

Summary
TRPV3 is a non-selective cation channel activated by warm to hot temperatures. In rodents, TRPV3 is highly expressed in basal keratinocytes of skin and oral/nasal epithelia. TRPV3 knockout mice showed impaired responses to innocuous and noxious heat but otherwise normal appearance and reactions to many sensory modalities. However, point mutations of TRPV3 at Gly573 to Ser and Cys have recently been linked to autosomal dominant hairless phenotypes and spontaneous dermatitis in mice and rats, implicating an important role for TRPV3 in alopecia and skin diseases. Exactly how the mutations affect TRPV3 function was unexplained. Here we show that both G573S and G573C mutations of murine TRPV3 are constitutively active in heterologous systems. In HEK293 cells, expression of the TRPV3 mutants causes cell death. In Xenopus oocytes, the constitutively active mutant channel is irresponsive to thermal and chemical stimuli but it reduces the temperature threshold and enhances the responses to heat and TRPV3 agonists of the wild type channel when they are co-expressed. We conclude that the G573S and G573C substitutions render the TRPV3 channel spontaneously active under normal physiological conditions, which in turn alters ion homeostasis and membrane potentials of skin keratinocytes, leading to hair loss and dermatitis-like skin diseases.
Introduction
The mammalian TRP superfamily contains about 28 members of non-allelic genes. They are further divided into TRPC (Canonical), TRPV (Vanilloid), TRPM (Melastatin), TRPA (ANKTM1), TRPP (Polycystin), and TRPML (MucoLipin) subfamilies [1]. Based on their temperature responsiveness, TRPA1, TRPM8, TRPV1, TRPV2, TRPV3, and TRPV4 are also classified as thermal sensitive channels. The combined actions of these channels allow the body to distinguish a broad temperature range from <17°C to >52°C [2,3]. Among them, TRPV3 and TRPV4 are activated at temperatures higher than 33 and 27°C, respectively, implicating that they are activate, at least partially, at the normal body temperature of mammalian species.
Amongst all mammalian TRP channels, only TRPV3 and TRPV4 have been shown to be highly expressed and functional in skin keratinocytes [4-8]. Most studies have focused on the sensory functions of these channels, primarily the sensation of warm and hot temperatures. Being the first defense system of the body, the skin is perfectly situated to sense environmental temperature changes and relay this information to primary afferents, which innervate the skin layer. Indeed, both TRPV3 and TRPV4 knockout mice show impaired performance in selecting the most appropriate temperatures as compared to the wild type animals [8,9]. However, the exact mechanism(s) by which the activated keratinocytes transmit information to sensory nerves remains unknown at the present time.
Like many other TRPV and sensory TRP channels, in addition to thermal stimulus, TRPV3 and TRPV4 are activated by a number of endogenous and exogenous chemical and physical stimuli. TRPV4 is activated by osmolarity changes [10,11], by the epoxygenase products of arachidonic acid [12], and by bisandrographolide from the medicinal plant, Andrographis Paniculata [13]. TRPV3 is activated by a number of plant extracts, such as camphor [8], carvacrol (from oregano) eugenol (from clove), and thymol (from thyme) [14]. All these compounds have low affinities to TRPV3 and are nonspecific in that they activate other TRP channels as well. Nonetheless, their uses as flavor enhancers, skin irritants or sensitizers are consistent with the predominant expression of TRPV3 in skin as well as oral and nasal epithelia. It has been speculated that TRPV3 may be involved in the response to allergens exposed to the body surface [14]. However, an endogenous ligand has yet to be found for TRPV3. Another chemical commonly used to activate TRPV3 is 2-aminoethoxydiphenyl borate (2APB) [7,14-19].
Consistent with TRPV3 having a role in sensory processes, TRPV3 null mice showed a reduced tendency to migrate towards warm surfaces and a defect in noxious heat sensation but are otherwise normal in appearance and behavior [8]. A surprise came from a recent genetic study, which reports that two independent autosomal dominant spontaneous hairless rodent mutants, the DS-Nh mice and WBN/Kob-Ht rats, share a common single amino acid substitution at Gly573 of their TRPV3 proteins [19]. In the DS-Nh mice, the Gly was changed to Ser while in WBN/Kob-Ht rats, it was changed to Cys. In addition to hair loss, these two mutant strains spontaneously develop atopic dermatitis (AD)-like skin rash in the presence of Staphylococcus aureus. Linkage analysis suggests that the genes responsible for dermatitis and hairlessness are tightly linked and inseparable from each other. Thus, the TRPV3 mutations at Gly573 appear to be associated with both hair loss and certain forms of dermatitis, suggesting that TRPV3 may be a novel theraputic target for treating these skin diseases.
The potential therapeutic value of TRPV3 calls for more detailed analysis of the G573 mutations in cellular function. Primary keratinocytes isolated from the DS-Nh mice carrying the G537S mutation of TRPV3 showed an enhanced response to heat (33 °C) in the presence of 100 μM 2APB as compared to wild type cells, indicating that the mutant may be more active than the wild type channel [19]. However, the original report did not include a detailed functional characterization of the mutant channels. We have created the G573S and G573C mutations in mouse TRPV3 and studied the function of these mutants after expressing them in HEK293 cells and Xenopus oocytes. Here, we show that both G573S and G573C mutants are constitutively active, leading to cell death under normal culture conditions. Co-expression of the mutant with the wild-type TRPV3 greatly lowered the temperature threshold of channel activation and the response to 2APB to camphor.
Materials and Methods
cDNA constructs and mutagenesis
The full-length cDNA for mouse TRPV3 was obtained and placed in the pAGA3 and pIRES2EGFP vectors as previously described [15]. To express the green fluorescence protein (GFP) fusion protein of TRPV3, the open-reading frame of the TRPV3 cDNA was subcloned into pEGFPC3 vector (BD-Clontech, Palo Alto, CA) at the EcoRI/SalI sites, in which the EcoRI site was filled in with Klenow fragment of DNA polymerase I and fused with the second codon of TRPV3 (AAT for Asn) via blunt-end ligation. The G573S and G573C mutations were created by PCR using a sense primer, 5′-TGTCCTCATCTGGGCCAC-3′, and antisense primers, 5′-TGACGCTGTACATGCTCATAGACTGGAAGCC-3′ and 5′-TGACGCTGTACATGCACATAGACTGGAAGCC-3′, for G573S and G573C, respectively. The PCR products were introduced back to TRPV3 in pAGA3 at the NsiI/BsrGI sites. After verification of the mutated site by sequencing, the full-length mutants were subcloned to the pIRES2EGFP and pEGFPC3 vectors. For inducible expression under the control of tetracycline promoter, the GFP-tagged TRPV3 and mutant constructs were excised from the pEGFPC3 vector using the AgeI/ApaI sites and ligated into the BspEI/ApaI sites of pcDNA4/TO vector (Invitrogen, Carlsbad, CA). The accuracy of all constructs at the mutated and junction regions was confirmed by DNA sequencing.
Mammalian cell culture and transfection
HEK 293 and T-Rex 293 (Invitrogen) cells were grown at 37°C, 5% CO2 in Dulbecco’s minimal essential medium containing 4.5 mg/ml glucose, 10% heat-inactivated fetal bovine serum, 50 units/ml penicillin, and 50 μg/ml streptomycin. For T-Rex 293, blasticidin S (10 μg/ml) was also included. Transfections were performed in wells of a 96-well plate using Lipofectamine 2000 (Invitrogen) as previously described [15]. The pIRES2EGFP vector was used for the expression in HEK 293 cells whereas pcDNA4/TO was used for the expression in T-Rex 293 cells. To induce TRPV3 expression, the T-Rex 293 cells were washed twice with the culture medium 16 hrs after transfection and doxycycline (20 ng/ml) was added to the desired wells. The condition of the cells and the expression of the GFP-tagged TRPV3 were monitored with the use of an epifluorescence microscope.
cRNA synthesis and expression in Xenopus oocytes
TRPV3 and the G573 mutants in pAGA3 vector were linearized using Xho1. Complementary RNAs were synthesized using mMessage mMachine reagents and protocols obtained from Ambion (Austin, TX). The resulting cRNAs were dissolved in diethylpyrocarbonate-treated H2O. Sexually mature female Xenopus laevis of older than 2.5 years of age were purchased from Xenopus Express, Inc. (Plant City, FL). For oocyte isolation, small pieces of ovarian lobe were dissected out from anesthetized frogs and shaken gently at 19 °C for 90 min in sterile OR2 solution containing 82 mM NaCl, 2 mM KCl, 1 mM MgCl2, 5 mM HEPES, pH 7.6, and supplemented with 1 mg/ml collagenase (Worthington Biochem, Lakewood, NJ). Denuded, healthy looking oocytes of more than 1 mm in diameter were selected and injected in a volume of 50 nl/cell with a total of 5 ng of cRNA. The injected oocytes were incubated at 19 °C for 2-3 days in sterile ND96 solution containing: 96 mM NaCl, 2 mM KCl, 1.8 mM CaCl2, 1 mM MgCl2, 5 mM HEPES, pH 7.6, supplemented with 275 μg/ml pyruvic acid and 20 μg/ml Gentamycin. The solution was changed daily.
Two-electrode voltage clamp
For two-electrode voltage clamp recordings, cRNA-injected oocytes were placed in a RC-3Z Oocyte Recording Chamber (Warner Instruments, Hamden, CT) and perfused with a bath solution that contained (in mM) 100 NaCl, 2.5 KCl, 1 MgCl2, 5 Hepes, pH 7.4. The oocytes were impaled with two intracellular glass electrodes filled with 3 M KCl connected to an OC-725C Oocyte Clamp amplifier (Warner Instruments, Hamden, CT). Voltage commands were made from the Pulse+Pulsefit program (HEKA Instruments, Southboro, MA) via an ITC-18 Computer Interface (Instrutech Co. Port Washington, NY). Oocytes were clamped at -20 mV, stepped to -100 mV for 20 ms, followed by a voltage ramp of 200 ms from -100 mV to +100 mV once every 0.56 second. Currents were recorded at the sampling rate of 1 kHz. Temperature changes were made using a CL-100 Bipolar temperature controller connected to a SC-20 dual in-line solution heater/cooler (Warner Instruments).
Drugs and chemicals
2APB was from Cayman Chemical Co (Ann Arbor, MI) and dissolved in DMSO at 0.5 M for stock solution. dl-Camphor was from Acros Organics (Morris Plains, NJ) and a 1 M stock solution was made using DMSO. The concentrations of DMSO used in the recording (<1%) did no cause any current change. Ruthenium red and doxycycline hyclate were from Sigma-Aldrich (St. Louis, MO) and stock solutions were made to 20 mM and 2 mg/ml, respectively, in water. The stock solution of blasticidin S HCl (5 mg/ml) was from Invitrogen.
Data analysis
Data are presented as means ± S.E.M. Temperature threshold and the 10-degree temperature coefficient (Q10) values were determined by plotting the logarithmic values of the absolute current amplitude at -100 mV as a function of temperature. The Q10 values were estimated from the slopes of the linear portions of the curve and the temperature threshold obtained from the intersection of the two linear fits. Statistical analyses were performed using Student’s t test.
Results
Constitutive activity and cell killing effect of G573 mutants of TRPV3 in mammalian cells
To understand the molecular mechanism underlying the hair loss and atopic dermatitis-like phenotype of the DS-Nh mice and WBN/Kob-Ht rats [19], we introduced the G573C and G573S substitutions in mouse TRPV3 with an intention to analyze their channel function by whole-cell patch clamping in HEK 293 cells, a method successfully used for the wild type TRPV3 [15,16]. The mutant cDNA was subcloned in the pIRES2EGFP vector and transfected to HEK 293 cells. This method allows the identification of transfected cells using fluorescence microscope, aid by the bicistronic expression of TRPV3 and GFP together. One to two days after transfection, about 70% of cells showed green fluorescence in cells that received the wild type TRPV3 cDNA while only less than 5% cells showed very weak green signal in cells transfected with either of the G573 mutants. There was substantial cell death in the mutant-transfected samples. Whole-cell voltage clamp recording of the few green cells revealed large leakage-like currents that were partially blocked by ruthenium red and N-methyl-D-glucamine (data not shown), suggestive of TRPV-like cation conductance. These data indicate that the two TRPV3 mutants are constitutively active and the activity is toxic to the cell.
To confirm that the G573 mutants are indeed cytotoxic, we transferred the wild type and G573C and G573S mutants of TRPV3 to pcDNA4/TO vector for the Tet-On inducible expression. To facilitate the identification of expressed channel protein, we inserted the coding sequence of EGFP in-frame before the second codon of the TRPV3 cDNA so that the expressed protein contains a GFP tag at the N-terminus. The cDNAs were transfected into T-Rex 293 cells and treated with doxycycline (20 ng/ml) 16 hrs post transfection. As shown in Fig. 1, massive cell death was seen after 26 hrs of treatment with 20 ng/ml doxycycline in cells that expressed G573C and G573S but not those that expressed the wild type TRPV3. The untreated cells remained normal with very few dead cells. The fluorescence of GFP-TRPV3 wild type was readily detectable in doxycycline-treated but not untreated cells. The fluorescence signal is much weaker in cells that expressed the mutant constructs, indicating that only a relatively low amount of the mutant protein is enough to cause cell death and consequently prevent further synthesis of the GFP-fusion protein. Consistent with this idea, substantial cell death is already clearly evident 7 hrs after the addition of doxycycline (Fig. 2A).
Figure 1. Expression of G573C and G573S mutant of TRPV3 in mammalian cells causes cell death.

T-Rex293 cells transfected with GFP-TRPV3 wild type (V3WT) and the mutants (V3G573C and V3G573S) in pcDNA4/TO vector were treated or not with 20 ng/ml doxycycline. Bright field (middle) and fluorescence (sides) images were taken at 26 hrs. Fluorescence images were taken using the same exposure time, gain and other camera settings. Substantial cell death (round-up cells) is seen in both the G573C and G573S expressing cells treated with doxycycline. Only few dead cells are present in untreated cells and cells expressing V3WT.
Figure 2. G573S expression and cell survival are partially rescued by ruthenium red but not by lowering extracellular Ca2+.

T-Rex293 cells were transfected with GFP-V3WT or GFP-V3G573S as in figure 1 and fusion protein expression was induced by 20 ng/ml doxycycline for 7 hrs. A, phase contrast (middle) and fluorescence (sides) images of cells untreated or treated with 10 μM ruthenium red (RR) for the duration of doxcycline induction. Note, the larger number of normal looking cells and more prominent fluorescence signal in the RR-treated than untreated group. B, images of cells with GFP-V3WT (left) and GFP-V3G573S (right) expression induced in a low Ca2+ medium for 8 hrs. Severe cell death and low fusion protein expression are still obvious with the mutant. For both conditions shown in A and B, GFP-V3G573C had a similar effect as GFP-V3G573S (not shown). Fluorescence images were taken using the same exposure time, gain and other camera settings.
To verify whether cell death and the low expression levels resulted from spontaneous channel activity during the culture period when the expression of the mutant TRPV3 was induced, a TRPV channel blocker, ruthenium red (RR at 10 μM), was added together with doxycycline. RR did not cause any obvious change in the expression of the wild type TRPV3, but it decreased the severity of cell death and increased the expression level of the mutant proteins (as seen by the number of cells that had higher intensity green signal than those not treated with RR) within the first 6-8 hrs of the induction (Fig. 2A). However, this rescue was incomplete. After 24 hrs, most cells in the G573S or G573C-transfected groups were still dead although the GFP signal was still much higher than cells that were not treated with RR (not shown).
A potential detrimental consequence of the elevated TRPV3 basal activity in the transfected cells is Ca2+ overload due to continued Ca2+ influx through the open channel. To test whether Ca2+ influx is responsible for the cell death effect of the mutant channels, extracellular Ca2+ concentration in the culture medium was reduced to ∼0.5 μM by the addition of 2 mM EGTA. Under this condition, induction of G573S or G573C expression still caused severe cell death, just like in the normal medium that contains 1.8 mM Ca2+ (Fig. 2B right for G573S). By contrast, cells expressing the wild type TRPV3 survived for the duration of the experiment (<30 hrs) despite the round-up appearance, which is typical for HEK 293 cells in the low Ca2+ medium (Fig. 2B left). These results demonstrate that the cell killing effect and impaired protein expression of the G573 mutants are consequences of their constitutive activity; however, the increased Ca2+ influx is not the only reason for these damages.
Enhanced basal activity and altered response to ligand stimulation of the G573 mutants expressed in Xenopus oocytes
Because TRPV3 is a heat-activated channel, the constitutive activity could result from a reduced temperature threshold that allows G573 mutants to be much more active than the wild type channel under the normal incubation temperature of 37 °C for mammalian cell culture. In previous studies, we have successfully expressed TRPV3 and measured its function in Xenopus oocytes [15,16]. We reasoned that because the oocytes are routinely incubated at 19 °C, cells expressing the G573 mutants might be able to survive at this low temperature.
The oocytes were injected with the cRNA for the wild type TRPV3 and the G573S and G573C mutants and incubated at 19 °C. Typically, at one day (18-24 hrs) after cRNA injection, only less than 20% of the wild type TRPV3-injected oocytes showed some level of discoloring of the animal and vegetal poles, which is likely due to damages caused by the injection alone. By contrast, about 75% of the mutant TRPV3-injected oocytes showed obvious discoloring, indicating that they are either dead or very unhealthy. Of the remaining ones, two-electrode voltage clamp experiments were performed. Unlike those injected with cRNA for the wild type, the oocytes injected with the cRNA for G573S and G573C showed very large basal currents (-30.5 ± 7.6 and -32.7 ± 9.1 μA at -100 mV, for G573C and G573S, respectively, n=11 and 18), which are much higher than the wild type TRPV3-expressing cells (-0.52 ± 0.11 μA, n=16) and uninjected control oocytes (-0.31 ± 0.07 μA, n=7) (see Fig. 3A-C for sample trace and 3D for all individual data points). The wild type-expressing cells responded to 2APB (300 μM), a TRPV3 agonist [15], and the response was blocked by ruthenium red (10 μM) (Fig. 3A). However, the mutant-expressing cells did not respond to 2APB, although the constitutively active basal current was inhibited partially by ruthenium red (Fig. 3B).
Figure 3. Constitutive activity of TRPV3 G573 mutants expressed in Xenopus oocytes.

Wild type TRPV3 (WT) and the G573C (G/C) or G573S (G/S) mutants were expressed in oocytes by cRNA injection. Currents were recorded by two-electrode voltage clamp. A, time course of current changes at -100 mV for WT in response to the application of 2APB at the room temperature (22 °C) and to temperature change from 22 °C to 40 °C as indicated. The 2APB-evoked response was blocked by ruthenium red (RR, right panel). Insets show I-V curves obtained by voltage ramps under conditions as indicated. B, similar to A but the oocyte expressed G573C. The basal current was partially blocked by RR (10 μM, right panel). Note the similar response to heat in the presence of RR. The current represents unblocked mutant channel activity as well as leak. The increase is due to normal thermodynamic effect as explained in Fig. 5. C, the response of uninjected oocyte to 2APB and heating. D, summary of basal current at -100 mV for all cells. Data points for individual cells are plotted. Mean values are indicated by the thick bars. Controls (Cntl) are uninjected oocytes.
To further evaluate the ligand response of the mutants, we tested the effect of camphor. At 10 mM, camphor caused a large current increase in oocytes that expressed the wild type TRPV3 but not those that expressed the G573S mutant or uninjected control cells (Fig. 4A). Interesting, oocytes coexpressing the wild type and the mutant also showed a robust response to camphor, as well as to 2APB (see Fig. 5A). The summary data (Fig. 4B) show that for both camphor and 2APB, cells coexpressing the wild type and the G573S mutant together had significantly larger current than those expressing the wild type TRPV3 alone, suggesting that although the mutant is irresponsive to ligand stimulation, its presence, presumably through heteromerization, enhanced the ligand response of the wild type TRPV3.
Figure 4. Ligand-invoked response in oocytes that expressed TRPV3 and the G573 mutants.
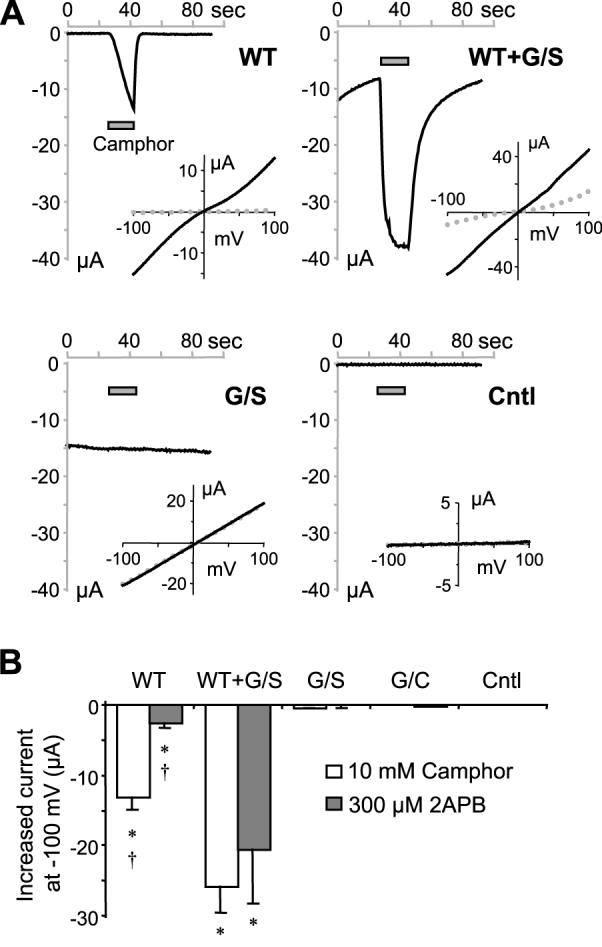
A, representative current traces at -100 mV for oocytes injected with cRNA for wild type TRPV3 (WT), G573S (G/S), WT plus G/S and uninjected controls (Cntl). Camphor (10 mM) was applied as indicated by the gray bars. Insets show I-V curves at basal (dotted gray lines) and at 20 s after camphor application (solid black lines). B, means ± SEM of currents at -100 mV evoked by 10 mM camphor (open bars) and 300 μM 2APB (gray bars), * p < 0.05 different from control, † p < 0.05 different from WT+G/S.
Figure 5. Heat-evoked activity in oocytes that expressed TRPV3 and the G573S mutant.

A, representative current traces at -100 mV for uninjected control (Cntl) and oocytes injected with cRNA for wild type TRPV3 (WT), WT plus G573S (G/S), and G/S alone in response to 300 μM 2APB (open bar), a temperature drop to 10°C, as well as a temperature ramp from 10°C to 40°C as indicated. Note a different scale is used for G/S alone than for other traces. B, I-V relationships for oocytes shown in A at 22°C, 22°C with 2APB, 10°C, and 40°C. C, current vs temperature plots for individual cells grouped by the injected cRNA. Note the large cell-to-cell variation within each group and the different scales used for currents for different groups. D, the same data in C were normalized to current at 40 °C. E, log current vs temperature plot for a representative oocyte that coexpressed WT and G/S. The intersection of the two linear fit lines gives the temperature threshold. Q10 values are determined by the antilog of 10*slope (1010*sploe). F, summary of response/basal (means ± SEM) for temperature rise from 22 to 40°C. * p < 0.05 different from control, † p < 0.05 different from WT+G/S.
Altered temperature response of the G573 mutants expressed in Xenopus oocytes
The temperature response of mutant-expressing cells was obvious, with the current decreased upon lowering the temperature and increased upon raising the temperature. However, the presence of ruthenium red did not block the temperature response (Fig. 3B). In order to quantify the temperature responsiveness, we first reduced the bath temperature to 10 °C for 2 min to allow the current to stabilize at this low temperature. Then the temperature was increased to 40 °C using a temperature ramp (Fig. 5A). Plotting the current at -100 mV as a function of temperature revealed linear relationships for all oocytes injected with the mutant cRNA between 10 to 40 °C. The 10-degree temperature coefficient (Q10) values were 1.245 ± 0.006 for G573S (n = 7) and 1.249 ± 0.007 for G573C (n = 5), suggesting that the responses are due to normal thermodynamic changes rather than temperature sensitivity of the channel. It appears that the G573 mutants are fully active at as low as 10 °C, the lowest temperature we have achieved in our system. The near linear current-voltage (I-V) relationships seen for these mutants at all temperatures (Fig. 5B) are in support of this notion because fully activated TRPV3 channels always display a linear I-V curve (R.X. and M.X.Z. unpublished observation). It is possible that the temperature thresholds of these mutants are lower than 0°C, which would be impractical to measure experimentally by any conventional method.
An interesting observation is that for the wild type TRPV3, the heat response is rather small and not significantly different from that obtained from the uninjected oocytes (Figs. 3A, 3C, 5C, and 5D). In response to temperature rise from 22 to 40 °C, the net current increase at -100 mV was 0.20 ± 0.07 μA for controls (n = 7) and 0.72 ± 0.57 μA for TRPV3 (n = 15). Raising the temperature higher to 46 °C resulted in much larger current increases to 15.6 ± 5.8 μA for control (n = 6) and 28.8 ± 7.6 μA for TRPV3 (n = 10). Although in both cases, the increase appeared larger for TRPV3 than for uninjected controls, the p values, 0.1 for 40 °C and 0.24 for 46 °C, did not reach statistic difference. We were unable to test higher temperatures because all oocytes were severely damaged after exposing to 47-51 °C. Therefore, the actual temperature threshold for TRPV3 expressed in Xenopus oocytes remains undermined. Since the heat response of TRPV3 expressed in oocytes is greatly enhanced in the presence of 2APB [15] and it is clearly detectable by whole-cell patch clamp recording in the absence of 2APB when the same channel is expressed in CHO and HEK 293 cells [4,18], it is possible that the temperature threshold of TRPV3 in oocytes is slightly higher than those determined in mammalian cells (33-39 °C). However, the large endogenous response to heat of higher than 40 °C makes this measurement very difficult.
To recapitulate the condition in keratinocytes of the hairless mutant rodents, which are autosomal dominant and express about equal amount of the wild type and the mutant TRPV3 [19], we coexpressed the wild type TRPV3 and the G573 mutants in oocytes. Although the basal activity at the room temperature is still high (-10.1 ± 3.7 μA at -100 mV, n=15, see Fig. 3D for individual data points), the current shows outward rectification and is blocked almost completely by lowering the temperature to 10 °C (Figs. 5A and 5B).
The current-temperature plots showed a clear biphasic response to temperatures between 10 and 40 °C (Fig. 5C, upper right graph). This was more clearly seen after normalizing the currents to those at 40 °C. As shown in Fig. 5D, only the wild type and G573S coexpressing cells revealed biphasic relationships while all other cells showed linear relationship between 10 and 40 °C. To determine the Q10 values and the temperature threshold, the current at -100 mV in the log scale was plotted as a function of temperature and the two linear portions of the curve were fitted by linear regression as exemplified in Fig. 5E. The Q10 values before and after the transition are 1.45 ± 0.15 and 3.63 ± 0.33, respectively. The temperature threshold is 15 ± 1 °C (n = 5), which is significantly lower than the published value for the wild type TRPV3 (33-39°C) expressed in mammalian cells [4,21,22]. The Q10 value of 3.6 at above 15 °C for the coexpressed oocytes indicates a moderate temperature regulation. As such, the wild type plus G573S coexpressing cells are the only ones that showed heat-evoked activation (expressed as the current at 40 °C/basal current at 22 °C) that is significantly higher than uninjected control oocytes (Fig. 5F). Note: no leak subtraction was performed for this plot because temperature rise also increases the leak current in a manner that follows normal thermodynamic roles, which should form the baseline for the comparison across all samples.
The wild type TRPV3 sustained the expression of G573 mutants in mammalian cells
The increased temperature threshold and reduced basal activity suggest that coexpression with the wild type TRPV3 might protect cells against the damages caused by the G573 mutants. To examine this possibility in mammalian cells, the wild type TRPV3 cDNA in pcDNA3 vector was cotransfected with the inducible constructs encoding the GFP-tagged wild type, G573S, or G573C mutant into T-Rex 293 cells. In the presence of constitutive expression of the wild type TRPV3, while induction of the GFP-TRPV3 wild type for 8 hrs did not cause dramatic cell death, that of GFP-G573S or GFP-G573C still greatly damaged the cells (Fig. 6 for G573S), suggesting that coexpression of the wild type subunit is insufficient to rescue the cells from damages caused by the G573 mutants. However, even through the cells look damaged, the green fluorescence signal was readily detectable in many cells, indicating that the wild type subunit can either prolong the life of the mutant protein or allow more mutant protein to be made through a partial rescue of the protein synthesis machinery. Therefore, coexpression of the wild type TRPV3 protein is protective to some extent, but it is not sufficient for a complete rescue.
Figure 6. Partial rescue of G573 mutant expression in mammalian cells by the wild type protein.

T-Rex293 cells were cotransfected with GFP-V3WT or GFP-V3G573S together with either the wild type TRPV3 in pcDNA3 or the empty pcDNA3 vector (as a control). Expression of GFP-fusion proteins was induced by 20 ng/ml doxyclycline 16 hrs post transfection. Phase contrast (middle) and fluorescence (sides) images of cells were taken at 8 hrs after induction. Fluorescence images were taken using the same exposure time, gain and other camera settings. Note the more pronounced fluorescence signal with the coexpression of wild type TRPV3 with the G573S mutant even though there are still a large number of round-up cells. Similar results were obtained for GFP-V3G573C (not shown).
Discussion
The data reported herein demonstrate that both G573C and G573S mutants of mouse TRPV3 are constitutively active and the mutant channels are insensitive to heat and TRPV3 ligands, 2APB and camphor. In both HEK 293 cells and Xenopus oocytes, the expression of the mutant channels causes cell death, presumably due to the elevation of intracellular Ca2+ concentrations, as well as membrane depolarization, as a result of the constitutively open channel. On appearance, the ionic currents mediated by the mutant channels in oocytes responded to temperature changes greatly with large net increases and decreases in current amplitudes. However, our close examination of the temperature dependence between 10 and 40 °C revealed a low Q10 value of <2, indicating that there is no thermal gating within this temperature range for the mutant channel. It is quite possible that the temperature range of thermal gating was shifted to much lower levels (<10 °C) for homomeric mutant channels. Consistent with this notion is that when the G573S mutant was coexpressed with the wild type protein, the mean temperature threshold was moved to 15 °C and the Q10 value reached ∼3.6 between 15 and 30 °C. The rather moderate thermal sensitivity displayed by these cells most likely resulted from the heterogeneity of tetrameric channel species composed of the mutant and wild type in all possible combinations.
It is well-documented that TRPV3 is abundantly expressed in skin keratinocytes. The association of the G573S mutation of TRPV3 in DS-Nh mice and the G573C mutation in the G573C WBN/Kob-Ht rats with a hairless phenotype and predisposition to dermatitis-like skin lesions strongly suggests a pivotal role of TRPV3 in hair growth and general skin health [19]. Dysregulation of TRPV3 channel function appeared to be the initial trigger for a cascade of events at cell and tissue levels that led to the hairless and dermatitis phenotypes in these rodent strains. Among them, massive mast cell infiltration and enhanced histamine levels in the skin tissue are indicative of altered allergic and/or inflammatory functions. Histological analysis revealed increased but disarranged hair follicles in the affected skin, consistent with the notion that mast cells and histamine are important for hair cycle, a process of cyclic growth and regression of hair follicles [19]. Interestingly, skin allergen, such as eugenol, is an activator of TRPV3. Stimulation of primary keratinocytes by eugenol and other chemical allergens, presumably through activation of TRPV3, induces the release of the inflammatory cytokine interleukin-1α [14,23], which has been shown to enhance mast cell proliferation in the presence of fibroblasts [24]. Thus, enhanced TRPV3 function can indeed explain the allergic and inflammatory manifestations displayed by the affected skin tissues of the mutant rodents. Furthermore, a recent study has identified TRPV3 as a novel target of androgen receptors that displays enhanced expression in response to dihydrotestosterone [25], suggestive a potential link of TRPV3 upregulation in androgenetic alopecia, a common problem that affects approximately 50% of the male population.
Our data also demonstrate that when coexpressed with the wild type TRPV3, the spontaneous activity of the G573 mutants are significantly reduced and the responses to heat, camphor, and 2APB are not only recovered but also enhanced. However, this somewhat protective effect is insufficient to rid of the detriment caused by the mutation, thus still leaving significant basal activation under physiological conditions. This is consistent with the autosomal dominant inheritance pattern of the DS-Nh mice and WBN/Kob-Ht rats. In HEK 293 cells heterologous expression of the mutant TRPV3 led to cell death regardless whether the wild type protein was co-expressed or not. By contrast, skin keratinocytes in the mutant rodents survive despite the presence of the TRPV3 mutation. Additional factors in the native environment may protect the keratinocytes from death under conditions when the TRPV3 channel activity is constantly augmented. One of them may be the low Ca2+ environment in the epidermis, which reduces the possibility of Ca2+ overload because of the TRPV3-mediated Ca2+ influx. However, induction of the TRPV3 mutant expression in HEK 293 cells in a low Ca2+ medium did not prevent the cell death effect, indicating that other events downstream of TRPV3 activation/expression, e.g. membrane depolarization, may also be important. The partial rescue by RR demonstrates that the cell death and impaired protein expression are due to TRPV3 channel function. Nevertheless, although the native keratinocytes survive with the G573 mutations, their functions are clearly impaired by the hyperactivity of the mutant channel as shown by the lack of hair growth and the high susceptibility to dermatitis.
In conclusion, we have demonstrated that the naturally occurring G573S and G573C mutant of TRPV3 found in DS-Nh mice and WBN/Kob-Ht rats, respectively, are spontaneously active. In heterologous systems, the expression of the mutant TRPV3 causes cell death. The homomeric mutant channels do not show temperature gating between 10 and 40 °C and are not responsive to chemical ligands, camphor and 2APB. Coexpression of the mutant with the wild type TRPV3 produces heteromeric channels activated by warming with a much reduced temperature threshold (15 °C) and enhanced net ligand response. Overall, our data are consistent with the idea that an over-active TRPV3 function is the initial trigger for the hairless and dermatitis phenotypes of the DS-Nh mice and WBN/Kob-Ht rats. Given that TRPV3 is subject to complex regulation by multiple environmental and physiological factors, just like other TRPV channels, focusing on the functional regulation of this channel will not only help reveal mechanisms of pathogenesis but also offer new therapeutic approaches for alopecia and certain skin diseases. Furthermore, the mutated glycine is one of the three conserved residues amongst all members of the class I TRP superfamily, including TRPVs, TRPMs, and TRPCs. It would be interesting to see whether this residue plays a critical function in channel gating for all class I TRPs.
Acknowledgements
We thank Ms Dina Chuang-Zhu for technical assistant. The work was supported in part by US National Institutes of Health grants NS042183, NS056942 and NS045758.
The authors declare no conflict of interest
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference
- 1.Montell C, Birnbaumer L, Flockerzi V, et al. A unified nomenclature for the superfamily of TRP cation channels. Mol Cell. 2002;9:229–231. doi: 10.1016/s1097-2765(02)00448-3. [DOI] [PubMed] [Google Scholar]
- 2.Jordt SE, McKemy DD, Julius D. Lessons from peppers and peppermint: the molecular logic of thermosensation. Curr Opin Neurobiol. 2003;13:487–492. doi: 10.1016/s0959-4388(03)00101-6. [DOI] [PubMed] [Google Scholar]
- 3.Patapoutian A, Peier AM, Story GM, Viswanath V. ThermoTRP channels and beyond: mechanisms of temperature sensation. Nat Rev Neurosci. 2003;4:529–539. doi: 10.1038/nrn1141. [DOI] [PubMed] [Google Scholar]
- 4.Peier AM, Reeve AJ, Andersson DA, et al. A heat-sensitive TRP channel expressed in keratinocytes. Science. 2002;296:2046–2049. doi: 10.1126/science.1073140. [DOI] [PubMed] [Google Scholar]
- 5.Chung MK, Lee H, Caterina MJ. Warm temperatures activate TRPV4 in mouse 308 keratinocytes. J Biol Chem. 2003;278:32037–32046. doi: 10.1074/jbc.M303251200. [DOI] [PubMed] [Google Scholar]
- 6.Chung MK, Lee H, Mizuno A, Suzuki M, Caterina MJ. TRPV3 and TRPV4 mediate warmth-evoked currents in primary mouse keratinocytes. J Biol Chem. 2004;279:21569–21575. doi: 10.1074/jbc.M401872200. [DOI] [PubMed] [Google Scholar]
- 7.Chung MK, Lee H, Mizuno A, Suzuki M, Caterina MJ. 2-aminoethoxydiphenyl borate activates and sensitizes the heat-gated ion channel TRPV3. J Neurosci. 2004;24:5177–5182. doi: 10.1523/JNEUROSCI.0934-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moqrich A, Hwang SW, Earley TJ, et al. Impaired thermosensation in mice lacking TRPV3, a heat and camphor sensor in the skin. Science. 2005;307:1468–1472. doi: 10.1126/science.1108609. [DOI] [PubMed] [Google Scholar]
- 9.Lee H, Iida T, Mizuno A, Suzuki M, Caterina MJ. Altered thermal selection behavior in mice lacking transient receptor potential vanilloid 4. J Neurosci. 2005;25:1304–1310. doi: 10.1523/JNEUROSCI.4745.04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liedtke W, Choe Y, Marti-Renom MA, et al. Vanilloid receptor-related osmotically activated channel (VR-OAC), a candidate vertebrate osmoreceptor. Cell. 2000;103:525–535. doi: 10.1016/s0092-8674(00)00143-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Strotmann R, Harteneck C, Nunnenmacher K, Schultz G, Plant TD. OTRPC4, a nonselective cation channel that confers sensitivity to extracellular osmolarity. Nat Cell Biol. 2000;2:695–702. doi: 10.1038/35036318. [DOI] [PubMed] [Google Scholar]
- 12.Watanabe H, Vriens J, Prenen J, Droogmans G, Voets T, Nilius B. Anandamide and arachidonic acid use epoxyeicosatrienoic acids to activate TRPV4 channels. Nature. 2003;424:434–438. doi: 10.1038/nature01807. [DOI] [PubMed] [Google Scholar]
- 13.Smith PL, Maloney KN, Pothen RG, Clardy J, Clapham DE. Bisandrographolide from Andrographis paniculata activates TRPV4 channels. J Biol Chem. 2006;281:29897–29904. doi: 10.1074/jbc.M605394200. [DOI] [PubMed] [Google Scholar]
- 14.Xu H, Delling M, Jun JC, Clapham DE. Oregano, thyme and clove-derived flavors and skin sensitizers activate specific TRP channels. Nat Neurosci. 2006;9:628–635. doi: 10.1038/nn1692. [DOI] [PubMed] [Google Scholar]
- 15.Hu HZ, Gu Q, Wang C, et al. 2-aminoethoxydiphenyl borate is a common activator of TRPV1, TRPV2, and TRPV3. J Biol Chem. 2004;279:35741–35748. doi: 10.1074/jbc.M404164200. [DOI] [PubMed] [Google Scholar]
- 16.Hu HZ, Xiao R, Wang C, et al. Potentiation of TRPV3 channel function by unsaturated fatty acids. J Cell Physiol. 2006;208:201–212. doi: 10.1002/jcp.20648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gu Q, Lin RL, Hu HZ, Zhu MX, Lee LY. 2-aminoethoxydiphenyl borate stimulates pulmonary C neurons via the activation of TRPV channels. Am J Physiol Lung Cell Mol Physiol. 2005;288:L932–L941. doi: 10.1152/ajplung.00439.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chung MK, Guler AD, Caterina MJ. Biphasic currents evoked by chemical or thermal activation of the heat-gated ion channel, TRPV3. J Biol Chem. 2005;280:15928–15941. doi: 10.1074/jbc.M500596200. [DOI] [PubMed] [Google Scholar]
- 19.Asakawa M, Yoshioka T, Matsutani T, et al. Association of a mutation in TRPV3 with defective hair growth in rodents. J Invest Dermatol. 2006;126:2664–2672. doi: 10.1038/sj.jid.5700468. [DOI] [PubMed] [Google Scholar]
- 20.Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D. The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature. 1997;389:816–824. doi: 10.1038/39807. [DOI] [PubMed] [Google Scholar]
- 21.Xu H, Ramsey IS, Kotecha SA, et al. TRPV3 is a calcium-permeable temperature-sensitive cation channel. Nature. 2002;418:181–186. doi: 10.1038/nature00882. [DOI] [PubMed] [Google Scholar]
- 22.Smith GD, Gunthorpe MJ, Kelsell RE, et al. TRPV3 is a temperature-sensitive vanilloid receptor-like protein. Nature. 2002;418:186–190. doi: 10.1038/nature00894. [DOI] [PubMed] [Google Scholar]
- 23.Corsini E, Primavera A, Marinovich M, Galli CL. Selective induction of cell-associated interleukin-1alpha in murine keratinocytes by chemical allergens. Toxicology. 1998;129:193–200. doi: 10.1016/s0300-483x(98)00088-2. [DOI] [PubMed] [Google Scholar]
- 24.Kameyoshi Y, Morita E, Tanaka T, Hiragun T, Yamamoto S. Interleukin-1α enhances mast cell growth by a fibroblast-dependent mechanism. Arch Dermatol Res. 2000;292:240–247. doi: 10.1007/s004030050481. [DOI] [PubMed] [Google Scholar]
- 25.Jariwala U, Prescott J, Jia L, et al. Identification of novel androgen receptor target genes in prostate cancer. Mol Cancer. 2007 Jun 6;39 doi: 10.1186/1476-4598-6-39. 6(1):39 [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]