K., and Hamilton, S.L. 2000. Regulation of RYR1 activity by Ca(2+) and calmodulin. Biochemistry 39 7807–7812. [DOI] [PubMed] [Google Scholar]
- Rodney, G.G., Moore, C.P., Williams, B.Y., Zhang, J.Z., Krol, J., Pedersen, S.E., and Hamilton, S.L. 2001a. Calcium binding to calmodulin leads to an N-terminal shift in its binding site on the ryanodine receptor. J. Biol. Chem. 276 2069–2074. [DOI] [PubMed] [Google Scholar]
- Rodney, G.G., Krol, J., Williams, B., Beckingham, K., and Hamilton, S.L. 2001b. The carboxy-terminal calcium binding sites of calmodulin control calmodulin’s switch from an activator to an inhibitor of RYR1. Biochemistry 40 12430–12435. [DOI] [PubMed] [Google Scholar]
- Saimi, Y. and Kung, C. 2002. Calmodulin as an ion channel subunit. Ann. Rev. Physiol. 64 289–311. [DOI] [PubMed] [Google Scholar]
- Schulman, H. and Braun, A. 1999. Calcium/calmodulin dependent kinases. In Calcium as cellular regulator (eds. E. Carafoli and C. Klee), pp. 311–343. Oxford University Press, Oxford.
- Shi, X., Chen, M., Huvos, P.E., and Hardwicke, P.M. 1998. Amino acid sequence of a Ca(2+)-transporting ATPase from the sarcoplasmic reticulum of the cross-striated part of the adductor muscle of the deep sea scallop: Comparison to serca enzymes of other animals. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 120 359–374. [DOI] [PubMed] [Google Scholar]
- Sobolev, V., Sorokine, A., Prilusky, J., Abola, E.E., and Edelman, M. 1999. Automated analysis of interatomic contacts in proteins. Bioinformatics 15 327–332. [DOI] [PubMed] [Google Scholar]
- Takeshima, H., Nishi, M., Iwabe, N., Miyata, T., Hosoya, T., Masai, I., and Hotta, Y. 1994. Isolation and characterization of a gene for a ryanodine receptor/calcium release channel in Drosophila melanogaster. FEBS Lett. 337 81–87. [DOI] [PubMed] [Google Scholar]
- Taylor, D.A., Sack, J.S., Maune, J.F., Beckingham, K., and Quiocho, F.A. 1991. Structure of a recombinant calmodulin from Drosophila melanogaster refined at 2.2-Å resolution. J. Biol. Chem. 266 21375–21380. [DOI] [PubMed] [Google Scholar]
- Tohtong, R., Yamashita, H., Graham, M., Haeberle, J., Simcox, A., and Maughan, D. 1995. Impairment of muscle function caused by mutations of phosphorylation sites in myosin regulatory light chain. Nature 374 650–653. [DOI] [PubMed] [Google Scholar]
- Tripathy, A., Xu, L., Mann, G., and Meissner, G. 1995. Calmodulin activation and inhibition of skeletal muscle Ca2+ release channel (ryanodine receptor). Biophys. J. 69 106–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Eldik, L. and Watterson, D.M. 1998. Calmodulin and signal transduction. Academic Press, San Diego, CA.
- VanScyoc, W.S. and Shea, M.A. 2001. Phenylalanine fluorescence studies of calcium binding to N-domain fragments of Paramecium calmodulin mutants show increased calcium affinity correlates with increased disorder. Protein Sci. 10 1758–1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanScyoc, W.S., Sorensen, B.R., Rusinova, E., Laws, W.R., Ross, J.B., and Shea, M.A. 2002. Calcium binding to calmodulin mutants monitored by domain-specific intrinsic phenylalanine and tyrosine fluorescence. Biophys. J. 83 2767–2780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, B., Sullivan, K.M.C., and Beckingham, K. 2003. Drosophila calmodulin mutants with specific defects in the musculature or in the nervous system. Genetics 165 1255–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong, L.W., Newman, R.A., Rodney, G.G., Thomas, O., Zhang, J.Z., Persechini, A., Shea, M.A., and Hamilton, S.L. 2002. Lobe-dependent regulation of ryanodine receptor type 1 by calmodulin. J. Biol. Chem. 277 40862–40870. [DOI] [PubMed] [Google Scholar]
- Xu, A. and Narayanan, N. 1999. Ca2+/calmodulin-dependent phosphorylation of the Ca2+-ATPase, uncoupled from phospholamban, stimulates Ca2+-pumping in native cardiac sarcoplasmic reticulum. Biochem. Biophys. Res. Commun. 258 66–72. [DOI] [PubMed] [Google Scholar]
- Xu, A., Hawkins, C., and Narayanan, N. 1993. Phosphorylation and activation of the Ca(2+)-pumping ATPase of cardiac sarcoplasmic reticulum by Ca2+/calmodulin-dependent protein kinase. J. Biol. Chem. 268 8394–8397. [PubMed] [Google Scholar]
- Zdarek, J. and Fraenkel, G. 1972. The mechanism of puparium formation in flies. J. Exp. Zool. 179 315–323. [Google Scholar]
- Zhang, M., Tanaka, T., and Ikura, M. 1995. Calcium-induced conformational transition revealed by the solution structure of apo calmodulin. Nat. Struct. Biol. 2 758–767. [DOI] [PubMed] [Google Scholar]
- Zhu, T., Beckingham, K., and Ikebe, M. 1998. High affinity Ca2+ binding sites of calmodulin are critical for the regulation of myosin I β motor function. J. Biol. Chem. 273 20481–20486. [DOI] [PubMed] [Google Scholar]
- Zuhlke, R.D., Pitt, G.S., Deisseroth, K., Tsien, R.W., and Reuter, H. 1999. Calmodulin supports both inactivation and facilitation of L-type calcium channels. Nature 399 159–162. [DOI] [PubMed] [Google Scholar]