

Abstract
LMP2A is consistently detected in Hodgkin's Lymphoma, Nasopharyngeal Carcinoma and has also been detected in Burkitt's Lymphoma. Interestingly, LMP2A is detected in the absence of the transcriptional activator EBNA2, suggesting an alternative mechanism is responsible for LMP2A expression. The intracellular domain of Notch (Notch-IC) and EBNA2 are functional homologues and recent microarray analysis indicates that LMP2A may constitutively activate the Notch pathway in vivo. Coupled with evidence that Notch-IC can bind to and activate the LMP2A promoter, we hypothesized that expression of LMP2A results in the constitutive activation of the Notch pathway to auto-regulate its promoter. Our data indicate that LMP2A constitutively activates the Notch pathway in B cells and epithelial cells. Expression of LMP2A alone is sufficient to activate its own expression and the amino-terminal signaling domain is required as LMP2B is unable to activate the LMP2A promoter. In addition, point mutations in tyrosines 31, 101 and 112 each results in a significant decrease in LMP2A promoter activation. Deletion of the RBP-Jκ consensus sequences results in a significant decrease in promoter activity. The observation that LMP2A activates its own promoter suggests that LMP2A exploits the Notch pathway in order to control its own expression and may explain EBNA2 independent expression of LMP2A in EBV associated malignancies.
Keywords: EBV, LMP2A, Notch, RBP-Jκ
Introduction
Epstein Barr virus (EBV) is a ubiquitous human herpesvirus that infects greater than 90% of the adult population (Kieff and Rickinson, 2007). Although most EBV infections are asymptomatic, disease arises in adolescents as infectious mononucleosis and in immunocompromised patients as lymphoproliferative disorders (Rickinson and Kieff, 2007; Thorley-Lawson, 2005; Thorley-Lawson and Gross, 2004). EBV is associated with malignancies of lymphoid and epithelial origin, including Hodgkin's Lymphoma (HL), Burkitt's Lymphoma (BL) and Nasopharyngeal Carcinoma (NPC) (Rickinson and Kieff, 2007). As is characteristic of herpesviruses, EBV is able to persist in the human host through the establishment of a lifelong latent infection. EBV establishes latency in vitro in B lymphocytes by limiting viral gene expression to a subset of genes which includes EBV nuclear antigens 1, 2, 3A, 3B, 3C and LP (EBNAs), latent membrane protein 1 (LMP1) and latent membrane protein 2 (LMP2A) (Kieff and Rickinson, 2007).
LMP2A contains a 27-amino acid carboxy-terminal cytoplasmic domain, a 119-amino acid tyrosine rich amino-terminal cytoplasmic domain and 12 hydrophobic transmembrane domains (Ikeda, 2005; Longnecker, 2000; Longnecker et al., 1991). In the plasma membrane of EBV infected B cells, LMP2A localizes to lipid rafts, where it interacts with Src family protein tyrosine kinases and the tyrosine rich amino terminus serves as a substrate for phosphorylation (Ikeda, 2005). LMP2A becomes highly tyrosine phosphorylated upon Lyn (Src family protein tyrosine kinase) interaction with tyrosine 112. This initial interaction is required for additional LMP2A tyrosine phosphorylation (Burkhardt et al., 1992; Fruehling et al., 1998). Tyrosines 74 and 85 form an immunoreceptor tyrosine-based activation motif (ITAM), which is required for LMP2A to constitutively interact with Syk (Fruehling and Longnecker, 1997). By sequestering Lyn and Syk to its amino-terminus, LMP2A is able to efficiently block B cell receptor signaling, which may be important for preventing activation of lytic replication (Ikeda, 2005; Longnecker, 2000). Preventing expression of highly immunogenic lytic proteins may allow EBV infected cells to evade the immune response.
EBV can also infect epithelial cells and LMP2A expression is consistently detected in EBV-associated malignancies of epithelial origin (Raab-Traub, 1992a; Raab-Traub, 1992b; Raab-Traub et al., 1987; Young et al., 1988). While the function of LMP2A in epithelial cells is still under investigation, evidence thus far indicates that the biology and signaling through LMP2A differs between cell types. This is likely due to differences in receptor and kinase expression between B cells and epithelial cells. LMP2A expression in epithelial cells similarly leads to the constitutive phosphorylation and activation of the PI3K/Akt pathway (Morrison, Klingelhutz, and Raab-Traub, 2003; Morrison and Raab-Traub, 2005; Scholle, Bendt, and Raab-Traub, 2000). However, while in B cells, Y112 and its interaction with Lyn Src family protein tyrosine kinase is critical for the phosphorylation and signaling functions of LMP2A (Fruehling et al., 1996; Fruehling et al., 1998), LMP2A in epithelial cells relies on Y101 as well as the ITAM motif (Y74/85) (Lu et al., 2006; Scholle, Longnecker, and Raab-Traub, 1999).
LMP2A has several roles in EBV latent infection, making it a key target for inhibition of EBV associated disease. LMP2A is consistently detected in all EBV-associated malignancies including HL and NPC. LMP2A transcript has also been detected in a wide variety of BL cell lines (Konishi et al., 2001; Rechsteiner et al., 2007; Tao et al., 1998) and three studies have demonstrated that LMP2A is expressed in fresh BL biopsies (Bell et al., 2006; Tao et al., 1998; Xue et al., 2002). Interestingly, in these malignancies, LMP2A is expressed in the absence of the viral transcriptional activator, EBNA2, indicating that an alternative mechanism is responsible for its expression (Rickinson and Kieff, 2007). Microarray analysis of B lymphocytes from transgenic mice expressing LMP2A has revealed LMP2A alters global transcriptional regulation in a similar manner to that seen in Hodgkin Reed Sternberg (HRS) cells, implicating a role for LMP2A in its pathogenesis. Of particular interest, LMP2A expression induces global changes in transcription factor usage in transgenic mice, downregulating the activities of E2A, early B cell factor (EBF), and Pax-5 (Portis, Dyck, and Longnecker, 2003; Portis and Longnecker, 2003). These transcription factors are essential for B cell development and are normally turned off when Notch signaling is active during lymphopoiesis (Osborne and Miele, 1999). Similar alterations in transcription factor usage have also been noted in HL (Cossman et al., 1999; Jundt et al., 2002a; Jundt et al., 2002b; Khan and Coates, 1994; Kuppers et al., 2002). The increased expression of Notch signaling components, RBP-Jκ, Enhancer of Split (Hes) and delta, with concomitant decreased expression of B cell specific transcription factors in LMP2A expressing B cells suggests that LMP2A is able to activate the Notch pathway in vivo (See Table 1) (Portis, Dyck, and Longnecker, 2003; Portis, Ikeda, and Longnecker, 2004; Portis and Longnecker, 2003). EBNA2 is a functional homolog of the cleaved form of Notch (Notch-IC) and each has the ability to bind and activate RBP-Jκ which allows for viral as well as cellular gene transcription. It has been shown previously that murine Notch1-IC can bind to and induce transcription from the LMP2A promoter in a reporter assay as well as using the endogenous promoter. The LMP2A promoter contains two consensus RBP-Jκ sites that are important for its activation by both Notch-IC and EBNA2 (Gordadze et al., 2001; Grossman et al., 1994; Hofelmayr et al., 2001; Hofelmayr et al., 1999; Hsieh et al., 1997; Ling et al., 1994; Strobl et al., 2000). In conjunction with these reports, the data from the microarray analysis led to the novel hypothesis that LMP2A constitutively activates the Notch pathway in vivo in order to regulate its own expression in the absence of EBNA2. This novel function of LMP2A would allow it to be expressed in EBV associated malignancies without expression of other immunogenic viral proteins.
Table 1.
Expression of genes associated with Notch signalinga
| Gene | Fold changeb |
|---|---|
| B cell transcription factor genes | |
| E2A* | −2.2 ± 0.2 |
| EBF* | −3.5 ± 1.0 |
| Pax-5* | −2.1 ± 0.2 |
| PU.1 | −3.0 ± 0.1 |
| Notch-associated genes | |
| Delta (Dlk-1 like) | 2.7 ± 1.0 |
| Enhancer of Split (Hes) | 2.2 ± 1.0 |
| Suppressor of Hairless (RBP-L) | 1.4 ± 0.2 |
| Sel 11 inhibitor of Notch | −3.0 ± 0.2 |
Data from Portis and Longnecker, 2003
Microarray experiments were performed in duplicate using CD19+ bone marrow (BM) B cells from wild-type and LMP2A transgenic mice (TgE). Average fold change in gene expression in cells from TgE versus wild-type mice is shown, along with standard deviation.
Data confirmed by RT-PCR and western blot (Portis and Longnecker, 2003).
Notch signaling is an evolutionarily conserved pathway that is necessary for development in a variety of systems, including lymphopoiesis. Notch receptors are single-pass transmembrane receptors that undergo proteolytic cleavage upon activation by ligand binding. Ligands for Notch are cell associated transmembrane proteins and include Delta and Jagged (Osborne and Miele, 1999). Once cleaved, the intracellular cytoplasmic domain (Notch-IC) translocates to the nucleus where it interacts with RBP-Jκ/CBF-1 and converts it from a transcriptional repressor to an activator (Kawamata et al., 2002; Maillard, Adler, and Pear, 2003; Osborne and Miele, 1999). Notch2 appears to be more important for B cell function, however both Notch1-IC and Notch2-IC are functional homologs of EBNA2 (Hsieh et al., 1997; Souabni et al., 2002). The release of the intracellular domain of Notch is mediated by γ-secretase (presenilin) activity, and γ-secretase inhibitors can effectively prevent cleavage and activation of the Notch pathway. Deregulated expression of wild type Notch receptors, ligands and downstream targets have been described in several cancers such as HL and large cell lymphomas (Jundt et al., 2002a; Mathas et al., 2006). Notch1 receptors are highly expressed in B cell derived HRS cells and activation dramatically increases the rate of proliferation and decreases susceptibility to chemically induced apoptosis (Jundt et al., 2002a; Mungamuri et al., 2006). Several γ-secretase inhibitors are currently in clinical trials for treatment of several lymphopoietic malignancies including HL.
To gain insight into EBV-associated malignancies, we sought to determine the mechanism by which LMP2A is expressed in the absence of EBNA2. Our data show that LMP2A activates the Notch pathway in both B cells and epithelial cells. Further, expression of LMP2A alone is sufficient to activate the LMP2A promoter. Signaling through the amino terminal domain of LMP2A is required for this function, as LMP2B is unable to confer LMP2A promoter activity. Point mutations of several conserved tyrosines in LMP2A also block the ability of LMP2A to auto-regulate its promoter and suggest a comple signaling cascade may be involved in Notch activation. While γ-secretase inhibitors are unable to block LMP2A promoter activity, deletion of the RBP-Jκ consensus sequences reduces activation, linking the Notch pathway to LMP2A auto-regulation. An auto-regulatory loop for LMP2A suggests a mechanism for EBNA2 independent LMP2A expression in EBV associated malignancies.
Results
Activation of the Notch pathway by LMP2A
Cleavage of the Notch receptor occurs following receptor activation, resulting in the release of the intracellular domain of Notch (Notch-IC). To determine whether LMP2A constitutively activates the Notch pathway, whole cell lysates from cells expressing LMP2A were analyzed using an antibody that specifically recognizes the intracellular domain of Notch1 following Notch cleavage. Previously described vector control and LMP2A expressing independent stable clones of BJAB B cells and HaCat epithelial cells were used (see Materials and Methods). HEK293 cells were transiently transfected with empty vector (pSG5), LMP2A, LMP2A-HA or Notch1-IC as a positive control. Western blot analysis of these cells shows that expression of LMP2A increases the amount of activated Notch1 significantly above basal levels for all three cell types tested (Figure 1A). As vector control cells express little to no activated Notch1, the result is specific to LMP2A expression. To further confirm that the Notch pathway is specifically activated by LMP2A, real time RT-PCR gene expression analysis of Hes-1, Dtx-1 and Notch1 was performed using RNA from BJAB cells stably expressing LMP2A (pMP2) or empty vector (pBamHyg). Hes-1 and Dtx-1 are downstream targets of an activated Notch receptor. As shown in figure 1B, the average change in gene expression for Hes-1 and Dtx-1 for BJAB cells expressing LMP2A compared with empty vector controls is 2.6 fold. This result is statistically significant (p<0.05 by student's t-test) and indicates that the Notch pathway is indeed activated in BJAB cells expressing LMP2A. The change in gene expression for Hes-1 is similar to that seen in the Microarray analysis shown in Table 1. Notch1 gene expression is similarly increased by 2.5 fold compared with vector control cells, however this result was not statistically significant by student's t-test (p=0.06).
Figure 1.
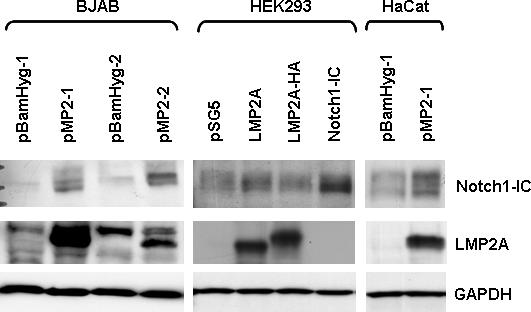
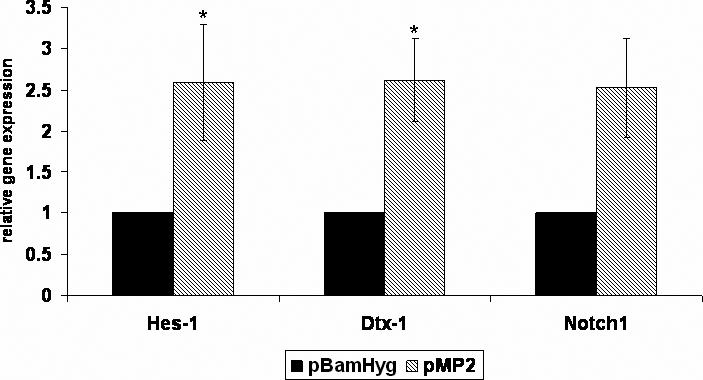
LMP2A constitutively activates the Notch pathway in B cells and epithelial cells. A) BJAB, HaCat and HEK293 cells were transfected with LMP2A or Notch1-IC as a control. Western blot analysis of whole cell lysates was performed using antibodies specific for LMP2A (14B7) or Notch1-IC (Cell Signaling). Equal protein loading was confirmed by probing blots for GAPDH. A representative blot from at least five independent experiments is shown. For the BJAB and HaCat stable cell lines, at least two independent clones were tested, and a representative blot is shown. A 10% gel was used for HEK293 and BJAB lysates, a 7.5% gel was used for HaCat lysates. B) Real time RT-PCR gene expression analysis was performed using total RNA isolated from BJAB cells stably expressing LMP2A (pMP2) or empty vector (pBamHyg). Primers specific for Hes-1, Dtx-1, and Notch1 were utilized along with GAPDH as a control for RNA levels. Change in gene expression was calculated using the ΔΔCt method. Experiments were repeated three times with at least two clones per experiment and the average +/− standard error is shown. * indicates p<0.05 compared with vector control. Notch1 p=0.06.
Auto-regulation of the LMP2A promoter
As Notch-IC and EBNA2 are functional homologs and both have been shown to activate the LMP2A promoter, we sought to determine whether LMP2A uses the ability to constitutively activate Notch to drive transcription from the LMP2A promoter. To this end, the full length LMP2A promoter, corresponding to base pairs 165696−166559 of the B95.8 EBV genome (Zimber-Strobl et al., 1991), was cloned into the pGL3 Basic reporter vector, rendering expression of firefly luciferase dependent upon the LMP2A promoter (LMP2p). HEK293 cells were transiently transfected with LMP2p along with empty vector (no activator), LMP2A, LMP2A-HA, LMP2B or vectors expressing the intracellular domain of Notch1 or Notch2 as controls. Protein expression from these vectors was confirmed by western blot, and a representative blot can be seen in the HEK293 panel of Figure 1A. Indeed, expression of LMP2A alone is sufficient to activate its own promoter (Figure 2). LMP2A and LMP2A-HA similarly induce a four fold increase in the relative luciferase activity. None of the activators induced firefly luciferase activity from the promoterless pGL3 Basic reporter vector (data not shown). LMP2B differs from LMP2A only in the first exon, with LMP2B lacking the exon encoding the amino-terminal tail attributed to the signaling capacity of LMP2A. Expression of LMP2B does not result in the activation of the LMP2p, indicating that auto-regulation is specific to signaling via the amino-terminus of LMP2A (Figure 2).
Figure 2.
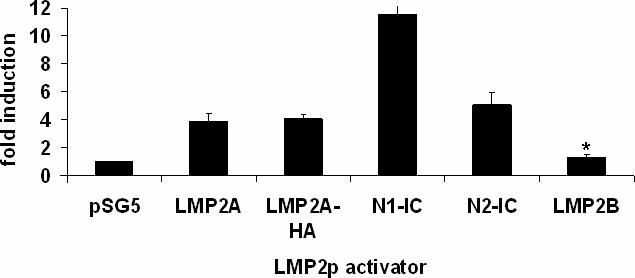
LMP2A can auto-activate its promoter in HEK293 cells. A reporter plasmid expressing firefly luciferase under the control of the full length LMP2A promoter (LMP2p) was transiently transfected into HEK293 cells with the indicated activators. Luciferase was quantified and normalized to an internal renilla luciferase control. Notch1-IC and Notch2-IC expressing plasmids were included as a positive control and LMP2B as a negative control. Average fold induction was calculated by dividing the normalized firefly luciferase value for each activator by the value obtained with no activator (empty vector). Data averaged from at least five independent experiments is shown +/− standard error. * indicates p<0.05 when compared with wild type LMP2A.
Inhibitors of Notch cleavage cannot block promoter activation by LMP2A
γ-secretase inhibitors block the proteolytic cleavage of the Notch intracellular domain from the membrane bound portion of the receptor and therefore block Notch mediated transcription in the nucleus. To determine whether cleavage of Notch is required for LMP2A auto-regulation, two γ-secretase inhibitors were examined. Varying concentrations of inhibitor were added concomitant with transfection and LMP2p activity assayed after 18 hours. As shown in figure 3A, addition of increasing concentrations of the inhibitor DAPT did not alter the ability of LMP2A to activate LMP2p. Similar results were seen using up to 50uM of γ-secretase inhibitor XII, Z-IL-CHO. The IC50 for DAPT is 115nM and the IC50 for Z-IL-CHO is 8−10uM, therefore the concentrations used for these experiments are sufficient to block Notch cleavage. These results were surprising; therefore we wanted to verify that the inhibitors were in fact blocking cleavage of Notch. Western blot analysis of Notch-IC expression was performed using the same lysates as were used for the luciferase assay. Figure 3B shows that the amount of Notch1-IC present in HEK293 cells treated with DAPT decreases as the concentration of DAPT increases. However, Notch1-IC does not become completely undetectable, even using an antibody with limited sensitivity. The bottom band seen in the western blot when LMP2A or Notch1-IC are expressed is still present when LMP2A expressing cells are treated with inhibitor, while the intensity of this band is less apparent in vector control cells, indicating that there is still some constitutively activated Notch in these cells.
Figure 3.
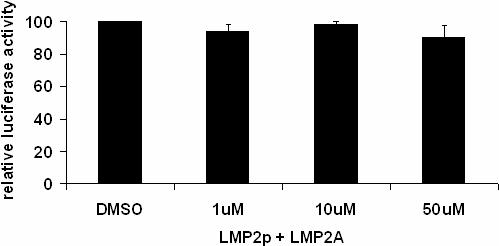
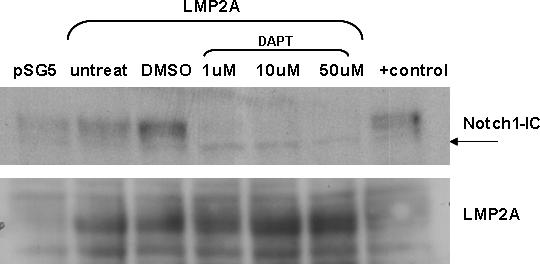
γ-secretase inhibitors are unable to inhibit LMP2A auto-regulation. HEK293 cells were transiently transfected with LMP2p and LMP2A in the presence of 1μM, 10 μM or 50 μM DAPT or DMSO vehicle control for 18 hours and lysed. Lysates were used to measure luciferase activity (A) or protein expression by Western blot (B). Luciferase activity for untreated cells was set to 100% and the luciferase activity of DMSO and inhibitor treated cells was normalized to this value. The average relative luciferase activity from three independent experiments is shown +/− standard error. There is no statistically significant difference between DMSO and inhibitor treated cells. Similar results were obtained using γ-secretase inhibitor XII (Z-IL-CHO), data not shown.
Analysis of the requirement for RBP-Jκ sites in LMP2A auto-regulation
It has been demonstrated that if the RBP-Jκ sites in the LMP2A minimal promoter are deleted, there is a significant decrease in responsiveness to murine Notch1-IC (Hofelmayr et al., 1999). Similar deletion mutations were generated in the context of the full length LMP2A promoter-firefly luciferase reporter plasmid to examine the requirement of the RBP-Jκ sites for LMP2A auto-regulation. The LMP2A promoter contains two RBP-Jκ sites, which consist of an 11 base pair consensus sequence, CGTGGGAA (Grossman et al., 1994; Ling et al., 1994). As shown in Figure 4, single deletions of the first or second RBP-Jκ site results in a modest yet statistically significant (p<0.05 by student's t-test) decrease in LMP2p activity via LMP2A and a more dramatic decrease in activity via Notch1-IC compared to the wild-type promoter. This activity is reduced further when both sites are deleted, and this decrease is statistically significant when compared with each single site deletion. These data indicate that in HEK293 cells, both RBP-Jκ sites are important for LMP2A auto-regulation. This is in contrast to previous data which demonstrates that the second RBP--IC to activate the minimal LMP2A promoter in B cells (Hofelmayr et al., 1999) and is likely due to the differences in cell types and promoter constructs used.
Figure 4.
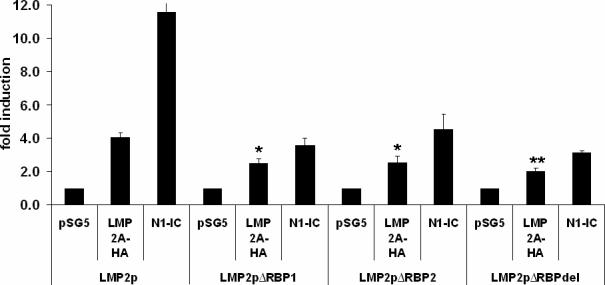
Both RBP-Jκ sites are important for auto-regulation. Each RBP-Jκ site was deleted from the full length LMP2p. LMP2pΔRBP1 lacks the first RBP-Jκ site (nucleotides 166244−166251 from the B95.8 genome), LMP2pΔRBP2 lacks the second RBP-Jκ site (166269−166276) and both RBP-jκ consensus are deleted in LMP2pΔRBPdel. Promoter constructs were transiently transfected into HEK293 cells with no activator, LMP2A or Notch1-IC as a control. Average fold induction was calculated as described in Figure 2 for each promoter. Data averaged from at least three independent experiments is shown +/− standard error. * indicates p<0.05 by student t-test compared with the wild type promoter. ** indicates p<0.05 by student t-test compared with each single deletion promoter.
Mutations of LMP2A inhibit auto-regulation
To begin to elucidate the mechanism by which LMP2A activates its own promoter, we took advantage of several point mutants in the amino terminus of LMP2A that have been characterized previously (Fruehling et al., 1998; Ikeda et al., 2000). Tyrosines in the amino-terminus of LMP2A have been shown to interact with signaling proteins that are important for the function of LMP2A in B cells and epithelial cells. Each mutant version of LMP2A-HA was transiently transfected into HEK293 cells and assayed for its ability to activate LMP2p. As mentioned above, LMP2B was unable to activate the promoter, demonstrating the requirement for signaling via the amino terminus of LMP2A. Point mutations of tyrosines 23, 60 and 64 as well as 74/85 (the ITAM motif) showed no statistically significant difference in LMP2p activation compared to wild type LMP2A-HA. The PY1PY2 mutant, which inhibits binding of Nedd4 family ubiquitin ligases, appears to have a slightly increased ability to activate the promoter. Although not statistically significant, this increase is likely due to the enhanced LMP2A expression of this mutant, as has been previously described (Ikeda et al., 2000; Ikeda, Ikeda, and Longnecker, 2001). In contrast, tyrosines 31, 101 and 112 have a statistically significant decrease in LMP2p activity relative to wild type LMP2A-HA (Figure 5A). Western blot analysis for mutant protein expression demonstrated that each LMP2A construct was present at equal levels, indicating that the decrease in promoter activity is not simply due to differences in protein expression. The bottom panel of Figure 5B demonstrates the protein expression levels for each tyrosine mutant typically seen in each experiment. Each of these tyrosines has been predicted to or demonstrated to activate signaling pathways and their lack of function in the luciferase assay indicates that multiple signaling events may converge on the LMP2p.
Figure 5.
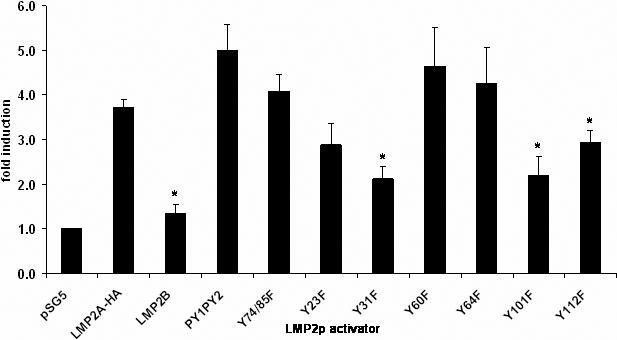
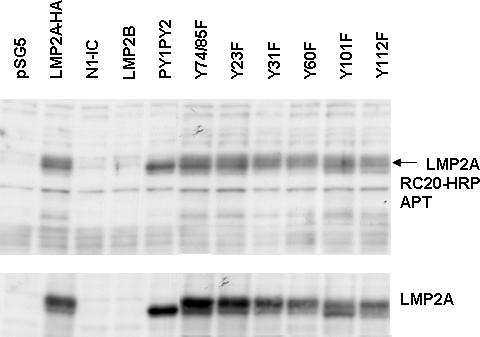
Signaling from the amino-terminus of LMP2A is required for promoter activation. A) LMP2p was transiently transfected into HEK293 with wild-type LMP2A-HA or various LMP2A point mutants as activators and analyzed for luciferase activity. Firefly luciferase values were normalized to an internal renilla luciferase control and fold induction calculated compared to no activator. Average fold induction from at least five independent experiments is shown +/− standard error. * indicates p<0.05 by student t-test. B) Whole cell lysates from HEK293 cells transfected with LMP2A-HA or LMP2A point mutants were used for Western blot analysis. Duplicate blots were probed with antibodies specific for LMP2A (14B7) or phospho-tyrosine (RC20-HRP). A representative blot of three independent experiments is shown.
LMP2A mutants are tyrosine phosphorylated in HEK293 cells
Tyrosine phosphorylation of LMP2A is required for many of the functions of LMP2A including the recruitment of various signaling molecules and activation of signal transduction pathways (Fruehling et al., 1996; Fruehling and Longnecker, 1997; Fruehling et al., 1998; Ikeda et al., 2000; Lu et al., 2006; Merchant, Caldwell, and Longnecker, 2000; Portis and Longnecker, 2004; Scholle, Bendt, and Raab-Traub, 2000; Swart et al., 2000). In order to determine whether tyrosine phosphorylation of LMP2A is important for activation of the LMP2p, mutant versions of LMP2A-HA were transiently transfected into HEK293 cells. Western blot analysis of whole cell lysates was performed with an anti-phosphotyrosine specific antibody. Figure 5B indicates there is no change in the tyrosine phosphorylation status of LMP2A-HA mutants in HEK293 cells, as previously observed (Scholle, Longnecker, and Raab-Traub, 2001). The arrow indicates the band corresponding to phosphorylated LMP2A based on the molecular weight. This data demonstrates that tyrosine phosphorylation is not necessarily required for LMP2A auto-regulation. Western blot analysis was also performed using an LMP2A specific antibody to ensure that each LMP2A mutant is properly expressed (bottom panel, Figure 5B).
Discussion
Prior to this study, the EBNA2-independent expression of LMP2A remained a mystery. These results demonstrate that LMP2A activates the Notch pathway in both B cells and epithelial cells, and exploits this pathway to regulate its own expression. Other groups have shown that Notch-IC can activate EBNA2 responsive promoters, including that of LMP2A, and gel shift analysis has demonstrated that Notch-IC can bind to RBP-Jκ sites in the LMP2A promoter (Hofelmayr et al., 1999). That LMP2A can activate the Notch pathway in vivo provides the missing link in an auto-regulatory loop. It is not surprising that LMP2A participates in an auto-regulatory loop, as another EBV oncogenic protein, LMP1, has been shown previously to activate its own expression through the Stat3 pathway (Chen et al., 2003; Huye et al., 2007; Ning et al., 2003; Ning, Huye, and Pagano, 2005). LMP2A auto-regulation through the Notch pathway may explain its EBNA2 independent expression in EBV associated malignancies such as HL, NPC and BL.
It is clear from mutational analysis of the LMP2A promoter that auto-regulation is dependent upon the presence of both RBP-Jκ sequences. This is in contrast to previous reports, which showed that in the context of the minimal LMP2A promoter, the second RBP-Jκ sequence is more important for activation by mNotch1-IC than the first in BL41-P3HR1 cells (Hofelmayr et al., 1999). Given several previous reports of differential signaling capacity of LMP2A depending on the cell context, it is logical that regulation of the LMP2A promoter also differs in the different cell types (Fruehling et al., 1996; Fruehling and Longnecker, 1997; Fruehling et al., 1998; Morrison, Klingelhutz, and Raab-Traub, 2003; Morrison and Raab-Traub, 2005). Additionally, the promoter constructs used here were different than those used by the Zimber-Strobl laboratory. Our promoter constructs contained the full length LMP2A promoter, corresponding to nucleotides 165696−166559 of the B95.8 genome, whereas the Zimber-Strobl laboratory used a construct containing the minimal LMP2A promoter (EBNA2 response element, −257 to −178 from the start site), thus it is possible that other elements in the full length promoter contribute to enhancement of promoter activity. Deletion of both RBP-Jκ does not completely abolish promoter activity in our experiments or those done previously (Hofelmayr et al., 1999), which supports the possibility of other regulatory elements in the LMP2p that are important for promoter regulation.
While auto-regulation requires the presence of the RBP-Jκ sites in LMP2p, indirectly demonstrating that the Notch pathway is indeed responsible for activation, the lack of inhibition in promoter activity using γ-secretase inhibitors is perplexing. There are several plausible explanations for this result. Previous reports have demonstrated that very low levels of activated Notch are sufficient to activate Notch responsive promoters (Schroeter, Kisslinger, and Kopan, 1998) and regardless of the quantity of exogenous Notch-IC transfected, the fold transactivation of a RBP-Jκ reporter construct remained the same even when Notch-IC protein becomes undetectable by western blot (Hofelmayr et al., 1999) . Western blot analysis of HEK293 cells transfected with LMP2A showed that even in the presence of 50uM DAPT, a small amount of Notch-IC protein remains in these cells and may be sufficient to maintain promoter activity. There is also evidence in the literature that other signaling pathways, such as the PI3K/Akt pathway, participate in crosstalk with the Notch signaling pathway and can lead to significantly enhanced transcriptional activation when compared with Notch signaling alone (Liu et al., 2006; McKenzie et al., 2006). In T cells, T cell receptor activation of PI3K results in a dramatic increase in the level of activation of downstream targets of the Notch pathway (McKenzie et al., 2006). It is tempting to speculate that LMP2A uses its ability to activate several signaling pathways to activate its own expression, and that while Notch activation is partially blocked by the γ-secretase inhibitors, low levels of Notch activity remain and transcriptional activity at the LMP2A promoter is enhanced by the ability of LMP2A to activate the PI3K pathway. This hypothesis is supported by the inability of the LMP2A Y31A mutant to activate the promoter. Tyrosine 31 is predicted to bind to PI3K based on predicted sequence homology (Longnecker, 2000), however prior to this report; a phenotype for the Y31A mutation has not been identified.
An alternative explanation for the lack of inhibition by the γ-secretase inhibitors is that LMP2A signaling may not induce an active cleavage event. Instead, it may stabilize cleaved Notch by sequestering away regulatory proteins such as cbl and AIP4/Itch/Nedd4 and preventing the degradation of Notch-IC by the proteasome. This explanation is not without precedence, as the LANA protein of KSHV, another γ-herpesvirus, has been recently demonstrated to induce activation of the Notch pathway by stabilizing the Notch-IC protein, which is normally rapidly turned over upon phosphorylation and binding of cbl and AIP4 (Carroll et al., 2006; Jehn et al., 2002). LMP2A is also known to bind to AIP4/Itch and is predicted to bind to cbl as well (Ikeda et al., 2000; Ikeda, Ikeda, and Longnecker, 2001; Longnecker, 2000). Both of these ubiquitin ligases are important for the regulation of Notch signaling and their binding to LMP2A and subsequent sequestration may result in a stable cleaved Notch protein that can readily activate responsive promoters. Although western blot analysis of cells treated with γ-secretase inhibitors show small amounts of cleaved Notch remaining, it remains to be determined whether Notch2 may be more important in this cell type, as reliable reagents to detect cleaved Notch2 are not available.
Given the complexity of LMP2A signaling and the context of an immortalized cell, it is likely that Notch activation is not necessarily the only mechanism for auto-regulation exploited by LMP2A. This is supported by our studies and those of the Zimber-Strobl laboratory which demonstrated that LMP2A promoter activation is not completely abrogated upon deletion of both RBP-Jκ sites in the presence of exogenous Notch-IC. In addition, the RBP-Jκ sites are not sufficient for Notch-IC to mediate transactivation of the LMP2p, and at least one other site in the minimal promoter is important for transactivation in both B cells and epithelial cells (Hofelmayr et al., 1999). Further support is provided by the mutational analysis of LMP2A, which indicated that multiple signaling pathways may play a role in auto-regulation. Signaling through the amino-terminus is required for LMP2A to mediate its auto-regulatory function because LMP2B is unable to confer promoter activity. Further, Y31, Y101 and Y112 each have a statistically significant decrease in activity, supporting the notion that there is a role for multiple signaling pathways in LMP2A auto-regulation. As mentioned above, Y31 may play a role in PI3K signaling. Y101 and Y112 are both important for binding of Src family protein tyrosine kinases, and therefore may be required to recruit and/or phosphorylate proteins in the LMP2A signalosome to potentiate Notch signaling.
As EBNA2 and Notch-IC are functional homologs, one might predict that LMP2A mediated activation of the Notch pathway in EBV associated malignancies would also lead to the activation of other EBNA2 responsive genes such as the Cp and Wp latency promoters. This is not the case, however, as other EBNA2 responsive genes are not detected in HL, NPC or BL (Bell et al., 2006; Kieff and Rickinson, 2007; Rickinson and Kieff, 2007). There is considerable evidence that the EBV genome is extensively methylated in cell lines exhibiting limited viral latency. More specifically, the Cp and Wp promoters are hypermethylated in tumor cell lines and in the latent memory B cells of seropositive healthy individuals (Ernberg et al., 1989; Masucci et al., 1989; Paulson et al., 2002; Robertson and Ambinder, 1997; Schaefer, Strominger, and Speck, 1997; Schaefer et al., 1991). Interestingly, these are the very same environments where the LMP2A transcript is often detected (Thorley-Lawson, 2005; Thorley-Lawson and Gross, 2004). Therefore, it is likely that the reason that LMP2A activation of the Notch pathway does not lead to activation of other EBNA2 responsive genes involves the methylation status of the promoters. This explanation assumes that the LMP2A promoter is hypomethylated, however the methylation status of the LMP2A promoter has not been determined, and mechanisms of EBV genome methylation are not yet clear. From our data, we propose a model for LMP2A expression in EBV associated malignancies where the LMP2A promoter remains hypomethylated and available for active transcription. Signaling cascades activated by LMP2A induce the activation of the Notch pathway which leads to LMP2A promoter activity in an auto-regulatory loop. This mechanism allows for LMP2A expression without triggering the expression of other immunogenic latent proteins and provides a means for LMP2A to maintain a latent EBV infection and promote survival of aberrantly proliferating cells.
Materials and Methods
Cell lines and culture conditions
BJAB is an EBV negative Burkitt's lymphoma cell line obtained from ATCC (Rockville, MD) and grown in RPMI 1640 medium supplemented with 10% fetal plex, 1000U of penicillin/mL and 1000ug of streptomycin/mL. Stable BJAB cell lines have been described previously (Ikeda et al., 2000) and were maintained in complete RPMI 1640 plus 400U/mL hygromycin. Stable HaCat cell lines were generated as described (Fukuda and Longnecker, 2007) and grown in DMEM supplemented with 10% heat-inactivated fetal bovine serum (FBS), 1000 U/mL penicillin, 1000 ug/mL streptomycin and 400U/mL hygromycin. pMP2 contains the LMP2A cDNA cloned into the pBamHyg retroviral vector. HEK293 cells were grown in DMEM supplemented with 10% fetal plex, 1000u/mL penicillin and 1000ug/mL streptomycin.
Plasmids
The LMP2A promoter (LMP2p), corresponding to nucleotides 165696−166559 of the B95.8 genome (Zimber-Strobl et al., 1991), was PCR amplified and cloned into the pGL3 Basic plasmid (Promega) as a KpnI/XhoI fragment. Deletion mutations of the RBP-jκ sites in the full length LMP2p were generated using the Quikchange Mutagenesis kit (Stratagene). All constructs and mutations were confirmed by sequence analysis. pRLCMV (Promega) expresses renilla luciferase under the control of the CMV promoter and is used as an internal transfection control. pSG5-Notch1-IC and pSG5-Notch2-IC were kindly provided by Paul Ling. pLMP2A and pLMP2A-HA have been described previously (Longnecker et al., 1991). pLMP2A-HA contains an HA tag at the carboxy-terminus of LMP2A. All point mutations were generated by PCR based mutagenesis in the context of pLMP2A-HA (Fruehling et al., 1998). All DNA used for experiments was banded twice on CsCl density gradients and purified by ethanol precipitation.
Transfection and Luciferase assays
HEK293 cells were plated in 6 well dishes and transfected using 15uL Polyethylenimine (PEI, 1ug/uL, Polysciences, Inc.) mixed with 200uL of serum-free DMEM and 0.1ug LMP2p, 0.01ug pRLCMV and 4ug activator DNA for ten minutes at room temperature. The transfection mixture was then added to each well containing cells in complete DMEM. 18 hours post-transfection, cells were washed with sterile PBS and lysed with passive lysis buffer (Promega) for 30 minutes rocking at room temperature. Relative luciferase activity was measured with the Promega Dual Luciferase Reporter Assay System in a Visibottom 96-well plate using a Victor plate reader according to the kit instructions. Firefly luciferase values were normalized for transfection efficiency based on Renilla luciferase activity.
Western blots
HEK293 cells were transfected for 18 hours with 4ug of each DNA as above. Cells were lysed in buffer containing 50mM Tris-HCl (pH 7.5), 150mM NaCl, 1% Triton X-100, 0.1% SDS, 10% glycerol, 10mM NaF, 1mM Na3VO4, 2mM EDTA, 0.5mM PMSF, 10ug/mL pepstatin and 10ug/mL leupeptin at 107 cells/mL lysis buffer. Lysates were mixed with 5× SDS sample buffer and heated to 70C for 10 minutes. Proteins were resolved by SDS-PAGE, transferred to Immobilon-P (Millipore, Bedford, Ma.), and blocked with 3% BSA in TBST. Primary antibodies- Rat monoclonal anti-LMP2A 14B7, rabbit polyclonal anti-cleaved Notch1 (Cell Signaling), and mouse monoclonal anti-GAPDH (Abcam) were incubated overnight. Blots were then probed with the appropriate HRP conjugated secondary antibodies and visualized using ECL (Amersham). To detect phosphotyrosine, RC20-HRP anti-phosphotyrosine antibody was used (Transduction Laboratories).
Inhibitors
γ-secretase inhibitor IX (DAPT IC50=115nM) and γ-secretase inhibitor XII (IC50 8−10uM) were obtained from Calbiochem and dissolved in DMSO. Subsequent dilutions were made in media containing 10% heat inactivated FBS and inhibitors were added concomitant with the transfection mixtures.
Real time RT-PCR
Total RNA from stable BJAB cell lines was isolated using the RT2qPCR-Grade RNA Isolation Kit (Superarray Bioscience Corporation, Frederick, MD). 5ug of RNA was reverse transcribed using the Superarray Bioscience RT2 First Strand Kit according to kit instructions. 2uL of template cDNA was then used in the RT2 qPCR Primer Assay- SYBR Green Kit from Superarray Bioscience with primers specific for human Hes-1, human Dtx-1, human Notch1 and human GAPDH. PCR was carried out using the BioRad iQ5 according to kit instructions. SYBR Green fluorescence from each well was detected and recorded from each well during the annealing step of each cycle. Ct values were normalized to GAPDH levels. Fold change in gene expression for each gene of interest was calculated using the ΔΔCt method. Three independent experiments were performed with at least two independent clones per experiment.
Acknowledgements
R.L. is a Stohlman Scholar of the Leukemia and Lymphoma Society of America and supported by the Public Health Service grants CA62234, CA73507, CA93444, and CA117794 from the National Cancer Institute and AI067048 from the National Institute of Allergy and Infectious Disease. L.A. is supported by the Training Program in Viral Replication (T32 AI060523) from the National Institute of Allergy and Infectious Disease.
We thank Paul Ling for kindly providing essential control plasmids for our experiments. We also thank Toni Portis and Michelle Swanson-Mungerson for helpful discussions and critical reading of the manuscript prior to submission. A special thanks to Nanette Susmarski for providing us with healthy cell lines. In addition, we also thank Ursula Zimber-Strobl for kindly sending us promoter constructs for preliminary experiments.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bell AI, Groves K, Kelly GL, Croom-Carter D, Hui E, Chan AT, Rickinson AB. Analysis of Epstein-Barr virus latent gene expression in endemic Burkitt's lymphoma and nasopharyngeal carcinoma tumour cells by using quantitative real-time PCR assays. J Gen Virol. 2006;87(Pt 10):2885–90. doi: 10.1099/vir.0.81906-0. [DOI] [PubMed] [Google Scholar]
- Burkhardt AL, Bolen JB, Kieff E, Longnecker R. An Epstein-Barr virus transformation-associated membrane protein interacts with src family tyrosine kinases. J Virol. 1992;66(8):5161–7. doi: 10.1128/jvi.66.8.5161-5167.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll KD, Bu W, Palmeri D, Spadavecchia S, Lynch SJ, Marras SA, Tyagi S, Lukac DM. Kaposi's Sarcoma-associated herpesvirus lytic switch protein stimulates DNA binding of RBP-Jk/CSL to activate the Notch pathway. J Virol. 2006;80(19):9697–709. doi: 10.1128/JVI.00746-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Hutt-Fletcher L, Cao L, Hayward SD. A positive autoregulatory loop of LMP1 expression and STAT activation in epithelial cells latently infected with Epstein-Barr virus. J Virol. 2003;77(7):4139–48. doi: 10.1128/JVI.77.7.4139-4148.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cossman J, Annunziata CM, Barash S, Staudt L, Dillon P, He WW, Ricciardi-Castagnoli P, Rosen CA, Carter KC. Reed-Sternberg cell genome expression supports a B-cell lineage. Blood. 1999;94(2):411–6. [PubMed] [Google Scholar]
- Ernberg I, Falk K, Minarovits J, Busson P, Tursz T, Masucci MG, Klein G. The role of methylation in the phenotype-dependent modulation of Epstein-Barr nuclear antigen 2 and latent membrane protein genes in cells latently infected with Epstein-Barr virus. J Gen Virol. 1989;70(Pt 11):2989–3002. doi: 10.1099/0022-1317-70-11-2989. [DOI] [PubMed] [Google Scholar]
- Fruehling S, Lee SK, Herrold R, Frech B, Laux G, Kremmer E, Grasser FA, Longnecker R. Identification of latent membrane protein 2A (LMP2A) domains essential for the LMP2A dominant-negative effect on B-lymphocyte surface immunoglobulin signal transduction. J Virol. 1996;70(9):6216–26. doi: 10.1128/jvi.70.9.6216-6226.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fruehling S, Longnecker R. The immunoreceptor tyrosine-based activation motif of Epstein-Barr virus LMP2A is essential for blocking BCR-mediated signal transduction. Virology. 1997;235(2):241–51. doi: 10.1006/viro.1997.8690. [DOI] [PubMed] [Google Scholar]
- Fruehling S, Swart R, Dolwick KM, Kremmer E, Longnecker R. Tyrosine 112 of latent membrane protein 2A is essential for protein tyrosine kinase loading and regulation of Epstein-Barr virus latency. J Virol. 1998;72(10):7796–806. doi: 10.1128/jvi.72.10.7796-7806.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda M, Longnecker R. Epstein-Barr virus latent membrane protein 2A mediates transformation through constitutive activation of the Ras/PI3-K/Akt Pathway. J Virol. 2007;81(17):9299–306. doi: 10.1128/JVI.00537-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordadze AV, Peng R, Tan J, Liu G, Sutton R, Kempkes B, Bornkamm GW, Ling PD. Notch1IC partially replaces EBNA2 function in B cells immortalized by Epstein-Barr virus. J Virol. 2001;75(13):5899–912. doi: 10.1128/JVI.75.13.5899-5912.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossman SR, Johannsen E, Tong X, Yalamanchili R, Kieff E. The Epstein-Barr virus nuclear antigen 2 transactivator is directed to response elements by the J kappa recombination signal binding protein. Proc Natl Acad Sci U S A. 1994;91(16):7568–72. doi: 10.1073/pnas.91.16.7568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofelmayr H, Strobl LJ, Marschall G, Bornkamm GW, Zimber-Strobl U. Activated Notch1 can transiently substitute for EBNA2 in the maintenance of proliferation of LMP1-expressing immortalized B cells. J Virol. 2001;75(5):2033–40. doi: 10.1128/JVI.75.5.2033-2040.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofelmayr H, Strobl LJ, Stein C, Laux G, Marschall G, Bornkamm GW, Zimber-Strobl U. Activated mouse Notch1 transactivates Epstein-Barr virus nuclear antigen 2-regulated viral promoters. J Virol. 1999;73(4):2770–80. doi: 10.1128/jvi.73.4.2770-2780.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh JJ, Nofziger DE, Weinmaster G, Hayward SD. Epstein-Barr virus immortalization: Notch2 interacts with CBF1 and blocks differentiation. J Virol. 1997;71(3):1938–45. doi: 10.1128/jvi.71.3.1938-1945.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huye LE, Ning S, Kelliher M, Pagano JS. Interferon Regulatory Factor 7 Is Activated by a Viral Oncoprotein through RIP-Dependent Ubiquitination. Mol. Cell. Biol. 2007;27(8):2910–2918. doi: 10.1128/MCB.02256-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda M, Ikeda A, Longan LC, Longnecker R. The Epstein-Barr virus latent membrane protein 2A PY motif recruits WW domain-containing ubiquitin-protein ligases. Virology. 2000;268(1):178–91. doi: 10.1006/viro.1999.0166. [DOI] [PubMed] [Google Scholar]
- Ikeda M, Ikeda A, Longnecker R. PY motifs of Epstein-Barr virus LMP2A regulate protein stability and phosphorylation of LMP2A-associated proteins. J Virol. 2001;75(12):5711–8. doi: 10.1128/JVI.75.12.5711-5718.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda M, Fukuda M, Longnecker R. Epstein Barr Virus. Function of Latent Membrane Protein 2A Caister Academic Press; 2005. [Google Scholar]
- Jehn BM, Dittert I, Beyer S, von der Mark K, Bielke W. c-Cbl binding and ubiquitin-dependent lysosomal degradation of membrane-associated Notch1. J Biol Chem. 2002;277(10):8033–40. doi: 10.1074/jbc.M108552200. [DOI] [PubMed] [Google Scholar]
- Jundt F, Anagnostopoulos I, Forster R, Mathas S, Stein H, Dorken B. Activated Notch1 signaling promotes tumor cell proliferation and survival in Hodgkin and anaplastic large cell lymphoma. Blood. 2002a;99(9):3398–403. doi: 10.1182/blood.v99.9.3398. [DOI] [PubMed] [Google Scholar]
- Jundt F, Kley K, Anagnostopoulos I, Schulze Probsting K, Greiner A, Mathas S, Scheidereit C, Wirth T, Stein H, Dorken B. Loss of PU.1 expression is associated with defective immunoglobulin transcription in Hodgkin and Reed-Sternberg cells of classical Hodgkin disease. Blood. 2002b;99(8):3060–2. doi: 10.1182/blood.v99.8.3060. [DOI] [PubMed] [Google Scholar]
- Kawamata S, Du C, Li K, Lavau C. Overexpression of the Notch target genes Hes in vivo induces lymphoid and myeloid alterations. Oncogene. 2002;21(24):3855–63. doi: 10.1038/sj.onc.1205487. [DOI] [PubMed] [Google Scholar]
- Khan G, Coates PJ. The role of Epstein-Barr virus in the pathogenesis of Hodgkin's disease. J Pathol. 1994;174(3):141–9. doi: 10.1002/path.1711740302. [DOI] [PubMed] [Google Scholar]
- Kieff E, Rickinson AB. Epstein-Barr virus and its replication. In: David PMH, Knipe M, editors. Fields Virology. Volume II Section II 68A Lippincott-Raven Publishers; Philadelphia, Pa: 2007. [Google Scholar]
- Konishi K, Maruo S, Kato H, Takada K. Role of Epstein-Barr virus-encoded latent membrane protein 2A on virus-induced immortalization and virus activation. J Gen Virol. 2001;82(Pt 6):1451–6. doi: 10.1099/0022-1317-82-6-1451. [DOI] [PubMed] [Google Scholar]
- Kuppers R, Schwering I, Brauninger A, Rajewsky K, Hansmann ML. Biology of Hodgkin's lymphoma. Ann Oncol 13 Suppl. 2002;1:11–8. doi: 10.1093/annonc/13.s1.11. [DOI] [PubMed] [Google Scholar]
- Ling PD, Hsieh JJ, Ruf IK, Rawlins DR, Hayward SD. EBNA-2 upregulation of Epstein-Barr virus latency promoters and the cellular CD23 promoter utilizes a common targeting intermediate, CBF1. J Virol. 1994;68(9):5375–83. doi: 10.1128/jvi.68.9.5375-5383.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z-J, Xiao M, Balint K, Smalley KSM, Brafford P, Qiu R, Pinnix CC, Li X, Herlyn M. Notch1 Signaling Promotes Primary Melanoma Progression by Activating Mitogen-Activated Protein Kinase/Phosphatidylinositol 3-Kinase-Akt Pathways and Up-regulating N-Cadherin Expression. Cancer Res. 2006;66(8):4182–4190. doi: 10.1158/0008-5472.CAN-05-3589. [DOI] [PubMed] [Google Scholar]
- Longnecker R. Epstein-Barr virus latency: LMP2, a regulator or means for Epstein-Barr virus persistence? Adv Cancer Res. 2000;79:175–200. doi: 10.1016/s0065-230x(00)79006-3. [DOI] [PubMed] [Google Scholar]
- Longnecker R, Druker B, Roberts TM, Kieff E. An Epstein-Barr virus protein associated with cell growth transformation interacts with a tyrosine kinase. J Virol. 1991;65(7):3681–92. doi: 10.1128/jvi.65.7.3681-3692.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Lin WH, Chen SY, Longnecker R, Tsai SC, Chen CL, Tsai CH. Syk tyrosine kinase mediates Epstein-Barr virus latent membrane protein 2A-induced cell migration in epithelial cells. J Biol Chem. 2006;281(13):8806–14. doi: 10.1074/jbc.M507305200. [DOI] [PubMed] [Google Scholar]
- Maillard I, Adler SH, Pear WS. Notch and the immune system. Immunity. 2003;19(6):781–91. doi: 10.1016/s1074-7613(03)00325-x. [DOI] [PubMed] [Google Scholar]
- Masucci MG, Contreras-Salazar B, Ragnar E, Falk K, Minarovits J, Ernberg I, Klein G. 5-Azacytidine up regulates the expression of Epstein-Barr virus nuclear antigen 2 (EBNA-2) through EBNA-6 and latent membrane protein in the Burkitt's lymphoma line rael. J. Virol. 1989;63(7):3135–3141. doi: 10.1128/jvi.63.7.3135-3141.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathas S, Janz M, Hummel F, Hummel M, Wollert-Wulf B, Lusatis S, Anagnostopoulos I, Lietz A, Sigvardsson M, Jundt F, Johrens K, Bommert K, Stein H, Dorken B. Intrinsic inhibition of transcription factor E2A by HLH proteins ABF-1 and Id2 mediates reprogramming of neoplastic B cells in Hodgkin lymphoma. Nat Immunol. 2006;7(2):207–15. doi: 10.1038/ni1285. [DOI] [PubMed] [Google Scholar]
- McKenzie G, Ward G, Stallwood Y, Briend E, Papadia S, Lennard A, Turner M, Champion B, Hardingham GE. Cellular Notch responsiveness is defined by phosphoinositide 3-kinase-dependent signals. BMC Cell Biol. 2006;7:10. doi: 10.1186/1471-2121-7-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merchant M, Caldwell RG, Longnecker R. The LMP2A ITAM is essential for providing B cells with development and survival signals in vivo. J Virol. 2000;74(19):9115–24. doi: 10.1128/jvi.74.19.9115-9124.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison JA, Klingelhutz AJ, Raab-Traub N. Epstein-Barr virus latent membrane protein 2A activates beta-catenin signaling in epithelial cells. J Virol. 2003;77(22):12276–84. doi: 10.1128/JVI.77.22.12276-12284.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison JA, Raab-Traub N. Roles of the ITAM and PY motifs of Epstein-Barr virus latent membrane protein 2A in the inhibition of epithelial cell differentiation and activation of {beta}-catenin signaling. J Virol. 2005;79(4):2375–82. doi: 10.1128/JVI.79.4.2375-2382.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mungamuri SK, Yang X, Thor AD, Somasundaram K. Survival signaling by Notch1: mammalian target of rapamycin (mTOR)-dependent inhibition of p53. Cancer Res. 2006;66(9):4715–24. doi: 10.1158/0008-5472.CAN-05-3830. [DOI] [PubMed] [Google Scholar]
- Ning S, Hahn AM, Huye LE, Pagano JS. Interferon Regulatory Factor 7 Regulates Expression of Epstein-Barr Virus Latent Membrane Protein 1: a Regulatory Circuit. J. Virol. 2003;77(17):9359–9368. doi: 10.1128/JVI.77.17.9359-9368.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ning S, Huye LE, Pagano JS. Interferon Regulatory Factor 5 Represses Expression of the Epstein-Barr Virus Oncoprotein LMP1: Braking of the IRF7/LMP1 Regulatory Circuit. J. Virol. 2005;79(18):11671–11676. doi: 10.1128/JVI.79.18.11671-11676.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborne B, Miele L. Notch and the immune system. Immunity. 1999;11(6):653–63. doi: 10.1016/s1074-7613(00)80140-5. [DOI] [PubMed] [Google Scholar]
- Paulson EJ, Fingeroth JD, Yates JL, Speck SH. Methylation of the EBV Genome and Establishment of Restricted Latency in Low-Passage EBV-Infected 293 Epithelial Cells. Virology. 2002;299(1):109–121. doi: 10.1006/viro.2002.1457. [DOI] [PubMed] [Google Scholar]
- Portis T, Dyck P, Longnecker R. Epstein-Barr Virus (EBV) LMP2A induces alterations in gene transcription similar to those observed in Reed-Sternberg cells of Hodgkin lymphoma. Blood. 2003;102(12):4166–78. doi: 10.1182/blood-2003-04-1018. [DOI] [PubMed] [Google Scholar]
- Portis T, Ikeda M, Longnecker R. Epstein-Barr virus LMP2A: regulating cellular ubiquitination processes for maintenance of viral latency? Trends Immunol. 2004;25(8):422–6. doi: 10.1016/j.it.2004.05.009. [DOI] [PubMed] [Google Scholar]
- Portis T, Longnecker R. Epstein-Barr virus LMP2A interferes with global transcription factor regulation when expressed during B-lymphocyte development. J Virol. 2003;77(1):105–14. doi: 10.1128/JVI.77.1.105-114.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portis T, Longnecker R. Epstein-Barr virus (EBV) LMP2A mediates B-lymphocyte survival through constitutive activation of the Ras/PI3K/Akt pathway. Oncogene. 2004;23(53):8619–28. doi: 10.1038/sj.onc.1207905. [DOI] [PubMed] [Google Scholar]
- Raab-Traub N. Epstein-Barr virus and nasopharyngeal carcinoma. Semin Cancer Biol. 1992a;3(5):297–307. [PubMed] [Google Scholar]
- Raab-Traub N. Epstein-Barr virus infection in nasopharyngeal carcinoma. Infect Agents Dis. 1992b;1(4):173–84. [PubMed] [Google Scholar]
- Raab-Traub N, Flynn K, Klein C, Pizza G, De Vinci C, Occhiuzzi L, Farneti G, Caliceti U, Pirodda E. EBV associated malignancies. J Exp Pathol. 1987;3(4):449–56. [PubMed] [Google Scholar]
- Rechsteiner MP, Berger C, Weber M, Sigrist JA, Nadal D, Bernasconi M. Silencing of latent membrane protein 2B reduces susceptibility to activation of lytic Epstein-Barr virus in Burkitt's lymphoma Akata cells. J Gen Virol. 2007;88(Pt 5):1454–9. doi: 10.1099/vir.0.82790-0. [DOI] [PubMed] [Google Scholar]
- Rickinson AB, Kieff E. Epstein-Barr virus. In: David PMH, Knipe M, editors. Fields Virology. Volume II Section II 68B Lippincott-Raven Publishers; Philadelphia, Pa: 2007. [Google Scholar]
- Robertson KD, Ambinder RF. Methylation of the Epstein-Barr virus genome in normal lymphocytes. Blood. 1997;90(11):4480–4. [PubMed] [Google Scholar]
- Schaefer BC, Strominger JL, Speck SH. Host-cell-determined methylation of specific Epstein-Barr virus promoters regulates the choice between distinct viral latency programs. Mol. Cell. Biol. 1997;17(1):364–377. doi: 10.1128/mcb.17.1.364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer BC, Woisetschlaeger M, Strominger JL, Speck SH. Exclusive expression of Epstein-Barr virus nuclear antigen 1 in Burkitt lymphoma arises from a third promoter, distinct from the promoters used in latently infected lymphocytes. Proc Natl Acad Sci U S A. 1991;88(15):6550–4. doi: 10.1073/pnas.88.15.6550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholle F, Bendt KM, Raab-Traub N. Epstein-Barr virus LMP2A transforms epithelial cells, inhibits cell differentiation, and activates Akt. J Virol. 2000;74(22):10681–9. doi: 10.1128/jvi.74.22.10681-10689.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholle F, Longnecker R, Raab-Traub N. Epithelial cell adhesion to extracellular matrix proteins induces tyrosine phosphorylation of the Epstein-Barr virus latent membrane protein 2: a role for C-terminal Src kinase. J Virol. 1999;73(6):4767–75. doi: 10.1128/jvi.73.6.4767-4775.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholle F, Longnecker R, Raab-Traub N. Analysis of the phosphorylation status of Epstein-Barr virus LMP2A in epithelial cells. Virology. 2001;291(2):208–14. doi: 10.1006/viro.2001.1197. [DOI] [PubMed] [Google Scholar]
- Schroeter EH, Kisslinger JA, Kopan R. Notch-1 signalling requires ligand-induced proteolytic release of intracellular domain. Nature. 1998;393(6683):382–6. doi: 10.1038/30756. [DOI] [PubMed] [Google Scholar]
- Souabni A, Cobaleda C, Schebesta M, Busslinger M. Pax5 promotes B lymphopoiesis and blocks T cell development by repressing Notch1. Immunity. 2002;17(6):781–93. doi: 10.1016/s1074-7613(02)00472-7. [DOI] [PubMed] [Google Scholar]
- Strobl LJ, Hofelmayr H, Marschall G, Brielmeier M, Bornkamm GW, Zimber-Strobl U. Activated Notch1 modulates gene expression in B cells similarly to Epstein-Barr viral nuclear antigen 2. J Virol. 2000;74(4):1727–35. doi: 10.1128/jvi.74.4.1727-1735.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swart R, Ruf IK, Sample J, Longnecker R. Latent membrane protein 2A-mediated effects on the phosphatidylinositol 3-Kinase/Akt pathway. J Virol. 2000;74(22):10838–45. doi: 10.1128/jvi.74.22.10838-10845.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao Q, Robertson KD, Manns A, Hildesheim A, Ambinder RF. Epstein-Barr virus (EBV) in endemic Burkitt's lymphoma: molecular analysis of primary tumor tissue. Blood. 1998;91(4):1373–81. [PubMed] [Google Scholar]
- Thorley-Lawson DA. EBV the prototypical human tumor virus--just how bad is it? J Allergy Clin Immunol. 2005;116(2):251–61. doi: 10.1016/j.jaci.2005.05.038. quiz 262. [DOI] [PubMed] [Google Scholar]
- Thorley-Lawson DA, Gross A. Persistence of the Epstein-Barr virus and the origins of associated lymphomas. N Engl J Med. 2004;350(13):1328–37. doi: 10.1056/NEJMra032015. [DOI] [PubMed] [Google Scholar]
- Xue SA, Labrecque LG, Lu QL, Ong SK, Lampert IA, Kazembe P, Molyneux E, Broadhead RL, Borgstein E, Griffin BE. Promiscuous expression of Epstein-Barr virus genes in Burkitt's lymphoma from the central African country Malawi. Int J Cancer. 2002;99(5):635–43. doi: 10.1002/ijc.10372. [DOI] [PubMed] [Google Scholar]
- Young LS, Dawson CW, Clark D, Rupani H, Busson P, Tursz T, Johnson A, Rickinson AB. Epstein-Barr virus gene expression in nasopharyngeal carcinoma. J Gen Virol. 1988;69(Pt 5):1051–65. doi: 10.1099/0022-1317-69-5-1051. [DOI] [PubMed] [Google Scholar]
- Zimber-Strobl U, Suentzenich KO, Laux G, Eick D, Cordier M, Calender A, Billaud M, Lenoir GM, Bornkamm GW. Epstein-Barr virus nuclear antigen 2 activates transcription of the terminal protein gene. J Virol. 1991;65(1):415–23. doi: 10.1128/jvi.65.1.415-423.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]