

Abstract
Sequences of three gene fragments (flaA, flaB, and vacA) from Helicobacter pylori strains isolated from patients in Germany, Canada, and South Africa were analyzed for diversity and for linkage equilibrium by using the Homoplasy Test and compatibility matrices. Horizontal genetic exchange in H. pylori is so frequent that different loci and polymorphisms within each locus are all at linkage equilibrium. These results indicate that H. pylori is panmictic. Comparisons with sequences from Escherichia coli, Neisseria meningitidis, and Drosophila melanogaster showed that recombination in H. pylori was much more frequent than in other species. In contrast, when multiple family members infected with H. pylori were investigated, some strains were indistinguishable at all three loci. Thus, H. pylori is clonal over short time periods after natural transmission.
Keywords: nucleotide sequencing/horizontal genetic exchange/evolution/linkage equilibrium
Helicobacter pylori is genetically one of the most diverse bacterial species so far reported. It also is subject to the highest known rate of intraspecific recombination. This paper presents the evidence for these two assertions and discusses their significance. In particular, is there a causal connection between high variability and high recombination rate?
Infection with H. pylori, a common human pathogen, causes chronic type B gastritis and is a prerequisite for the development of duodenal ulcers and most gastric ulcers (1). H. pylori infection is also an important risk factor for gastric malignancies such as adenocarcinoma (2) and mucosa-associated lymphoid tissue lymphoma (3). Most microbiological studies of H. pylori have concentrated on virulence factors (4) and much less is known about its population biology. DNA fingerprinting (5–7), multilocus enzyme electrophoresis (MLEE) (8) and DNA sequence analysis of the ureC/glmM and cagA genes (9–11) have revealed an unusually high degree of genetic variability within this species, whose origin is unclear.
Motility is essential for the virulence of H. pylori and is based on a flagellar apparatus with several unique features (12). The flagellar filament is composed of two flagellins, FlaA and FlaB, which are covered by a flagellar sheath and therefore thought to be shielded from antibody selection. Little has been published about the sequence variability of the flaA and flaB genes. VacA is a secreted vacuolating cytotoxin thought to be involved in ulcerogenesis (13), whose sequence variability seems to reflect mosaicism (14). During sequence analyses of the flaB gene directed to better understanding of its role in virulence, we noticed a degree of sequence variability which seemed unprecedented in the bacterial kingdom. We extended these analyses to data available in GenBank from gene fragments of the flaA and vacA genes and tested the sequence diversity of these three gene fragments among strains isolated from individuals in three different study groups. The sequence data were analyzed for evidence of recombination using a novel tool, the Homoplasy Test (15), which has been designed to test recombination in nucleotide sequence data sets derived from closely related organisms and by compatibility matrices, which can reveal reticulate evolution (16). The data provide overwhelming evidence that recombination in H. pylori is so much more frequent than mutation as to effectively randomize the sequences and generate linkage equilibrium. Clonal descent was observed only in strains isolated from paired family members.
MATERIALS AND METHODS
Bacterial Strains.
H. pylori bacteria were isolated from epidemiologically unrelated individuals who underwent gastroduodenal endoscopy within the Ruhr area in Germany (54 strains) and the “Cape-colored” population in Capetown, South Africa (22 strains) as well as from four families in Hessen, Germany (14 strains from 16 individuals).
DNA Sequences.
Thirty-three vacA and flaA sequences were from GenBank (accession nos. U63218–U63287, excluding U63244, and U63250–63252) and had been obtained by Robin N. Beech (McGill University, Montreal, Quebec, Canada) from strains isolated from 33 Canadians. Other sequences were obtained by direct sequencing of PCR products generated using the following primers: flaA, OLHPFlaA-4 (ATT GAT GCT CTT AGC GTC) and OLHPFlaA-9 (CAA GCG TTA TTG TCT GGT C); flaB, OLHPFlaB-9 (AAG GCA TGC TCG CTA GCG) and OLHPFlaB-10 (TAA TGT CTC TAG CGT CGG); and vacA, OLHPVacA-3 (ACA ACC GTG ATC ATT CCA GC) and OLHPVacA-4 (ATA CGC TCC CAC GTA TTG C). PCR reactions were performed by using a Perkin–Elmer GeneAmp 2400 thermal cycler as follows: denaturation, 94°C for 1 min; annealing 50°C for 1 min; extension, 72°C for 1 min; 35 cycles. Then 75 ng of PCR products purified by using the QIAquick PCR purification kit (Qiagen) were used in cycle sequencing reactions from both strands with the ABI Prism Dye Terminator cycle sequencing kit (Applied Biosystems) by using the primers listed and independent PCR products for each strand. All sequences were reduced to a common length consisting of nucleotides 634–1,104 (flaA, GenBank accession no. X60746), 798–1,136 (flaB, GenBank accession no. L08907), and 802–1,245 (vacA, GenBank accession no. Z26883).
Sequences from other species were as follows. Neisseria meningitidis: 11 unique gene fragments from the housekeeping genes abcZ, adk, aroE, gdh, mtg, pdhC, pgm, pilA, pip, ppk, and serC (17) (GenBank accession nos. AF037753–AF037981); Escherichia coli: icd (18) (AF017587–AF017603, coordinates 19–1182), mdh (19) (ECU04742–ECU04760 and ECU04770, coordinates 1–849), putP (20) (L01132, L01133 and L01150–L01159, coordinates 424-1890), and trpC (21) (U23489–U23500, U25884–U25886, U25417–U25423, and U25425–U25429, coordinates 15–1370); and Drosophila melanogaster: Adh (22) (M17827, M17828, and M17830–M17837, exons only), Amy (23) (L22716, L22719, L22721, L22725–L22727, L22729, L22731, and L22733, exons only), Est6 (24) (J01467, exons only), and white (informative sites from the whole locus as summarized in ref. 25).
Phylogenetic Analyses.
Sequences were aligned using seqlab and pileup from the Wisconsin Package Version 9.1, Genetics Computer Group (GCG), Madison, WI. Ka and Ks values using Jukes–Cantor distances (26) were calculated using dnasp 2.52 (27). The Homoplasy Test (15) was performed by using a modified, faster version of the homoplasy program (ftp://novell-del-valle.vz-berlin.mpg.de/software/homoplasy.zip), which incorporates a Win95/WinNT interface and accepts as input either MSF or MEGA files. This program extracts all polymorphic, synonymous first and third codon position sites plus all uniform third codon position sites, except for stop codons or codons encoding Met or Trp, for which there are no synonymous codons. It calculates Se values either against an outgroup, as described (15), or by multiplying the number of sites by the factor 1.0 (low expression), 0.83 (intermediate), or 0.73 (high). It also calculates homoplasy ratios as described (15). The data reported here were the mean values from at least five independent determinations without an outgroup; the variation between independent calculations were no more than a few percentage of the mean value. Se values were calculated assuming high expression of flaA, intermediate expression of vacA, and low expression of flaB. Analyses using sequences from H. mustelae as an outgroup for flaA and flaB yielded similar results. For sequences from the other species, intermediate expression was assumed and comparable results were obtained when sequences from Salmonella enterica were used as an outgroup for the E. coli analyses. Compatibility matrices and mean neighborhood similarity values were calculated by using the program reticulate (16).
All available sequences were used for the Homoplasy Test whereas only unique sequences were used for calculating Ka and Ks and for the compatibility matrices. For N. meningitidis, only unique sequences were used for all tests to avoid the bias introduced by sequencing gene fragments from multiple representatives of uniform clonal groupings (17).
RESULTS
Free Recombination at Three Loci in H. pylori.
A 339-bp fragment of the flaB gene encoding one of the two flagellins of H. pylori was sequenced from 54 strains isolated from patients with gastritis or gastroduodenal ulcers from the Ruhr region and elsewhere in Germany. Somewhat surprisingly, all sequences were unique, even within this single geographical region. Sequences of gene fragments from flaA (the second flagellin) and vacA (vacuolating cytotoxin) genes from strains isolated from 33 individuals in Canada had been submitted to GenBank by Robin N. Beech. These sequences too were all unique. For all three genes, ≈20% of the sites were polymorphic in different strains and almost all polymorphism resulted in synonymous substitutions, which did not affect the amino acid sequence. On average, pairs of strains differed by 15–21% of nucleotides at synonymous positions but only at 0.3–2.5% of nonsynonymous positions, which can result in an amino acid change (Table 1). These observations indicate that the function of all three genes must be under strong purifying selection because otherwise the proportion of synonymous to nonsynonymous mutations would have been closer to equality. We also performed similar analyses with genes or gene fragments from GenBank from the species N. meningitidis, D. melanogaster, and E. coli. The species N. meningitidis possesses considerable sequence diversity (17) and undergoes frequent recombination (28). D. melanogaster recombines at each generation during gamete formation, and much of the sequence variation between related isolates of E. coli has been attributed to recombination (29). The level of synonymous polymorphism was comparable between H. pylori and N. meningitidis and was considerably lower in E. coli and D. melanogaster (Table 1).
Table 1.
Sequence variability among unique sequences at the flaA, flaB, and vacA loci in H. pylori from different sources
| Gene, bp | No. of unique sequences, total | % Polymorphic | Mean % Ka, range | Mean % Ks, range |
|---|---|---|---|---|
| H. pylori | ||||
| vacA (444) | ||||
| Canada | 33 (33) | 21.8 | 2.5 ± 1.1 | 14.1 ± 5.5 |
| S. Africa | 21 (22) | 17.6 | 2.2 ± 1.6 | 14.7 ± 7.3 |
| German families | 9 (14) | 12.8 | 2.2 ± 1.6 | 17.4 ± 4.7 |
| All data | 63 (69) | 25.4 | 3.1 ± 1.8 | 16.8 ± 6.4 |
| flaA (471) | ||||
| Canada | 33 (33) | 17.0 | 0.5 ± 0.6 | 15.6 ± 3.9 |
| S. Africa | 21 (22) | 10.8 | 0.03 ± 0.08 | 13.2 ± 4.3 |
| German families | 9 (14) | 9.8 | 0.1 ± 0.2 | 16.5 ± 3.9 |
| All data | 63 (69) | 21.0 | 0.3 ± 0.5 | 15.6 ± 4.0 |
| flaB (339) | ||||
| Germany | 54 (54) | 18.9 | 0.3 ± 0.3 | 21.4 ± 6.4 |
| S. Africa | 19 (22) | 15.3 | 0.4 ± 0.3 | 24.3 ± 10.4 |
| German families | 9 (14) | 11.8 | 0.4 ± 0.5 | 20.8 ± 6.0 |
| All data | 82 (90) | 22.4 | 0.4 ± 0.3 | 23.0 ± 7.4 |
| D. melanogaster (Adh, Amy, Est6) | 0.2 (0.1–0.3) | 2.7 (2.1–3.3) | ||
| E. coli (putP, icd, mdh, trpC) | 0.2 (0.06–0.8) | 6.6 (4.1–9.7) | ||
| N. meningitidis (11 genes) | 0.7 (0.2–7.8) | 13.4 (5.9–26.8) | ||
Phylogenetic analysis based on tree algorithms was inappropriate for the three genes from H. pylori because the arrangement of the clusters resembled a bush rather than a tree (data not shown), suggesting that frequent recombination had distorted any evidence of phylogenetic descent. The importance of recombination was estimated by using the Homoplasy Test (15). The logic of this test is as follows. If the same site changes twice in the ancestry of a set of sequences, this is called a homoplasy. A parsimonious tree for a set of sequences is constructed, and the number of observed homoplasies, obsh, is calculated. We also need to calculate exph, the number expected if the population is clonal, and sh, the number expected if recombination is so frequent that the population is in linkage equilibrium. One can then calculate the “homoplasy ratio,”
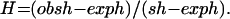 |
The homoplasy ratio, H, is a number whose expectation is 0 if the population is clonal and 1.0 if it is in linkage equilibrium. For simplicity, analysis is confined to potentially synonymous sites. The value of exph depends on the number of polymorphic sites, and on Se, the “effective site number.” In general, because of codon bias, Se will be less than S, the number of potentially synonymous sites. This effect, estimated as 0.73 × S for highly expressed genes and 0.83 × S for genes with medium expression (15), has been implemented in the homoplasy program. The values of sh were calculated by randomly shuffling the columns of the strains X sites matrix, while retaining the observed number of bases at each site. The values used are the means from ten such shuffles.
The results of such homoplasy analyses are shown in Table 2. Among the strains of H. pylori isolated from the German and Canadian patients, all three genes yielded values of H which are close to 1.0 and considerably higher than values of H observed with the three control species. These results show that sequence polymorphism within H. pylori genes is close to linkage equilibrium, both in Canadian and German populations. Homoplasy tests with other (housekeeping) genes from H. pylori also have yielded high values of H (unpublished data). In contrast, at least some degree of linkage disequilibrium is indicated by the data from the other species. For the gene with the highest H value, the D. melanogaster white locus, Kirby and Stephens (25) identified an 800-bp region that is not compatible with the neutral equilibrium model. Excluding this region from the analysis only raised the H value to 0.66. The other genes from D. melanogaster yielded still lower H values, indicating that selection has caused a departure from linkage equilibrium even in a species in which recombination occurs at each generation.
Table 2.
Analysis of sequences from various species with the Homoplasy Test and by compatibility matrices
| Species, gene | No. of sequences | Sites
|
Homoplasy Test
|
Similarity | ||||
|---|---|---|---|---|---|---|---|---|
| syn. | var. | inf. | Se | obsh | H | |||
| H. pylori | ||||||||
| flaA | ||||||||
| Canada | 33 | 144 | 59 | 40 | 105 | 124 | 0.8 | 0.44 |
| S. Africa | 22 | 156 | 49 | 34 | 114 | 55 | 0.55 | 0.52 |
| flaB | ||||||||
| Germany | 54 | 110 | 61 | 47 | 110 | 261 | 0.83 | 0.35 |
| S. Africa | 22 | 112 | 50 | 38 | 112 | 66 | 0.41 | 0.38 |
| vacA | ||||||||
| Canada | 33 | 111 | 41 | 29 | 92 | 83 | 0.93 | 0.42 |
| S. Africa | 22 | 124 | 44 | 31 | 103 | 36 | 0.46 | 0.68 |
| D. melanogaster | ||||||||
| Adh | 15 | 253 | 16 | 12 | 210 | 4 | 0.16 | 0.81 |
| Amy | 10 | 487 | 26 | 23 | 404 | 9 | 0.27 | 0.72 |
| EST6 | 13 | 528 | 28 | 14 | 438 | 10 | 0.5 | 0.5 |
| white | 15 | 5972 | 72 | 52 | 4000 | 61 | 0.62 | 0.53 |
| geom. mean: 0.34 | 0.63 | |||||||
| E. coli | ||||||||
| putP | 12 | 482 | 100 | 64 | 400 | 50 | 0.39 | 0.72 |
| icd | 17 | 385 | 65 | 45 | 320 | 45 | 0.41 | 0.63 |
| mdh | 20 | 279 | 33 | 25 | 232 | 11 | 0.24 | 0.86 |
| trpC | 27 | 419 | 63 | 45 | 348 | 23 | 0.12 | 0.75 |
| geom. mean: 0.26 | 0.74 | |||||||
| N. meningitidis | ||||||||
| abcZ | 15 | 134 | 57 | 37 | 134 | 29 | 0.24 | 0.78 |
| pdhC | 24 | 146 | 61 | 56 | 146 | 73 | 0.34 | 0.63 |
| pilA | 36 | 143 | 49 | 42 | 119 | 70 | 0.35 | 0.51 |
| serC | 29 | 135 | 43 | 26 | 112 | 47 | 0.56 | 0.43 |
| geom. mean (11 genes): 0.34 | 0.57 | |||||||
syn., all polymorphic first and third codon position sites plus all uniform third codon position sites; var., variable sites; and inf., informative sites. Se, effective site number; obsh, the observed number of homoplasies; and H, homoplasy ratio. Similarity, mean neighborhood similarity for all pairs of informative sites; geom. mean, geometric mean. The number of sites for the white locus consisted of all sites sequenced since most of the locus consists of untranslated introns. The four genes shown for N. meningitidis are those with the lowest and highest H values plus two genes with intermediate values. Data for the other seven genes is available on request from the authors.
These sets of sequences also were examined by compatibility matrices (16), which score pairs of informative sites for compatibility within maximal parsimony trees. Only very small blocks were compatible within the three genes from H. pylori, and most pairs of sites were incompatible whereas large numbers of sites and larger blocks were compatible within the genes from the three other species (Fig. 1, Table 2). The mean neighborhood similarities correlated significantly with the H ratios (r = −0.72). These data provide further evidence for an unusually high degree of recombination within H. pylori.
Figure 1.

Representative compatibility matrices of alleles from different species. White spaces represent pairs of informative sites that are compatible with a maximal parsimony tree and black spaces show pairs of sites that could not be accommodated by such a tree. The genes selected from each species were broadly representative of the other genes examined and were chosen as including approximately the same number of informative sites. Quantitative mean neighborhood similarities are given in Table 2.
Clonal Descent Over Short Time Periods.
The observation that none of the strains from Germany and Canada possessed identical alleles at any of the three genes raised the possibility that special mechanisms might accelerate the rate of mutation or recombination in H. pylori. It even might be impossible to isolate identical strains from different individuals. We identified four families among industrial laborers in the Hessen region of Germany where multiple family members were infected with H. pylori. All three gene fragments were sequenced from 14 strains isolated from these infected individuals. Within each of the families, one or two pairs of strains were indistinguishable at all three gene loci and only 4/14 strains were singletons (Table 3). Four of the five pairs of strains were isolated from a parent and its child and the fifth from two parents within one family. The sequence diversity between the nine unique allele combinations was comparable with that in the former study population (Table 1). These results suggest that transmission of bacterial clones had occurred within families and also that each of the families investigated had been colonized with more than one unrelated strain of H. pylori. No detectable genetic changes had accumulated within the gene fragments since transmission, showing that H. pylori is clonal over the short term.
Table 3.
Sources of strains with identical flaA, flaB, and vacA gene fragments from four German families
| Family | Identical strains | Unique strains |
|---|---|---|
| 1 | F-C1, M-C2 | |
| 2 | F-M | C1 |
| 3 | M-C1 | F |
| 4 | M-C1 | F, C2 |
F, father; M, mother; and C1, C2: children. The data summarizes the sources of pairs of identical strains within families. All alleles differed between strains from different families.
Identical Alleles in Bacteria from Unrelated Sources.
If H. pylori is clonal over the short term, it might be possible to recognize clonal groupings by analyzing bacteria from other geographical areas. Many unrelated strains of N. meningitidis are isolated from individuals in Europe and the U.S., but the diversity of isolates from Africa is low even in the absence of epidemic disease (30). We sequenced the same three genes from 22 H. pylori strains isolated from Cape-colored patients with gastroduodenal disease in Capetown, South Africa. The Cape-colored population is anthropologically distinct from other South African populations and is descended from Western Europeans, South East Asians, and South Africans (mainly Hottentots) (31). The sequence diversity of the three genes was comparable with that in strains from the other sources (Table 1). As hoped, identical alleles were found in certain strains for each of the genes: one flaA allele was present in two of the 22 strains, three flaB alleles were each present in two strains, and one vacA allele also was present in two strains. However, each pair of strains that was identical at one locus differed at both other loci, suggesting that these pairs of bacteria may have descended from common ancestors but that sufficient recombination had occurred to result in linkage equilibrium between the genes.
Analysis of these sequences with the Homoplasy Test showed values of H that are intermediate between that expected for clonality and for random assortment and that were closer to the results with the other species (Table 2). These intermediate values of H could be explained if, as is likely, the ancestors of the Cape-colored population harbored genetically distinct H. pylori populations, which have not yet had time to reach linkage equilibrium. In agreement, larger blocks of sites and more pairs of sites were compatible with maximal parsimony trees than was the case with the sequences from Canada or Germany (Fig. 1, Table 2).
DISCUSSION
Linkage Equilibrium and Panmixis.
The data presented here indicate that recombination is so frequent in H. pylori that remnants of clonal descent are difficult to discern within individual genes from unrelated bacteria. Furthermore, recombination is so frequent that alleles at independent loci are rarely coinherited for long time periods (linkage equilibrium).
Many other bacterial species consist of clones or clonal groupings whose members have inherited identical alleles at most loci from a common ancestor (17, 32, 33). Within clonal bacteria, the importance of recombination for disrupting clonal relationships has been documented repeatedly (29, 34, 35) but normally only local areas of the chromosome are perturbed (36). In other, less clonal species, including H. pylori (8), linkage equilibrium due to frequent recombination is indicated by MLEE data (37–38). However, MLEE is based on amino acid changes which result in charge differences and yields no information on the degree of sequence variability at synonymous sites. Almost all of the sequence variation described here was at synonymous sites and would not have been detected by MLEE. Nonsynonymous variation resulting in amino acid changes was rare and therefore the sequence diversity is very unlikely to reflect pressure due to selection.
The sequence data presented here show that all strains randomly isolated from individuals in Germany and Canada possessed unique alleles at the flaA, flaB, and vacA loci, that almost all strains isolated from Cape-colored patients possessed unique alleles, and that clonal spread was only found within families. Similarly, all 29 clinical isolates from France possessed unique sequences at the ureC locus (10). Furthermore, although paired alleles can be found in strains from certain human populations, such as the Cape-colored patients, none of the pairs of strains which possessed identical alleles at one locus were identical for the other two alleles tested. These results indicate that the frequency of mixing of alleles at different loci by horizontal genetic exchange is sufficient in H. pylori to rapidly disrupt clonal groupings and that the population structure of the species is panmictic (34). Like other bacterial species such as N. gonorrhoeae (34) and Bacillus subtilis (39) that have a panmictic population structure, H. pylori is naturally competent for DNA transformation (40), which can result in frequent recombination.
Recombination in H. pylori.
The results indicate that H. pylori possesses a pool of alleles sufficiently large that identical sequences are unlikely to occur in random samples of 50 strains. This degree of sequence diversity is exceedingly high for bacteria, in which comparative sequencing has been usually performed within species with a clonal or epidemic structure in which identical alleles are found repeatedly in different isolates (17). The sequence diversity in H. pylori is only partially due to extensive nucleotide variation. Indeed, the percentage of synonymous sites that was polymorphic for the three genes analyzed here was comparable to that of 11 housekeeping genes from N. meningitidis (Table 1). However, the number of unique sequences in H. pylori is apparently much higher than in N. meningitidis, in which numerous strains possess identical alleles.
The Homoplasy Test and compatibility matrices provided evidence for extremely high levels of recombination affecting the three genes from Canadian and German strains and somewhat lower levels in strains from the Cape-colored patients. Thus, shuffling of sequence diversity by extensive intragenic recombination is so frequent in H. pylori that it can generate a much larger number of unique sequences than in species where recombination is less frequent.
H. pylori grows deep in the gastric mucus, an ecological niche free of other bacterial species. Our conclusion that recombination is frequent implies that mixed colonization with different strains of H. pylori occurs repeatedly, as supported by the observation that patients can be simultaneously colonized with strains that differ in randomly amplified polymorphic DNA pattern (41, 42). Even if most individuals were infected with H. pylori early in childhood and harbored their individual strain thereafter, occasional mixed colonization and transformation would result in extensive genetic rearrangements with time. In other bacteria, mechanisms such as selective sweeps (43, 44) or sequential bottlenecks (45) purify populations of genetic variants, resulting in uniform genes or in clonal groupings. Such purification mechanisms must be rare or inefficient in H. pylori.
Selective sweeps depend on competition between bacteria and the overgrowth of less fit variants by fitter variants. If a favorable mutation arises in bacteria with an intermediate frequency of recombination, that mutation will spread through the population together with linked chromosomal DNA, resulting in homogeneity of that portion of the chromosome throughout the population affected by the selective sweep. Thus, the relatively low variation of most bacterial populations is explained by selective sweeps, which are possibly ineffective in H. pylori due to frequent recombination that destroys genetic linkage. These considerations imply that the genetic diversity seen in H. pylori reflects frequent recombination during a long evolutionary history in the relative absence of purification mechanisms.
Sequential bottlenecks depend on founder effects during geographical spread to areas where the bacteria can multiply extensively and/or overgrow the local bacterial population. If the founder population is small, this will result in a reduction in genetic diversity. In H. pylori, such a reduction in diversity has not occurred. However, sufficient geographic spread has occurred that the same polymorphic sites and sequence diversity were present when data were pooled from bacteria from different countries (Table 1). Unless sequence diversity were caused by repeated mutations at the same positions, we must conclude that individual polymorphisms have spread globally during the long evolutionary history of H. pylori and have led to unprecedented allelic diversity. The reason why geographic spread, and the replacement of local by invading populations, has not led to genetic homogeneity is that genetic recombination has ensured the maintenance of individual point mutations present in both populations.
What is the causal connection between high variability and high recombination rate in H. pylori? Genes from both N. meningitidis and H. pylori showed comparable, high levels of sequence polymorphism (Table 1) but differed in the frequency of recombination according to both the Homoplasy Test and compatibility matrix analysis (Table 2). Recombination in N. meningitidis was only slightly more frequent than in E. coli, which is characterized by considerably lower levels of sequence polymorphism. We note that identical alleles were repeatedly found in unrelated strains of N. meningitdis (17), indicating that complete genes are often exchanged in this species, whereas identical alleles were very rare in H. pylori, supporting our conclusion that recombination is much more frequent in that species. Alternatively, much smaller DNA fragments are integrated after recombination in H. pylori than in N. meningitidis.
Recombination by itself does not generate sequence polymorphisms. However, recombination does prevent the reduction in variability caused by selective sweeps and sequential bottlenecks, thus increasing the polymorphism in a population. DNA transformation can affect variability in another way: occasional horizontal transfer from related species can be an important source of sequence polymorphism, possibly even more important than mutation (29, 46). Currently, at least one other species is known to occasionally inhabit the same habitat as H. pylori, namely H. heilmannii (47). Of course, all variation must ultimately arise from mutation, but interspecies gene pools (35) encompass a much larger source of genetic polymorphisms than do single species.
Acknowledgments
We gratefully acknowledge being directed to compatibility matrices by T. S. Whittam and the helpful comments of two anonymous reviewers. We also gratefully acknowledge expert technical assistance by Susanne Friedrich, Michaela Stieglitz-Rumberg, and Kerstin Zurth. This work was supported by Grant Su 133/2-2 from the Deutsche Forschungsgemeinschaft (to S.S.). N.H.S. was supported by a Wellcome Trust grant to B. G. Spratt, and E.K. was supported by Grant Ku 1168/1-1 from the Deutsche Forschungsgemeinschaft.
ABBREVIATION
- MLEE
multilocus enzyme electrophoresis
Footnotes
References
- 1. Peterson W L. N Engl J Med. 1991;324:1043–1048. doi: 10.1056/NEJM199104113241507. [DOI] [PubMed] [Google Scholar]
- 2.Parsonnet J, Friedman G D, Vandersteen D P, Chang Y, Vogelman J H, Orentreich N, Sibley R K. N Engl J Med. 1991;325:1127–1131. doi: 10.1056/NEJM199110173251603. [DOI] [PubMed] [Google Scholar]
- 3.Bayerdörffer E, Neubauer A, Rudolph B, Thiede C, Lehn N, Eidt S, Stolte M. Lancet. 1995;345:1591–1594. doi: 10.1016/s0140-6736(95)90113-2. [DOI] [PubMed] [Google Scholar]
- 4.Labigne A, de Reuse H. Infect Agents Dis. 1996;5:191–202. [PubMed] [Google Scholar]
- 5.Majewski S I, Goodwin C S. J Infect Dis. 1988;157:465–471. doi: 10.1093/infdis/157.3.465. [DOI] [PubMed] [Google Scholar]
- 6.Oudbier J H, Langenberg W, Rauws E A, Bruin-Mosch C. J Clin Microbiol. 1990;28:559–565. doi: 10.1128/jcm.28.3.559-565.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Akopyanz N, Bukanov N O, Westblom T U, Kresovich S, Berg D E. Nucleic Acids Res. 1992;20:5137–5142. doi: 10.1093/nar/20.19.5137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Go M F, Kapur V, Graham D Y, Musser J M. J Bacteriol. 1996;178:3934–3938. doi: 10.1128/jb.178.13.3934-3938.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Garner J A, Cover T L. J Infect Dis. 1995;172:290–293. doi: 10.1093/infdis/172.1.290. [DOI] [PubMed] [Google Scholar]
- 10.Kansau I, Raymond J, Bingen E, Courcoux P, Kalach N, Bergeret M, Braimi N, Dupont C, Labigne A. Res Microbiol. 1996;147:661–669. doi: 10.1016/0923-2508(96)84023-x. [DOI] [PubMed] [Google Scholar]
- 11.Van der Ende A, Pan Z-J, Bart A, van der Hulst R W M, Feller M, Xiao S-D, Tytgat G N J, Dankert J. Infect Immun. 1998;66:1822–1826. doi: 10.1128/iai.66.5.1822-1826.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Suerbaum S. Trends Microbiol. 1995;3:168–170. doi: 10.1016/s0966-842x(00)88913-1. [DOI] [PubMed] [Google Scholar]
- 13.Cover T L. Mol Microbiol. 1996;20:241–246. doi: 10.1111/j.1365-2958.1996.tb02612.x. [DOI] [PubMed] [Google Scholar]
- 14.Atherton J C, Cao P, Peek R M J, Tummuru M K, Blaser M J, Cover T L. J Biol Chem. 1995;270:17771–17777. doi: 10.1074/jbc.270.30.17771. [DOI] [PubMed] [Google Scholar]
- 15.Maynard Smith J, Smith N H. Mol Biol Evol. 1998;15:590–599. doi: 10.1093/oxfordjournals.molbev.a025960. [DOI] [PubMed] [Google Scholar]
- 16.Jakobsen I B, Easteal S. Comput Appl Biosci. 1996;12:291–295. doi: 10.1093/bioinformatics/12.4.291. [DOI] [PubMed] [Google Scholar]
- 17.Maiden M C J, Bygraves J A, Feil E, Morelli G, Russell J E, Urwin R, Zhang Q, Zhou J, Zurth K, Caugant D A, et al. Proc Natl Acad Sci USA. 1998;95:3140–3145. doi: 10.1073/pnas.95.6.3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang F S, Whittam T S, Selander R K. J Bacteriol. 1997;179:6551–6559. doi: 10.1128/jb.179.21.6551-6559.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boyd E F, Nelson K, Wang F-S, Whittam T S, Selander R K. Proc Natl Acad Sci USA. 1994;91:1280–1284. doi: 10.1073/pnas.91.4.1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nelson K, Selander R K. J Bacteriol. 1992;174:6886–6895. doi: 10.1128/jb.174.21.6886-6895.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Milkman R, Bridges M M. Genetics. 1993;133:455–468. doi: 10.1093/genetics/133.3.455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Laurie C C, Bridgham J T, Choudhary M. Genetics. 1998;129:489–499. doi: 10.1093/genetics/129.2.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Inomata N, Shibata H, Okuyama E, Yamazaki T. Genetics. 1995;141:237–244. doi: 10.1093/genetics/141.1.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cooke P H, Oakeshott J G. Proc Natl Acad Sci USA. 1989;86:1426–1430. doi: 10.1073/pnas.86.4.1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kirby D A, Stephan W. Genetics. 1996;144:635–645. doi: 10.1093/genetics/144.2.635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jukes T H, Cantor C R. In: Mammalian Protein Metabolism. Munro H N, editor. New York: Academic; 1969. pp. 21–132. [Google Scholar]
- 27.Rozas J, Rozas R. Comput Appl Biosci. 1997;13:307–311. [PubMed] [Google Scholar]
- 28.Morelli G, Malorny B, Müller K, Seiler A, Wang J, del Valle J, Achtman M. Mol Microbiol. 1997;25:1047–1064. doi: 10.1046/j.1365-2958.1997.5211882.x. [DOI] [PubMed] [Google Scholar]
- 29.Guttman D S, Dykhuizen D E. Science. 1994;266:1380–1383. doi: 10.1126/science.7973728. [DOI] [PubMed] [Google Scholar]
- 30.Achtman M. In: Meningococcal Disease. Cartwright K, editor. New York: Wiley; 1995. pp. 159–175. [Google Scholar]
- 31.Botha M C. S Afr Med J, Suppl. 1. 1972;4:1–28. [Google Scholar]
- 32.Selander R K, Musser J M, Caugant D A, Gilmour M N, Whittam T S. Microb Pathog. 1987;3:1–7. doi: 10.1016/0882-4010(87)90032-5. [DOI] [PubMed] [Google Scholar]
- 33.Selander R K, Li J, Nelson K. In: Escherichia coli and Salmonella. Curtiss III R, Ingraham J L, Lin E C C, Low K B, Magasanik B, Reznikoff W S, Riley M, Schaechter M, Umbarger H E, editors. Washington, DC: Am. Soc. Microbiol.; 1996. pp. 2691–2707. [Google Scholar]
- 34.Maynard Smith J, Smith N H, O’Rourke M, Spratt B G. Proc Natl Acad Sci USA. 1993;90:4384–4388. doi: 10.1073/pnas.90.10.4384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maiden M C J, Malorny B, Achtman M. Mol Microbiol. 1996;21:1297–1298. doi: 10.1046/j.1365-2958.1996.981457.x. [DOI] [PubMed] [Google Scholar]
- 36.Milkman R, Bridges M M. Genetics. 1990;126:505–517. doi: 10.1093/genetics/126.3.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Souza V, Nguyen T T, Hudson R R, Pinero D, Lenski R E. Proc Natl Acad Sci USA. 1992;89:8389–8393. doi: 10.1073/pnas.89.17.8389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Duncan K E, Ferguson N, Kimura K, Zhou X, Istock C A. Evolution. 1994;48:1995–2025. doi: 10.1111/j.1558-5646.1994.tb02229.x. [DOI] [PubMed] [Google Scholar]
- 39.Istock C A, Duncan K E, Ferguson N, Zhou X. Mol Ecol. 1992;1:93–103. doi: 10.1111/j.1365-294x.1992.tb00161.x. [DOI] [PubMed] [Google Scholar]
- 40.Nedenskov-Sörensen P, Bukholm G, Bøvre K. J Infect Dis. 1990;161:365–366. doi: 10.1093/infdis/161.2.365. [DOI] [PubMed] [Google Scholar]
- 41.Taylor N S, Fox J G, Akopyants N S, Berg D E, Thompson N, Shames B, Yan L, Fontham E, Janney F, Hunter F M, et al. J Clin Microbiol. 1995;33:918–923. doi: 10.1128/jcm.33.4.918-923.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Berg D E, Gilman R H, Lelwala-Guruge J, Srivastava K, Valdez Y, Watanabe J, Miyagi J, Akopyants N S, Ramirez-Ramos A, Yoshiwara T H, et al. Clin Infect Dis. 1997;25:996–1002. doi: 10.1086/516081. [DOI] [PubMed] [Google Scholar]
- 43.Guttman D S, Dykhuizen D E. Genetics. 1994;138:993–1003. doi: 10.1093/genetics/138.4.993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dykhuizen D. Encyclopedia of Microbiology. New York: Academic; 1992. pp. 351–355. [Google Scholar]
- 45.Achtman M. Trends Microbiol. 1995;3:186–192. doi: 10.1016/s0966-842x(00)88918-0. [DOI] [PubMed] [Google Scholar]
- 46.Zhou J J, Bowler L D, Spratt B G. Mol Microbiol. 1997;23:799–812. doi: 10.1046/j.1365-2958.1997.2681633.x. [DOI] [PubMed] [Google Scholar]
- 47.Stolte M, Kroher G, Meining A, Morgner A, Bayerdorffer E, Bethke B. Scand J Gastroenterol. 1997;32:28–33. doi: 10.3109/00365529709025059. [DOI] [PubMed] [Google Scholar]