

Abstract
NAD+-dependent glycerol-3-phosphate dehydrogenase (G3PDH) is generally absent in archaea, because archaea, unlike eukaryotes and eubacteria, utilize glycerol-1-phosphate instead of glycerol-3-phosphate for the biosynthesis of membrane lipids. Surprisingly, the genome of the hyperthermophilic archaeon Archaeoglobus fulgidus comprises a G3PDH ortholog, gpsA, most likely due to horizontal gene transfer from a eubacterial organism. Biochemical characterization proved G3PDH-like activity of the recombinant gpsA gene product. However, unlike other G3PDHs, the up to 85°C thermostable A. fulgidus G3PDH exerted a 15-fold preference for NADPH over NADH. The A. fulgidus G3PDH bears the hallmarks of adaptation to halotolerance and thermophilicity, because its 1.7-Å crystal structure showed a high surface density for negative charges and 10 additional intramolecular salt bridges compared to a mesophilic G3PDH structure. Whereas all amino acid residues required for dihydroxyacetone phosphate binding and reductive catalysis are highly conserved, the binding site for the adenine moiety of the NAD(P) cosubstrate shows a structural variation that reflects the observed NADPH preference, for example, by a putative salt bridge between R49 and the 2′-phosphate.
Keywords: glycerol-3-phosphate dehydrogenase, crystal structure, diglycerol phosphate, Archaeoglobus fulgidus, Leishmania mexicana, thermophilic enzymes
Two different classes of enzymes, which are not phylogenetically related, are known that catalyze the reversible reduction of dihydroxyacetone phosphate (DHAP) with NAD(P)H. These enzymes are NAD+-dependent glycerol-3-phosphate dehydrogenases (G3PDH) and NAD(P)+-dependent glycerol-1-phosphate dehydrogenases (G1PDH). The product of the reaction catalyzed by the former enzyme is glycerol-3-phosphate (G3P) and that of the latter enzyme is the enantiomeric glycerol-1-phosphate (G1P) (Koga et al. 1998; Nishihara et al. 1999).
So far, G3PDH is mainly found in eubacteria and eukaryotes. The cytoplasmic enzyme is composed of only one type of subunit of molecular mass near 40 kDa and lacks a prosthetic group. All G3PDHs analyzed until now are strictly NAD+ dependent or show preference for NADH over NADPH in DHAP reduction (Schomburg et al. 1995). The physiological function of G3PDHs in bacterial and eukaryotic cells is mainly the formation of G3P for the synthesis of phospholipids, which in bacteria and eukaryotes have a G3P backbone. In yeast, mammals, and plants, this enzyme additionally performs a crucial role in the shuttling of reducing equivalents from the cytosol into mitochondria (Shen et al. 2003). In trypanosomes, eukaryotic parasites, G3PDH is located in a specialized organelle, the glycosome, in which glycolysis takes place. The bloodstream form of trypanosomes is totally dependent on glycolysis for energy metabolism. In this situation, G3PDH is especially important, because it is directly responsible for maintaining the NAD+/NADH balance in the glycosome by the reoxidation of NADH produced by glyceraldehyde-3-phosphate dehydrogenase (GAPDH) during glycolysis (Suresh et al. 2000). Because of its importance, the glycosomal enzyme has been extensively studied and its crystal structure was recently solved (Kohl et al. 1996; Marche et al. 2000; Suresh et al. 2000; Choe et al. 2003).
G1PDH is only found in archaea. It is composed of a single type of subunit with a molecular mass of 38 kDa and also lacks a prosthetic group (Nishihara and Koga 1995, 1997; Noguchi et al. 1998). Its function is to catalyze the formation of G1P for the synthesis of the membrane lipids, which in archaea have a G1P backbone rather than one derived from G3P (Morii et al. 2000). The enzyme shows significant sequence similarity to glycerol dehydrogenase from eubacteria, archaea, and eukaryotes, for which several crystal structures are known (Ruzheinikov et al. 2001; Daiyasu et al. 2002; Brinen et al. 2003; de Vries et al. 2003).
Archaeoglobus fulgidus is a hyperthermophilic archaeon with a growth temperature optimum of 83°C (Stetter et al. 1987; Stetter 1988). It thrives best on lactate and sulfate as energy sources. Under salt stress the organism synthesizes diglycerol phosphate as compatible solute most probably with G3P as a precursor (Martins et al. 1997; Lamosa et al. 2000; Goncalves et al. 2003). The sequenced genome of A. fulgidus harbors a gene gpsA (AF0871) putatively encoding for G3PDH and a gene egsA (AF1674) for a G1PDH (Klenk et al. 1997). Whereas egsA gene orthologs are present in all archaeal genomes sequenced so far, a gpsA gene was only found in the genomes of A. fulgidus, Methanothermobacter thermautotrophicus, and Aeropyrum pernix (Smith et al. 1997; Kawarabayasi et al. 1999). The putative gpsA gene in the genome of A. pernix is predicted not to encode for a G3PDH, because the deduced primary structure lacks the characteristic substrate binding site. The gpsA gene product from A. fulgidus shares less than 35% sequence identity to the other G3PDHs. Therefore, it had first to be proven that the gpsA gene in A. fulgidus indeed encodes for a functional G3PDH.
Here, we report the heterologous overexpression of the gpsA gene from A. fulgidus in Escherichia coli. The overproduced enzyme was purified, biochemically characterized, and crystallized. The 1.7-Å crystal structure of this archaeal, NADP+-dependent G3PDH was finally compared with the only other known G3PDH structure, namely, the glycosomal NAD+-dependent G3PDH from the eukaryote Leishmania mexicana.
Results and Discussion
Purification of heterologously produced G3PDH from A. fulgidus
E. coli strain Rosetta (DE3) transformed with the expression vector pEGPD1 carrying gpsA from A. fulgidus exhibited high and heat-stable G3PDH activity (Table 1). After a heat treatment step (70°C for 45 min) the enzyme was purified to homogeneity by chromatography on Resource Q, hydroxyapatite, and Source 15 Phe columns. About 10 mg of the purified enzyme were obtained from a 2-L culture. SDS-PAGE analysis revealed the presence of a polypeptide of 36 kDa apparent molecular mass (Fig. 1 ▶). The N-terminal amino acid sequence was found to be identical to that deduced from the gpsA gene of A. fulgidus. The molecular mass of the gpsA gene product is predicted to have a molecular mass of 36,784 Da and an isoelectric point at pH 5.0.
Table 1.
Purification of NADP+-dependent G3PDH from A. fulgidus heterologously produced in E. coli
| Step | Protein (mg) | Total activity (U) | Specific activity (U/mg) | Yield (%) |
| Cell extract | 730 | 1000 | 1.4 | 100 |
| Heat treatment | 91 | 980 | 11 | 98 |
| Resource Q | 58 | 820 | 14 | 82 |
| Hydroxyapatite | 23 | 570 | 25 | 57 |
| Source 15 Phe | 9.9 | 300 | 30 | 30 |
The cell extract was prepared from 3.2 g (wet mass) cells. One unit was defined as 1 μmole NADPH oxidation per minute with dihydroxyacetone phosphate at pH 6.6 and 70°C.
Figure 1.
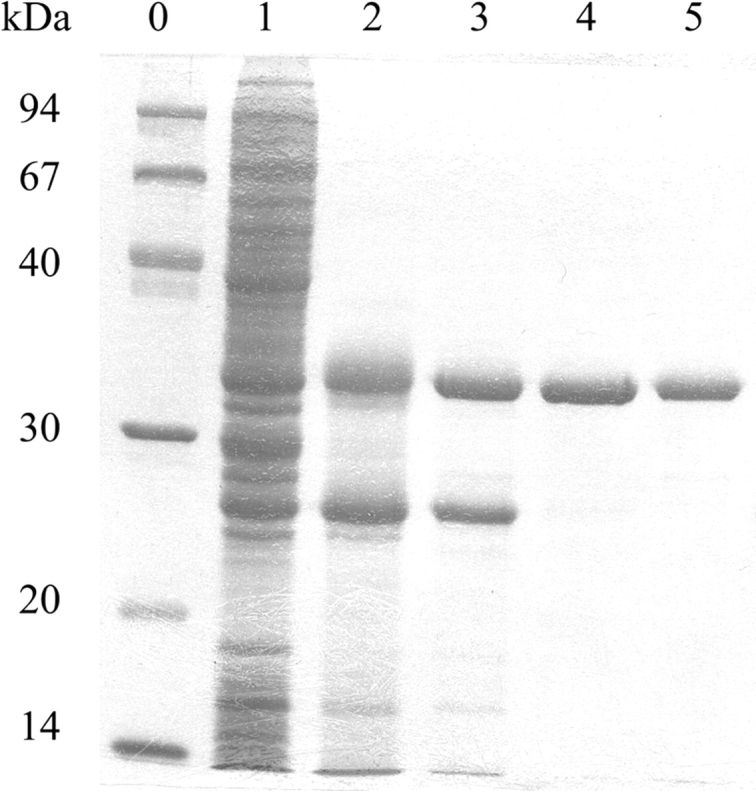
SDS-PAGE analysis of NADP+-dependent G3PDH from A. fulgidus heterologously produced in E. coli. About 5 μg protein were loaded on a 12% gel. After electrophoresis, the gels were stained with Coomassie Blue R250. (Lane 0) molecular mass standard; (lane 1) cell extract; (lane 2) supernatant after heat treatment; (lane 3) G3PDH fraction after chromatography on Resource Q; (lane 4) G3PDH fraction after chromatography on Hydroxyapatite; (lane 5) G3PDH fraction after chromatography on Source 15 Phe.
Kinetic properties
The purified G3PDH from A. fulgidus was 15 times more active with NADPH than with NADH in catalyzing the reduction of DHAP. Unlike bacterial and eukaryotic G3PDH, which are strictly NADH dependent or prefer NADH over NADPH, the archaeal G3PDH exhibits a strong preference for NADPH. In the energy metabolism of A. fulgidus, all redox reactions involve F420 or ferredoxin rather than NAD+ and NADP+ as electron acceptor/donor (Kunow et al. 1993). A. fulgidus contains an active F420H2:NADP+ oxidoreductase, which catalyzes the regeneration of NADPH for biosynthetic purposes (Kunow et al. 1993; Warkentin et al. 2001). However, an enzyme catalyzing hydride transfer from F420H2 to NAD+ appears to be absent in A. fulgidus. Therefore, the preference of G3PDH from A. fulgidus for NADPH appears to be physiologically relevant.
Initial velocities of DHAP reduction with NADPH at pH 6.6 and 70°C were determined at several fixed levels of NADPH and variable concentrations of DHAP. Double reciprocal plots of the initial velocities versus the DHAP concentrations gave straight lines that intersected in one point on the abscissa to the left of the ordinate. A similar pattern was obtained when the initial velocities were plotted versus the NADPH concentrations. From the intercepts on the ordinate apparent Vmax values were obtained. Double reciprocal plots of the apparent Vmax values versus the corresponding NADPH and DHAP concentrations yielded straight lines that intersect in one point on the Y-axis. From the intercepts on the abscissa and the intercept on the ordinate the KM and Vmax were calculated. The KM and Vmax values for G3P oxidation with NADP+ were determined in the same manner. The KM and Vmax values obtained are summarized in Table 2.
Table 2.
Kinetic properties of NADP+-dependent G3PDH from A. fulgidus
| A. fulgidus G3PDH | |
| DHAP reduction | |
| Vmax with NADPH (μmol/min/mg) | 44 |
| Km DHAP (mM) | 1 |
| Km NADPH (mM) | 0.04 |
| Ki L-α-G3P (mM)a | 17 |
| Ki NADP+ (mM)b | 0.005 |
| V with NADPH (μmol/min/mg)c | 30 |
| V with NADH (μmol/min/mg)c | 2 |
| L-α-G3P oxidation | |
| Vmax with NADP+ (μmol/min/mg) | 4 |
| Km L-α-G3P (mM) | 0.1 |
| Km NADP+ (mM) | 0.8 |
| Ki DHAP (mM)d | 0.1 |
| V with NADP+ (μmol/min/mg)c | 3 |
| V with NAD+ (μmol/min/mg)c | 4 |
Michaelis constants were determined by two-substrate kinetic analysis from the initial rates at pH 6.6 and 70°C. The substrate and cofactor concentrations ranged from 0.1 × Km to 10 × Km. Ki values were calculated from Dixon plots. The concentrations of substrates and cofactors used were:
a 0–15 mM of glycerol-3-phosphate (G3P) at several concentrations of dihydroxyacetone phosphate (DHAP) (0.20–0.75 mM) and 0.10 mM of NADPH.
b 0–100 μM of NADP+ at several concentrations of NADPH (20–50 μM) and 2.0 mM DHAP.
c Specific activity under standard assay conditions.
d 0–0.50 mM of DHAP at several concentrations of G3P (0.5–2.0 mM) and 2.0 mM NADP+.
The formation of NADPH coupled to G3P oxidation at pH 9.0 could only be detected spectrofluorometrically (excitation at 340 nm, emission at 460 nm). Despite the relatively high concentration of the substrates NADP+ (5 mM) and G3P (2 mM), the scavenging of DHAP by hydrazine and the low proton concentration (10−9 M) in the assay, the concentrations of NADPH produced were nevertheless too low to be measured by the increase in absorbance at 340 nm. This might be the result of product inhibition. The Ki for NADPH was, however, too low to be determined (<5 μM). G3PDH from A. fulgidus is thus clearly committed to catalyze G3P formation rather than G3P oxidation. This is another example that NADP+ is confined, with few exceptions, to reactions of reductive biosynthesis (Carugo and Argos 1997).
Purified G3PDH from A. fulgidus showed no activity with dihydroxyacetone, glycerol, glycerol-2-phosphate, D-glyceraldehyde-3-phosphate, DL-glyceraldehyde, D-ery-throse-4-phosphate, D-fructose-6-phosphate, β-D-glucose-6-phosphate, or α-D-galactose-1-phosphate.
Temperature and pH optima
The activity of DHAP reduction with NADPH increased up to a temperature of approximately 70°C from where it again decreased (Fig. 2A ▶). The Arrhenius plot (Fig. 2B ▶) showed two-phase linearity having a transition point around 50°C, which is generally interpreted as being due to conformational flexibility changes. The pH optimum at 70°C for DHAP reduction was below 5.0 and that for G3P oxidation above pH 9.5. The temperature and pH optima probably in part reflect the instability of the substrates rather than the activity of the enzyme under the experimental conditions.
Figure 2.

Effect of temperature on NADP+-dependent G3PDH activity of A. fulgidus. (A) Activity vs. temperature (°C). (B) Arrhenius plot of the same data. The activity was determined by following NADPH oxidation with dihydroxyacetone phosphate at pH 6.6 in 50 mM potassium phosphate buffer at the temperatures indicated. From the slopes of the linear parts in the Arrhenius plot an activation energy of 51 kJ/mol was calculated for the temperature range between 50°C and 75°C and of 22 kJ/mol for the temperature range between 30°C and 45°C.
Thermostability
In 200 mM potassium phosphate (pH 7.0) or 100 mM NaCl (not shown) or 50 mM ammonium sulfate (pH 7.0) the G3PDH from A. fulgidus was completely stable for up to 30 min at a temperature of 85°C (Fig. 3 ▶). At lower salt concentrations the enzyme was less thermostable (shown for potassium phosphate in Fig. 3 ▶) but more stable than any other G3PDH reported so far. For example, desalted E. coli G3PDH loses 50% of its activity in 4 h even at 0°C (Schryvers and Weiner 1981) and the enzyme from Ceratitis capitata is completely inactivated by incubation at 60°C for 5 min (Fernández-Sousa et al. 1977).
Figure 3.

Effect of salts on the thermostability of NADP+-dependent G3PDH from A. fulgidus. (○) 5 mM K2HPO4, (▵) 50 mM K2HPO4, (□) 200 mM K2HPO4, (•) 50 mM (NH4)2SO4. The pH was 7.0. Aliquots of 0.1 mg enzyme/mL were incubated in sealed tubes for 30 min at the indicated temperatures. After incubation, all tubes were rapidly cooled in an ice bath and analyzed for activity by following NADPH oxidation with dihydroxy-acetone phosphate under standard assay conditions.
Crystal structure
The gpsA gene product from A. fulgidus has a calculated isoelectric point of pH 5.0 and shares only 32% sequence identity to the structurally characterized glycosomal G3PDH from L. mexicana. Like many other enzymes from the trypanosomal glycosome, the L. mexicana G3PDH exhibits a high isoelectric point of 8.7 (Kohl et al. 1996; Suresh et al. 2000). Thus, the overall difference for the charge distribution in these two G3PDHs was predicted to be very large. The structure of the A. fulgidus enzyme was solved by multiple isomorphous replacement (MIR) and finally refined at 1.7 Å resolution.
The electron density defined G3PDH from A. fulgidus as a homodimer, which has a size of about 88 Å × 49 Å × 47 Å. The monomers of the dimer are linked by a twofold noncrystallographic axis (Fig. 4A ▶). Each monomer is composed of 335 amino acid residues and can be subdivided into two domains, an N-terminal dinucleotide-binding domain with a typical Rossmann fold (1–180)(Rossmann et al. 1974) and a C-terminal, helical domain (181–335) (Fig. 4B ▶). The interface between the monomers is predominantly hydrophobic (73% nonpolar and 27% polar residues) and buries 12% of the surface (1703 Å2) of each monomer. Along the protein surface four sulfate anions were found to contact each monomer. Three sulfate-binding sites are commonly occupied in both monomers. The occupation of the remaining sulfate-binding sites is apparently affected by crystal packing restraints, so that one can postulate up to five sulfate-binding sites per undisturbed monomer. The glycerole molecule that is bound by each monomer is not located in the active site, but along a cleft on the protein surface surrounded by mostly basic residues (Arg 203, Lys 217, Arg 256).
Figure 4.

Structure of NADP+-dependent G3PDH from A. fulgidus. (A) Ribbon diagram of the G3PDH dimer when viewed perpendicular to the twofold noncrystallographic axis. One monomer is colored in green, one in blue. The N-terminal dinucleotide binding domains are shown in light green and in light blue; the C-terminal helix domains are shown in dark green and dark blue. The figure was produced with MOLSCRIPT (Kraulis 1991) and Raster3D (Merritt and Murphy 1994). (B) Fold topology diagram of the G3PDH monomer.
Structural comparison with Leishmania G3PDH
Despite the large difference in isoelectric points and the relatively low sequence identity (Fig. 5 ▶; Suresh et al. 2000) the quaternary structures of the archaeal and trypanosomal G3PDHs resemble each other, as the superposition of the G3PDH dimers from A. fulgidus and L. mexicana resulted in an root mean square deviation of 2.0 Å for 346 common Cα positions (Fig. 6 ▶). This deviation is mainly due to a 9° rotational offset between the monomers of the two dimers (not shown). The rotation axis is arranged almost perpendicularly (94°) to the dimer axis. Despite this large structural similarity, a comparison of the dimer interfaces of G3PDH from A. fulgidus and L. mexicana revealed that the interface residues exhibited less than 13% sequence identity (5 of 39 residues).
Figure 5.

Alignment of the primary structures of G3PDH from A. fulgidus and L. mexicana. The residues, which are involved in substrate binding including the dinucleotide binding site GXGXXG (residues 7,9,12), are depicted in red. The conserved substrate binding sites are highlighted in green. The secondary structure assignment of G3PDH from A. fulgidus is shown above the sequence alignment.
Figure 6.

Superposition (A) of the NADP+-dependent G3PDH monomer from A. fulgidus (blue) and of the G3PDH monomer from L. mexicana (green). The overall r.m.s.d. value between these models was 2.0 Å for 314 structurally equivalent residues. The loop between the indicated α-helices α14 and α15 is visible in the structure of G3PDH from A. fulgidus but is disordered in the structure of G3PDH from L. mexicana, the only other structurally solved G3PDH (Suresh et al. 2000), if the substrate binding site is not fully occupied. The substrates NADPH and dihydroxyacetone phosphate were modeled into the structure of the A. fulgidus G3PDH at the corresponding binding sites known from the L. mexicana G3PDH structure (Choe et al. 2003). The structures of the L. mexicana enzyme and of the enzyme substrate complex were taken from the RCSB Protein Data Bank (accession codes 1N1E and 1EVY). (B) Active site region of A. fulgidus G3PDH showing the predicted binding modes of the substrates glycerol-3-phosphate and NADP+. The substrate molecules were modeled into the structure using the SCULPT option of the program PyMOL, which permits interactive energy minimization.
The most significant difference in tertiary structure appears to be in the loop segment connecting helix α14 and α15, which was visible as a highly flexible region in the structure of the A. fulgidus enzyme and not resolvable in the structure of the substrate-free L. mexicana G3PDH (Suresh et al. 2000). In the latter enzyme it was reported that this loop segment became ordered only upon substrate binding (Choe et al. 2003).
Another significant difference between the structures of the archaeal and eukaryotic G3PDHs is the distribution of surface charges. A comparison of the surface potential maps reveals that G3PDH from A. fulgidus has more negative groups on its surface than the enzyme from L. mexicana (Fig. 7 ▶) reflecting that the A. fulgidus enzyme has its isoelectic point at pH 5.0 and the L. mexicana enzyme has its one at pH 8.7. The surface of the two enzymes slightly differs also in the percentage of nonpolar groups, which is 52% for the A. fulgidus G3PDH and 58% for the L. mexicana enzyme. The average value for soluble proteins is 57% (Miller et al. 1987). The predominantly negatively charged hydrophilic surface of the archaeoglobal G3PDH is known as a general hallmark of halophilic proteins (Madern et al. 2000; Mamat et al. 2002). Correspondingly, the observed salt-dependent thermostability of the G3PDH from A. fulgidus (see above), that is also reported for other enzymes purified from this archaeon, is also a characteristic of halophilic enzymes (Mamat et al. 2002).
Figure 7.

Surface charge comparison of NAD+-dependent G3PDH from L. mexicana (A) and NADP+ dependent G3PDH from A. fulgidus (B). The surface potential maps were calculated in the presence of 100 mM salt. This figure was prepared with GRASP (Nicholls et al. 1993).
The G3PDH from A. fulgidus harbors 10 additional intramolecular salt bridges compared to the L. mexicana enzyme (39 vs. 29, or 0.12 vs. 0.08 ion pairs per residue). The differences in surface charge, in surface polarity, and in number of intramolecular ion pairs probably reflect the adaptation of the archaeal and trypanosomal enzymes to different growth temperatures, 83°C for the A. fulgidus enzyme and 37°C for L. mexicana. Such a contribution of salt bridges to thermostability has been extensively discussed before (Yip et al. 1995; Sterner and Liebl 2001; Hagemeier et al. 2003).
In the crystal structure of L. mexicana G3PDH, which was heterologously produced in E. coli, additional electron density was found at the hydrophobic pocket created by side chains from the C-terminal domain of one monomer and the N-terminal domain of the other dimer subunit (Suresh et al. 2000). In the crystal structure of the A. fulgidus G3PDH such extra electron density was not found. In this regard it is of importance that the primary structure in this region is not conserved between the two enzymes.
Active site structure
The ribbon diagram in Figure 6 ▶ shows that the two domains of the monomer form a cleft. Complexes between G3PDH of A. fulgidus and its substrates could not be structurally analyzed so far, although several crystals were soaked with different substrate combinations and concentrations. However, in no case was substrate binding in the active site observed after X-ray analysis, which might be caused either by the rather high KM values of the substrates or by a requirement of slight domain movement upon substrate binding. From the recent structure of a complex between L. mexicana G3PDH and a bisubstrate adduct, it is known that both substrates, NAD+ (the enzyme is NAD(H) specific) and glycerol-3-phosphate, bind within the cleft between the two domains (Choe et al. 2003). The superposition of the two structures revealed that the substrate-binding sites in both G3PDH are well conserved, so that it was easily possible to model DHAP and NADPH into the structure of the A. fulgidus G3PDH (Fig. 6 ▶). Despite this fact the following conclusions remain largely speculative.
The highest sequence and structural conservation is found around the binding site for the substrate glycerole-3-phosphate. Here, the strictly conserved residues Lys 105, Lys 192, and Asp 250 (L. mexicana G3PDH: Lys 125, Lys 210, and Asp 263; see also sequence alignment in Fig. 5 ▶) are in H-bonding distance to the 2-hydroxy group of G3P. One of these residues deprotonates the 2-hydroxy group during the reaction course for promoting hydride transfer from the C2 atom onto the C4 carbon of the nicotinamide ring. Likewise, the binding site around the 3-phosphate is highly preserved with the H-bonding residues Asn 193, Thr 254, and Asn 260 (L. mexicana G3PDH: Asn 211, Thr 267, and Asn 275). Interestingly, one of the two sulfates that are bound to the active site region is closely positioned to the expected binding site of the 3-phosphate, whereas the second sulfate forms two salt bridges with the side chains of Lys 105 and Lys 192. Korndörfer et al. (1995) similarly reported that in the crystal structure of GAPDH from Thermotoga maritima, which was also crystallized at high ammonium sulfate concentrations, two sulfate anions were bound within the active site. One of these sulfate anions was likewise proposed to be bound at the substrate’s C-3 phosphate binding site (Korndörfer et al. 1995). Hence, competition between sulfate and substrate binding might be the reason why the A. fulgidus G3PDH is inhibited in the presence of ammonium sulfate (data not shown).
Concerning the NAD(P)+-binding site, the glycine-rich loop with the recognition sequence GXGXXG between the secondary strucure elements β1 and α1 is highly conserved. Some conservative changes are found around the nicotin-amide binding site, for example, Met 11 makes planar interactions with the ring plane of the nicotinamide (L. mexicana: Phe 26). The unusual specificity of G3PDH from A. fulgidus for NADPH rather than NADH can now be structurally explained by a predicted ionic interaction between the 2′-phosphate of NADPH and the side chain of Arg 49 (Fig. 6B ▶). Arg 49 is also suitably located to form simultaneously a salt bridge with the 5′-phosphate of the adenosine group of NADP+. As expected for a residue determining the NADP+ preference of the A. fulgidus enzyme, Arg 49 is not conserved among eubacterial and eukaryotic G3PDH enzymes. For example, the NAD+-dependent G3PDH from L. mexicana contains a phenylalanine at this position (Phe 63) that makes some hydrophobic interactions with the ribose of the adenosine group in NADP+. The introduction of a positive charge interacting with the 2′-phosphate of NADP+ has been reported before for other NADP+-dependent enzymes (Charron et al. 2000; Ermler et al. 2002). For example, in NADP+-dependent GAPDH from Methanothermus fervidus cocrystallized with NADP+, it was shown that the 2′-phosphate of the adenosine moiety of NADP+ is likewise stabilized by a tight electrostatic interaction with the positively charged side chain of lysine (Charron et al. 2000). Almost no structural conservation is observed at the most distal site of the NAD(P)+ binding site, where the adenine moiety is bound. Here, Phe 33 forms π-stackering interactions with the adenine, whereas in the L. mexicana enzyme these interactions are made from the other side of the adenine by a phenylalanine that is replaced by Gly 83 in the archaeal G3PDH.
Toward a physiological function
The most important physiological function of G3PDH in bacterial and eukaryotic cells is the formation of the G3P backbone for the synthesis of the phospholipid membrane. Archaeal cells do not require this activity for membrane biosynthesis because their phospholipids have a G1P backbone (Koga et al. 1998). Then, what could be the physiological role of archaeoglobal G3PDHs?
Nishihara et al. (1999) suggested that G3PDH of A. fulgidus is involved in a pathway of glycerol metabolism leading to G1P. Accordingly, exogenously supplied glycerol is taken up and phosphorylated by ATP to form G3P, which is converted to DHAP by G3PDH. Then DHAP is subsequently converted to G1P by G1PDH. Genes putatively encoding a glycerol kinase (AF0866) and a glycerol uptake facilitator (AF1426) were found in the genome of A. fulgidus (Klenk et al. 1997). However, considering the kinetically favored direction and the equilibrium of the reaction catalyzed by G3PDH, this enzyme appears not to be suitable for the formation of DHAP from G3P.
 |
As Nishihara et al. have already pointed out, a flavin-dependent G3PDH encoded by another gene (AF1328) might be a better candidate for the production of DHAP from G3P in A. fulgidus under glycerol-assimilating conditions. The flavin-dependent G3PDH catalyzes the oxidation of G3P to DHAP (Eo′ = −190 mV) with menaquinone (Eo′= −75 mV) or ubiquinone (Eo′= +113 mV) as electron acceptors, which are exergonic rather than endergonic reactions. For the thermodynamic reasons mentioned above, also a function of the NADP+-dependent G3PDH of A. fulgidus in the regeneration of NADPH and therefore in redox control can be excluded.
Under salt stress conditions A. fulgidus synthesizes di-glycerol phosphate as the principal compatible solute in the cells (Martins et al. 1997). Although the stereoconfiguration of diglycerol phosphate and its biosynthesis pathway have not been reported yet, the compatible solute is most probably synthesized from G3P (Lamosa et al. 2000). G3P is proposed to be generated from DHAP via G3PDH, which in turn would be generated from glyceraldehyde-3-phosphate by isomerization. A putative triose phosphate isomerase gene (AF1304) is present in the genome of A. fulgidus (Klenk et al. 1997). If G3PDH is involved in this stress response mechanism in A. fulgidus, the G3PDH gene should be inducible rather than constitutive. It might be important to note that A. fulgidus cell extracts obtained under standard culture conditions did not exhibit any NADP+-dependent G3PDH activity (data not shown).
Pyridine nucleotide dependent G3PDH genes were found, until now, only in the genomes of A. fulgidus and M. thermautotrophicus and not in the other genomes of archaea already sequenced (http://www.ncbi.nlm.nih.gov/genomes/static/a.html). Sequence comparison reveals that the G3PDH from A. fulgidus is most similar to the enzyme from Bacillus subtilis followed by other eubacterial G3PDHs. The lack of the gpsA gene in most archaea suggests that this gene was aquired by A. fulgidus via lateral gene transfer, although it also has to be considered that most archaea lost G3PDH during evolution. Lateral gene transfer has been proposed to be a major force in the evolution of prokaryotic genomes (Boucher et al. 2003). Recently, Ruepp et al. (2000) reported that a significant number of genes in the thermoacidophilic archaeon Thermoplasma acidophilum might have been obtained by lateral gene transfer and that these “lifestyle genes,” which are crucial for adaptation of the organism to certain environmental conditions, are shared between phylogenetically distant organisms within one ecological niche.
Materials and methods
E. coli strain TOP10 and plasmid DNA pET101/TOPO were obtained from Invitrogen. E. coli strain Rosetta (DE3) was from Novagen. Resource Q, Source 15 Phe, and Sephadex G-25 Superfine were from Amersham Biosciences.
Cloning of the gpsA gene from A. fulgidus
The following oligonucleotide primers were used to amplify the A. fulgidus (VC16, DSMZ 304) gpsA gene (AF0871) by PCR: 5′-CACCATGATTGTTTCGATACTGGGAGC-3′ (sense) and 5′-TTATTTAAATGTAGCGAGTTCAAACAGCACTTCC-3′ (anti-sense). To enable directional cloning, the third base from the 5′ end has been changed. The amplified 1-kb fragment was purified and ligated with the expression vector pET101/TOPO to generate pEGPD1 according to the instruction manual. E. coli strain TOP10 was transformed with pEGPD1 and was spread onto LB agar plate containing 50 mg/L ampicillin. Positive colonies were selected by the colony PCR method using T7 primers. A selected positive transformant was cultured in liquid LB medium containing 50 mg/L ampicillin, and the plasmid pEGPD1 carrying gpsA was isolated.
Overproduction and purification of G3PDH from A. fulgidus
E. coli strain Rosetta (DE3) was transformed with the pEGPD1 carrying gpsA, and then the transformant was cultivated at 37°C in liquid LB medium containing 50 mg/L ampicillin and 38 mg/L chloramphenicol until the absorbance at 600 nm reached 0.4. Induction was carried out by the addition of 1 mM isopropyl-β-D-thiogalactopyranoside to the medium, and the cultivation was continued for 4 h at the same temperature. The cells, which produced G3PDH from A. fulgidus, were harvested by centrifugation, suspended in 20 mM Tris-HCl (pH 9.0) and disrupted by ultrasonication. The lysate was heated for 45 min at 70°C and the denatured protein was then removed by centrifugation (15,000g, 45 min). The supernatant was loaded onto a Resource Q column equilibrated with 20 mM Tris-HCl (pH 9.0), and G3PDH activity was eluted with a linear gradient of 0 to 0.5 M KCl. The active fractions were pooled, concentrated by 30 kDa centrifugal filter device (Millipore) and desalted by passage through a Sephadex G-25 Superfine column equilibrated with 10 mM potassium phosphate (pH 7.0). The desalted enzyme solution was then put on a hydroxyapatite column equilibrated with the same buffer, and the enzyme was eluted with a linear gradient of 10 to 500 mM potassium phosphate (pH 7.0). The fractions with G3PDH activity were collected, solid KCl was added (3 M), and the solution was put on a Source 15 Phe column equilibrated with 20 mM Tris-HCl (pH 8.0) containing 3 M KCl. The enzyme was eluted with a linear KCl gradient from 3 to 0 M. The active fractions were concentrated and desalted by passage through a Sephadex G-25 Superfine column equilibrated with 10 mM MOPS-KOH (pH 7.0). All chromatographic steps were performed in the cold room (4°C). The thus purified G3PDH was used for all experiments. Preparation of the cell extracts from A. fulgidus was described previously (Mander et al. 2002).
Determination of G3PDH specific activity
The activity of G3PDH from A. fulgidus was routinely determined at 70°C. The reduction of DHAP with NADPH (standard assay) or NADH was followed spectrophotometrically at 340 nm in a reduced cuvette with d = 1 cm (ɛ340 = 6.2 mM−1cm−1). The 1-mL reaction mixture contained 50 mM Tris-HCl (pH 6.6 at 70°C), 0.15 mM NADPH or NADH, 1 mM DHAP, and 10–50 mU enzyme. The oxidation of G3P with NADP+ or NAD+ was monitored spectrofluorometrically at 460 nm with excitation at 340 nm. The 1-mL reaction mixture contained 50 mM glycine-NaOH (pH 9.0 at 70°C), 2 mM G3P, 5 mM NADP+ or NAD+, 100 mM hydrazine (pH 9.0, 25°C), and 0.1–0.5 U enzyme. The reactions were started by the addition of the enzyme. One unit of enzyme was defined as the amount of enzyme catalyzing the oxidation of 1 μmol of NADPH per min at pH 6.6 and 70°C. Protein was determined by the Bradford dye-binding method using the BioRad protein assay kit. The standard protein of the assay was bovine serum albumin.
The pH optimum of the G3PDH from A. fulgidus was determined in the following buffers (pH at 70°C): MES-NaOH (pH 5.0–6.0), potassium phosphate (pH 6.0–7.5), and glycine-NaOH (pH 8.0–9.5). The temperature optimum was measured in 50 mM potassium phosphate (pH 6.6 at 25°C) instead of 50 mM Tris-HCl, because the pH of phosphate buffer changes less with temperature than that of Tris buffer.
Crystallization and data collection
Crystals were obtained using the hanging-drop vapor diffusion method at 4°C, in which 1 μL of 12 mg/mL protein solution was mixed with an equal volume of mother liquor, consisting of 0.6–1.6 M (NH4)2SO4, 0–3% (v/v) dioxane and 100 mM MES-NaOH (pH 6.0). The crystals were transferred to a cryo-solution composed of the reservoir solution and 25% glycerol. After 20 min soaking in the cryo-solution at 4°C, the crystals were frozen in liquid nitrogen or under a stream of gaseous nitrogen and X-ray data were collected at 100 K. Heavy atom derivatives were prepared by soaking the crystals in a reservoir solution containing 1 mM uranyl nitrate (3 h, 4°C) or 10 mM HgCl2 (7 days, 4°C). These crystals were frozen in the same solution containing 25% glycerol. Prior to freezing, the uranylnitrate-treated crystals were back soaked for 0.5 min in reservoir solution containing 25% glycerol. The enzyme crystallized in the space group P21 with cell constants of a = 63.22 Å, b = 67.59 Å, c = 81.16 Å, and β = 106.54°, best compatible with two monomers per asymmetric unit. Diffraction data of native and derivative crystals were collected on a CuKα rotating anode (Bruker/Nonius FR591, 50 kV, 80 mA) equipped with an OSMIC focusing system and a 345-mm imaging plate (MAR Research) and at the beam line BW7A at the Deutsches Elektronen Synchroton DESY in Hamburg. The collected data (Table 3) were scaled and interpreted using DENZO and SCALEPACK (Otwinowski and Minor 1996) or XDS (Kabsch 1993). The structure was solved using the multiple isomorphous replacement (MIR) method for phase determination and the CCP4 suite (Bailey 1994) for performing the calculations.
Table 3.
Summary of crystallographic analysis
| Data collection statistics | ||||||
| Data set | Cell | Resolution (Å) | Measured, unique reflections | Rmergea | I/σ(I)b | Completeness |
| Native | 63.2 Å, 67.6 Å, 81.2 Å, 106.5° | 17–1.63 | 333,108, 75,658 | 0.067 (0.506) | 17.2 (2.3) | 0.926 (0.887) |
| HgCl2 | 62.8 Å, 67.7 Å, 81.0 Å, 106.6° | 17–2.40 | 95,871, 25,017 | 0.067 (0.157) | 20.3 (6.4) | 0.985 (0.940) |
| Ethyl-Hg-phosphate | 62.8 Å, 67.7 Å, 81.0 Å, 106.6° | 17–2.30 | 105,225, 29,046 | 0.054 (0.145) | 19.2 (7.4) | 0.997 (0.999) |
| Gadolinium acetate | 63.2 Å, 67.5 Å, 81.1 Å, 106.5° | 17–2.20 | 99,151, 28,984 | 0.042 (0.164) | 25.2 (7.2) | 0.917 (0.917) |
| Uranyl acetate | 62.9 Å, 67.7 Å, 80.8 Å, 107.0° | 17–2.40 | 91,831, 25,405 | 0.064 (0.173) | 20.1 (5.0) | 0.995 (0.992) |
| Phasing statistics | |||||||
| 17–7.1 Å | − 4.5 Å | −3.6 Å | −3.1 Å | −2.7 Å | −2.5 Å | Total | |
| FOM (after SOLVE)c | 0.72 | 0.61 | 0.47 | 0.43 | 0.43 | 0.39 | 0.46 |
| FOM (after RESOLVE)c | 0.87 | 0.79 | 0.77 | 0.68 | 0.62 | 0.62 | 0.71 |
| Refinement statistics | ||||||
| Data set | Resolution (Å) | Reflections (F > O) | Rfactor (Rfree)d | Number of atoms protein, water, ions | r.m.s.d. bonds (Å) | r.m.s.d. bond angles (°) |
| aRmerge = ∑hkl ∑i|Ii(hkl) − 〈I(hkl) 〉|/∑hkl ∑iIi(hkl); values in parentheses correspond to the highest resolution shell. | ||||||
| b As calculated with the program TRUNCATE (Bailey 1994). | ||||||
| c The figure of merit (FOM) is defined as the estimated cosine of the phase error. | ||||||
| dRfactor = ∑|Fobs − Fcalc|/∑Fobs; Rfree calculated with 9% of randomly selected data. | ||||||
| Native | 17–1.7 | 63,641 | 0.149 (0.194) | 5194, 753, 53 | 0.019 | 1.641 |
Refinement and structure analysis
After phase improvement by the solvent flattening procedure of DM (Bailey 1994) the quality of the electron density was sufficient to automatically build 80% of the model using the program MAID (Levitt 2001). The missing residues were manually incorporated within O (Jones et al. 1991). The resulting model was refined with CNS (Brünger et al. 1998) and REFMAC5 (Murshudov et al. 1997) to 1.7 Å resolution. The final Rcryst and Rfree factors were 14.9% and 19.3%, respectively. The model consists of 2 × 335 amino acid residues, eight sulfate anions, one ammonium ion, two glycerole molecules, and 753 H2O molecules. The overall B factor is 17 Å2. Due to asymmetric packing of side chains in the dimer interface, the side chains of the residues Phe 248 and Ile 252 were modeled in two alternative conformations. According to PROCHECK (Laskowski et al. 1993), no dihedral angle of non-glycine residues was located in disallowed regions. The accessible protein surfaces were calculated using the programs NACCESS (Miller et al. 1987) and MSMS (Sanner et al. 1996) using a probe radius of 1.4 Å. Fold similarities were identified with the DALI server (Holm and Sander 1993). Ion–ion interactions were defined as an ion pair when the distance between the charged atoms was below 4 Å (Table 3).
Accession number
The structure factors and atomic coordinates of G3PDH from A. fulgidus have been deposited in the RCSB Data Bank with the accession code 1TXG.
Acknowledgments
This work was supported by the Max Planck Society, by the Asahi Kasei Pharma Corp., and by the Fonds der Chemischen Industrie. We thank Alex Tucker from beam line BW7A, EMBL, in Hamburg; and Tobias Klar for helpful suggestions and Erica Lyon for reading the manuscript.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.04980304.
References
- Bailey, S. 1994. The CCP4 suite: Programs for protein crystallography. Acta Crystallogr. D 50 760–763. [DOI] [PubMed] [Google Scholar]
- Boucher, Y., Douady, C.J., Papke, R.T., Walsh, D.A., Boudreau, M.E., Nesbo, C.L., Case, R.J., and Doolittle, W.F. 2003. Lateral gene transfer and the origins of prokaryotic groups. Annu. Rev. Genet. 37 283–328. [DOI] [PubMed] [Google Scholar]
- Brinen, L.S., Canaves, J.M., Dai, X., Deacon, A.M., Elsliger, M.A., Eshaghi, S., Floyd, R., Godzik, A., Grittini, C., Grzechnik, S.K., et al. 2003. Crystal structure of a zinc-containing glycerol dehydrogenase (TM0423) from Thermotoga maritima at 1.5 Å resolution. Proteins 50 371–374. [DOI] [PubMed] [Google Scholar]
- Brünger, A., Adams, P.D., Clore, G.M., Delano, W.L., Gros, P., Grosse-Kunstleve, R., Jiang, J.-S., Kuszewski, J., Nilges, M., Pannu, N.S., et al. 1998. Crystallography and NMR system: A new software suite for macro-molecular structure determinations. Acta Crystallogr. D 54 905–921. [DOI] [PubMed] [Google Scholar]
- Carugo, O. and Argos, P. 1997. NADP-dependent enzymes. II: Evolution of the mono- and dinucleotide binding domains. Proteins 28 29–40. [DOI] [PubMed] [Google Scholar]
- Charron, C., Talfournier, F., Isupov, M.N., Littlechild, J.A., Branlant, G., Vitoux, B., and Aubry, A. 2000. The crystal structure of d-glyceraldehyde-3-phosphate dehydrogenase from the hyperthermophilic archaeon Methanothermus fervidus in the presence of NADP+ at 2.1 Å resolution. J. Mol. Biol. 297 481–500. [DOI] [PubMed] [Google Scholar]
- Choe, J., Guerra, D., Michels, P.A., and Hol, W.G. 2003. Leishmania mexicana glycerol-3-phosphate dehydrogenase showed conformational changes upon binding a bi-substrate adduct. J. Mol. Biol. 329 335–349. [DOI] [PubMed] [Google Scholar]
- Daiyasu, H., Hiroike, T., Koga, Y., and Toh, H. 2002. Analysis of membrane stereochemistry with homology modeling of sn-glycerol-1-phosphate dehydrogenase. Protein Eng. 15 987–995. [DOI] [PubMed] [Google Scholar]
- de Vries, R.P., Flitter, S.J., van de Vondervoort, P.J., Chaveroche, M.K., Fontaine, T., Fillinger, S., Ruijter, G.J., d’Enfert, C., and Visser, J. 2003. Glycerol dehydrogenase, encoded by gldB is essential for osmotolerance in Aspergillus nidulans. Mol. Microbiol. 49 131–141. [DOI] [PubMed] [Google Scholar]
- Ermler, U., Hagemeier, C.H., Roth, A., Demmer, U., Grabarse, W., Warkentin, E., and Vorholt, J.A. 2002. Structure of methylene-tetrahydromethanopterin dehydrogenase from Methylobacterium extorquens AM1. Structure 10 1127–1137. [DOI] [PubMed] [Google Scholar]
- Fernández-Sousa, J.M., Gavilanes, J.G., Municio, A.M., and Pérez-Aranda, A. 1977. L-glycerol-3-phosphate dehydrogenase from the insect Ceratitis capitata purification, physicochemical and enzymic properties. Biochim. Bio-phys. Acta 481 6–24. [DOI] [PubMed] [Google Scholar]
- Goncalves, L.G., Huber, R., da Costa, M.S., and Santos, H. 2003. A variant of the hyperthermophile Archaeoglobus fulgidus adapted to grow at high salinity. FEMS Microbiol. Lett. 218 239–244. [DOI] [PubMed] [Google Scholar]
- Hagemeier, C.H., Shima, S., Thauer, R.K., Bourenkov, G., Bartunik, H.D., and Ermler, U. 2003. Coenzyme F420-dependent methylenetetrahydromethanopterin dehydrogenase (Mtd) from Methanopyrus kandleri: A methanogenic enzyme with an unusual quarternary structure. J. Mol. Biol. 332 1047–1057. [DOI] [PubMed] [Google Scholar]
- Holm, L. and Sander, C. 1993. Secondary structure comparison by alignment of distance matrices. J. Mol. Biol. 233 123–138. [DOI] [PubMed] [Google Scholar]
- Jones, T.A., Zou, J.Y., Cowan, S.W., and Kjeldgaard, M. 1991. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A 47 110–119. [DOI] [PubMed] [Google Scholar]
- Kabsch, H. 1993. Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J. Appl. Cryst. 26 795–800. [Google Scholar]
- Kawarabayasi, Y., Hino, Y., Horikawa, H., Yamazaki, S., Haikawa, Y., Jinno, K., Takahashi, M., Sekine, M., Baba, S., Ankai, A., et al. 1999. Complete genome sequence of an aerobic hyperthermophilic crenarchaeon, Aeropyrum pernix K1. DNA Res. 6 83–101. [DOI] [PubMed] [Google Scholar]
- Klenk, H.P., Clayton, R.A., Tomb, J.F., White, O., Nelson, K.E., Ketchum, K.A., Dodson, R.J., Gwinn, M., Hickey, E.K., Peterson, J.D., et al. 1997. The complete genome sequence of the hyperthermophilic, sulphate-reducing archaeon Archaeoglobus fulgidus. Nature 390 364–370. [DOI] [PubMed] [Google Scholar]
- Koga, Y., Kyuragi, T., Nishihara, M., and Sone, N. 1998. Did archaeal and bacterial cells arise independently from noncellular precursors? A hypothesis stating that the advent of membrane phospholipid with enantiomeric glycerophosphate backbones caused the separation of the two lines of descent. J. Mol. Evol. 46 54–63. [DOI] [PubMed] [Google Scholar]
- Kohl, L., Drmota, T., Thi, C.D., Callens, M., Van Beeumen, J., Opperdoes, F.R., and Michels, P.A. 1996. Cloning and characterization of the NAD-linked glycerol-3-phosphate dehydrogenases of Trypanosoma brucei and Leishmania mexicana and expression of the trypanosome enzyme in Escherichia coli. Mol. Biochem. Parasitol. 76 159–173. [DOI] [PubMed] [Google Scholar]
- Korndörfer, I., Steipe, B., Huber, R., Tomschy, A., and Jaenicke, R. 1995. The crystal structure of hologlyceraldehyde-3-phosphate dehydrogenase from the hyperthermophilic bacterium Thermotoga maritima at 2.5 Å resolution. J. Mol. Biol. 246 511–521. [DOI] [PubMed] [Google Scholar]
- Kraulis, P.J. 1991. MOLSCRIPT: A program to produce both detailed and schematic plots of protein structures. J. Appl. Cryst. 24 946–950. [Google Scholar]
- Kunow, J., Schwörer, B., Stetter, K.O., and Thauer, R.K. 1993. A F420-dependent NADP reductase in the extremely thermophilic sulfate reducing Archaeoglobus fulgidus. Arch. Microbiol. 160 199–205. [Google Scholar]
- Lamosa, P., Burke, A., Peist, R., Huber, R., Liu, M.Y., Silva, G., Rodrigues-Pousada, C., LeGall, J., Maycock, C., and Santos, H. 2000. Thermostabilization of proteins by diglycerol phosphate, a new compatible solute from the hyperthermophile Archaeoglobus fulgidus. Appl. Environ. Microbiol. 66 1974–1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski, R.A., MacArthur, M.W., Moss, D.S., and Thornton, J.M. 1993. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Cryst. 26 283–291. [Google Scholar]
- Levitt, D.G. 2001. A new software routine that automates the fitting of protein X-ray crystallographic electron-density maps. Acta Crystallogr. D 57 1013–1019. [DOI] [PubMed] [Google Scholar]
- Madern, D., Ebel, C., and Zaccai, G. 2000. Halophilic adaptation of enzymes. Extremophiles 4 91–98. [DOI] [PubMed] [Google Scholar]
- Mamat, B., Roth, A., Grimm, C., Ermler, U., Tziatzios, C., Schubert, D., Thauer, R.K., and Shima, S. 2002. Crystal structures and enzymatic properties of three formyltransferases from archaea: Environmental adaptation and evolutionary relationship. Protein Sci. 11 2168–2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mander, G.J., Duin, E.C., Linder, D., Stetter, K.O., and Hedderich, R. 2002. Purification and characterization of a membrane-bound enzyme complex from the sulfate-reducing archaeon Archaeoglobus fulgidus related to heterodisulfide reductase from methanogenic archaea. Eur. J. Biochem. 269 1895–1904. [DOI] [PubMed] [Google Scholar]
- Marche, S., Michels, P.A., and Opperdoes, F.R. 2000. Comparative study of Leishmania mexicana and Trypanosoma brucei NAD-dependent glycerol-3-phosphate dehydrogenase. Mol. Biochem. Parasitol. 106 83–91. [DOI] [PubMed] [Google Scholar]
- Martins, L.O., Huber, R., Huber, H., Stetter, K.O., Dacosta, M.S., and Santos, H. 1997. Organic solutes in hyperthermophilic archaea. Appl. Environ. Microbiol. 63 896–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merritt, E.A., and Murphy, M.E.P. 1994. Raster3D Version 2.0—A program for photorealistic molecular graphics. Acta Crystallogr. D 50 869–873. [DOI] [PubMed] [Google Scholar]
- Miller, S., Lesk, A.M., Janin, J., and Chothia, C. 1987. The accessible surface area and stability of oligomeric proteins. Nature 328 834–836. [DOI] [PubMed] [Google Scholar]
- Morii, H., Nishihara, M., and Koga, Y. 2000. CTP:2,3-di-O-geranylgeranyl-sn-glycero-1-phosphate cytidyltransferase in the methanogenic archaeon Methanothermobacter thermoautotrophicus. J. Biol. Chem. 275 36568–36574. [DOI] [PubMed] [Google Scholar]
- Murshudov, G.N., Vagin, A.A., and Dodson, E.J. 1997. Refinement of macro-molecular structures by the maximum-likelihood method. Acta Crystallogr. D 53 240–255. [DOI] [PubMed] [Google Scholar]
- Nicholls, A., Bharadwaj, R., and Honig, B. 1993. GRASP: Graphical representation and analysis of surface properties. Biophys. J. 64 166–170. [Google Scholar]
- Nishihara, M. and Koga, Y. 1995. sn-Glycerol-1-phosphate dehydrogenase in Methanobacterium thermoautotrophicum: Key enzyme in biosynthesis of the enantiomeric glycerophosphate backbone of ether phospholipids of archaebacteria. J. Biochem. (Tokyo) 117 933–935. [DOI] [PubMed] [Google Scholar]
- ———. 1997. Purification and properties of sn-glycerol-1-phosphate dehydrogenase from Methanobacterium thermoautotrophicum: Characterization of the biosynthetic enzyme for the enantiomeric glycerophosphate backbone of ether polar lipids of Archaea. J. Biochem. (Tokyo) 122 572–576. [DOI] [PubMed] [Google Scholar]
- Nishihara, M., Yamazaki, T., Oshima, T., and Koga, Y. 1999. sn-Glycerol-1-phosphate-forming activities in Archaea: Separation of archaeal phospholipid biosynthesis and glycerol catabolism by glycerophosphate enantiomers. J. Bacteriol. 181 1330–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noguchi, S., Maeda, M., Nishihara, M., Koga, Y., and Sone, N. 1998. Expression and use of Methanobacterium thermoautotrophicum sn-glycerol 1-phosphate dehydrogenase for the assay of sn-glycerol 1-phosphate in Archaea. J. Biosci. Bioeng. 86 266–270. [Google Scholar]
- Otwinowski, Z. and Minor, W. 1996. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276 307–326. [DOI] [PubMed] [Google Scholar]
- Rossmann, M.G., Moras, D., and Olsen, K.W. 1974. Chemical and biological evolution of a nucleotide-binding protein. Nature 250 194–199. [DOI] [PubMed] [Google Scholar]
- Ruepp, A., Graml, W., Santos-Martinez, M.L., Koretke, K.K., Volker, C., Mewes, H.W., Frishman, D., Stocker, S., Lupas, A.N., and Baumeister, W. 2000. The genome sequence of the thermoacidophilic scavenger Thermoplasma acidophilum. Nature 407 508–513. [DOI] [PubMed] [Google Scholar]
- Ruzheinikov, S.N., Burke, J., Sedelnikova, S., Baker, P.J., Taylor, R., Bullough, P.A., Muir, N.M., Gore, M.G., and Rice, D.W. 2001. Glycerol dehydrogenase. Structure, specificity, and mechanism of a family III polyol dehydrogenase. Structure 9 789–802. [DOI] [PubMed] [Google Scholar]
- Sanner, M.F., Olson, A.J., and Spehner, J.C. 1996. Reduced surface: An efficient way to compute molecular surfaces. Biopolymers 38 305–320. [DOI] [PubMed] [Google Scholar]
- Schomburg, D., Salzmann, M., and Stephan, D. 1995. Class 1.1: oxidoreductases. In Enzyme handbook (eds. D. Schoburg and D. Stephan), pp. 1–10. Springer-Verlag, Berlin.
- Schryvers, A. and Weiner, J.H. 1981. The anaerobic sn-glycerol-3-phosphate dehydrogenase of Escherichia coli. Purification and characterization. J. Biol. Chem. 256 9959–9965. [PubMed] [Google Scholar]
- Shen, W., Wei, Y., Dauk, M., Zheng, Z., and Zou, J. 2003. Identification of a mitochondrial glycerol-3-phosphate dehydrogenase from Arabidopsis thaliana: Evidence for a mitochondrial glycerol-3-phosphate shuttle in plants. FEBS Lett. 536 92–96. [DOI] [PubMed] [Google Scholar]
- Smith, D.R., Doucette-Stamm, L.A., Deloughery, C., Lee, H., Dubois, J., Aldredge, T., Bashirzadeh, R., Blakely, D., Cook, R., Gilbert, K., et al. 1997. Complete genome sequence of Methanobacterium thermoautotrophicum ΔH: Functional analysis and comparative genomics. J. Bacteriol. 179 7135–7155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterner, R. and Liebl, W. 2001. Thermophilic adaptation of proteins. Crit. Rev. Biochem. Mol. Biol. 36 39–106. [DOI] [PubMed] [Google Scholar]
- Stetter, K.O. 1988. Archaeoglobus fulgidus gen.nov, sp-nov: A new taxon of extremely thermophilic archaebacteria. Syst. Appl. Microbiol. 10 172–173. [Google Scholar]
- Stetter, K.O., Lauerer, G., Thomm, M., and Neuner, A. 1987. Isolation of extremely thermophilic sulfate reducers: Evidence for a novel branch of archaebacteria. Science 236 822–824. [DOI] [PubMed] [Google Scholar]
- Suresh, S., Turley, S., Opperdoes, F.R., Michels, P.A., and Hol, W.G. 2000. A potential target enzyme for trypanocidal drugs revealed by the crystal structure of NAD-dependent glycerol-3-phosphate dehydrogenase from Leishmania mexicana. Struct. Fold. Des. 8 541–552. [DOI] [PubMed] [Google Scholar]
- Warkentin, E., Mamat, B., Sordel-Klippert, M., Wicke, M., Thauer, R.K., Iwata, M., Iwata, S., Ermler, U., and Shima, S. 2001. Structures of F420H2:NADP+ oxidoreductase with and without its substrates bound. EMBO J. 20 6561–6569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yip, K.S.P., Stillman, T.J., Britton, K.L., Artymiuk, P.J., Baker, P.J., Sedelnikova, S.E., Engel, P.C., Pasquo, A., Chiaraluce, R., Consalvi, V., et al. 1995. The structure of Pyrococcus furiosus glutamate dehydrogenase reveals a key role for ion-pair networks in maintaining enzyme stability at extreme temperatures. Structure 3 1147–1158. [DOI] [PubMed] [Google Scholar]