

Abstract
Senile plaques in the cerebral parenchyma are a pathognomonic feature of Alzheimer’s disease (AD) and are mainly composed of aggregated fibrillar amyloid β (Aβ) proteins. The plaques are associated with neuronal degeneration, lipid membrane abnormalities, and chemical evidence of oxidative stress. The view that Aβ proteins cause these pathological changes has been challenged by suggestions that they have a protective function or that they are merely byproducts of the pathological process. This investigation was conducted to determine whether Aβ proteins promote or inhibit oxidative damage to lipid membranes. Using a mass spectrometric assay of oxidative lipid damage, the 42-residue form of Aβ (Aβ42) was found to accelerate the oxidative lipid damage caused by physiological concentrations of ascorbate and submicromolar concentrations of copper(II) ion. Under these conditions, Aβ42 was aggregated, but nonfibrillar. Ascorbate and copper produced H2O2, but Aβ42 reduced H2O2 concentrations, and its ability to accelerate oxidative damage was not affected by catalase. Lipids could be oxidized by H2O2 and copper-(II) in the absence of ascorbate, but only at significantly higher concentrations, and Aβ42 inhibited this reaction. These results indicate that the ability of Aβ42 to promote oxidative damage is more potent and more likely to be manifest in vivo than its ability to inhibit oxidative damage. In conjunction with prior results demonstrating that oxidatively damaged membranes cause Aβ42 to misfold and form fibrils, these results suggest a specific chemical mechanism linking Aβ42-promoted oxidative lipid damage to amyloid fibril formation.
There is an abundance of evidence suggesting that oxidative stress is involved in the pathogenesis of Alzheimer’s disease (AD);1 however, it is not yet possible to define a chemical mechanism linking any key feature of AD pathology to a specific oxidative process. In an effort to define such a mechanism, numerous associations have been described between AD and oxidative changes in proteins, nucleic acids, and lipids (1-5). Oxidatively damaged lipids have been a focus of study in this laboratory since finding that they promote misfolding and fibril formation by the 42-residue amyloid β protein (Aβ42) (6) and that they act synergistically with Aβ42 to promote fibril formation by the 40-residue amyloid β protein (Aβ40) (7). Others have implicated lipids in the pathogenesis of Alzheimer’s disease by demonstrating that oxidative lipid damage is increased (8-10), that the relative abundance of various lipid classes is altered (11-14), and that markers of oxidative damage such 4-hydroxy-2-nonenal (4-HNE) and isoprostanes (15-19) are increased in AD. It has also been demonstrated that membrane binding by Aβ proteins mediates neurotoxicity (20-22), that merely altering membrane lipid composition protects PC12 cells from Aβ-mediated toxicity (23), and that plasma membranes from human brain are particularly effective at accelerating Aβ fibrillogenesis (24). Thus, it is important to understand the nature of the interaction between Aβ proteins and lipid membranes, particularly membranes subject to oxidative damage.
With respect to the role of Aβ proteins and oxidative damage, two contrasting views are prevalent. On one hand, Aβ proteins are observed to promote oxidative damage in conjunction with redox-active metal ion species. For example, copper levels are significantly increased in AD (25) while Aβ binds Cu(II) with high affinity, reduces it to Cu(I), produces H2O2, and oxidizes compounds such as dopamine, cholesterol, and ascorbate (26-33). On the other hand, Aβ proteins appear to have antioxidant properties (34-41). The density of plaques containing Aβ protein correlates inversely with markers of oxidative damage (42), and in Downs syndrome, cortical deposition of Aβ proteins correlates with reduced oxidative damage (43). Moreover, Aβ proteins prevent lipoprotein oxidation (37) and metal-induced neuronal death in culture (44).
Given the evidence that lipid membranes are oxidatively damaged in AD and that they promote fibril formation, it becomes important to ascertain whether Aβ proteins exhibit prooxidant or antioxidant activity toward a lipid membrane. Prooxidant activity might lead to accelerated oxidative membrane damage, and this damage may further stimulate Aβ misfolding, whereas antioxidant activity may quench these processes and protect the membrane from damage. Accordingly, this investigation was undertaken to examine the circumstances in which prooxidant and antioxidant activities of Aβ were manifest toward lipids. 1-Steroyl-2-arachidonoyl-sn-glycero-3-phosphatidylcholine (SAPC) was chosen as an oxidizable substrate because it is one of the most common unsaturated lipid species in brain tissue (45) and because the chemistry of arachidonate oxidation is well understood. The most commonly used assays of oxidative damage rely on indirect measures of oxidative activity or on direct assay of only one or one class of oxidative degradation products. To avoid the ambiguities of such methods, the studies described herein employ a direct assay of oxidizable substrate concentration by mass spectrometry.
MATERIALS AND METHODS
Materials
1-Stearoyl-2-arachidonyl-sn-glycero-3-phosphocholine (SAPC) in chloroform and 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC) as powder were obtained from Avanti Polar Lipids (Alabaster, AL). SAPC was packaged in 10 mg quantities, under argon, in sealed glass ampules and stored at −80 °C until the day of use. Phenylethylamine (PA), dopamine (DA), BHT (2,6-di-tert-butyl-4-hydroxytoluene), bovine liver catalase, sodium hydrosulfite (dithionite), and diethylenetriaminepentaacetic acid (DTPA) were all purchased from Sigma (St. Louis, MO). Aβ40 and Aβ42 designate the 40- and 42-residue Aβ proteins, and Aβm–n designates a polypeptide segment corresponding to residues m through n of Aβ42. Lyophilized Aβ40 and Aβ42 of greater than 95% purity were obtained from American Peptide (Sunnyvale, CA) or rPeptide (Athens, GA). H13A and H14A variants of Aβ42 were obtained from rPeptide, while Aβ1–28, Aβ22–35, α-MSH, and neurotensin were obtained from American Peptide. The Aβ42 mutant M35V (21) as well as Aβ42 with an oxidized methionine (M35OX) (20) were kindly provided by Dr. Kevin Barnham. Aβ proteins were either stored desiccated at −20 °C or dissolved in HFIP at 0.5 mg/mL at −20 °C. Immediately prior to use, HFIP was evaporated, and proteins were redissolved in aqueous buffer at concentrations of 1–5 μM. DA and PA stock solutions (50 mM) were freshly prepared daily in 5% acetonitrile and 0.1% formic acid. These solutions were mixed to yield 10 and 1 μM DA and PA, respectively, in 5 mM HEPES at pH 7.4. Water was purified through an Elix and MilliQ A10 synthesis water purification system (Millipore, Bedford, MA) to 18 MΩ·cm and <3ppb organic carbon content.
Physical Characterization
To characterize the physical state of Aβ42, a 5 μM solution was preincubated with 0.5 μM Cu(II) for 30 min and then centrifugally filtered through a 10K MW cutoff Microcon filter (Millipore). Protein concentrations before and after filtration were measured by BCA assay (Pierce, Rockford, IL) with an albumin standard. Unfiltered solutions were also examined by infrared spectroscopy using a Horizon internal reflection cell (Harrick Scientific, Ossining, NY) with a germanium internal reflection crystal. A total of 512 scans were recorded with a Bio-Rad Digilab FTS-60A FTIR spectrometer and an MCT detector. They were processed with one level of zero filling and triangular apodization, but no smoothing, deconvolution, vapor subtraction, or nonlevel baseline correction was performed.
Lipid Vesicle Preparation and Oxidation
SAPC (lyophilized from chloroform) and DMPC were prepared as 10 mg/mL solutions in cyclohexane and lyophilized to produce a light powder. Quantities of 10 mg were resuspended in separate 1 mL aliquots of 5 mM argon-sparged HEPES at pH 7.4, bath-sonicated for 10 min, and extruded through 100 nm polycarbonate membranes to produce unilamellar vesicles. Lipid concentrations were determined by phosphate analysis (46), and 10 μg/mL cycloheximide was added to inhibit bacterial growth. Lipid vesicle stock suspensions were stored under argon for up to 2 weeks at 4 °C. Immediately prior to use, aliquots from these stocks were mixed in 5 mM HEPES to yield final concentrations of 10 μM SAPC and 25 μM DMPC. Reactants such as ascorbate (50 μM), Cu(II) (0.5 or 5 μM), H2O2 (200 μM), and proteins were added to these mixtures, with the final lipid concentrations as above. Anaerobic conditions were produced by continuous sparging of solutions with argon and were verified using a FOXY oxygen sensor (Ocean Optics, Dunedin, FL) and dithionitetreated water as an anaerobic standard. All reactions were conducted at room temperature (approximately 21 °C).
LC/MS/MS
Chromatographic separations were performed using an Agilent 1100 HPLC system modified with a low volume static mixer (Agilent, 01090-68702) and narrow bore tubing to minimize delay volumes. Solvents were pumped at 120 μL/min through an Agilent XDB C8 1 × 50 mm column (Agilent, Palo Alto, CA). Between experiments, the column was washed with several injections of 50% methanol in water, followed by an injection of 50 μL of 50 mM EDTA (in 1:1 methanol:water) to remove any adsorbed copper. Phospholipids were separated using a binary solvent system consisting of 50% methanol in water (solvent A) and 30:70 chloroform:methanol and 0.1% formic acid, pH adjusted to 5.6 using NH4OH (solvent B). Using a gradient running from 33% to 100% solvent B over 1.5 min and then holding at 100% solvent B for 2.5 min, DMPC and SAPC eluted at approximately 3.5 and 4 min, respectively. Amines were separated using a binary solvent system consisting of 5% acetonitrile and 0.1% formic acid (solvent A) and 95% acetonitrile and 0.1% formic acid (solvent B) (modified from refs 47 and 48). Using 15.7% solvent B, DA and PE eluted at approximately 0.5 and 0.7 min, respectively.
Phospholipid concentrations were assayed by LC/MS/MS determination of the phosphatidylcholine-specific collision-induced fragment at m/z 184 as previously described (49). Electrospray ionization mass spectrometry was performed with a turbo ion spray source on a QTrap (Applied Biosystems/MDS Sciex, Foster City, CA) operating in multiple reaction monitoring (MRM) mode with a declustering potential of 100 V, source voltage of 5500 V, and source temperature of 250 °C. DMPC was employed as the internal standard, and the following MRM transitions were monitored at the indicated collision energies (CE): SAPC at m/z 811→184 (CE 49) and m/z 811→86 (CE 81) and DMPC at m/z 679→184 (CE 35). Aqueous samples were loaded onto a C8 column, rather than using organic extraction, allowing us to directly measure the samples and preventing Na+ adducts from forming during electrospray ionization. Peak identification and quantification were performed using the Analyst Classic method in Analyst 1.4 (Applied Biosystems/MDS Sciex). Standard mixtures yielded linear results between 250 pmol (0.125 μM) and 30 μmol (15 μM) with a correlation coefficient (r2) of 0.993. Hydrolysis products of SAPC were measured at m/z 283 and m/z 311 using negative ion enhanced (ion trap) mode and direct infusion of sample.
DA concentrations were assayed by LC/MS/MS using PA as an internal standard. The declustering potential was 100 V, the source voltage was 5500 V, and the source temperature was 450 °C. MRM transitions were monitored for DA at m/z 154→137 (CE 13) and m/z 154→119 (CE 27) and for PA at m/z 122→105 (CE 13). Standard mixtures yielded linear results between 0.625 pmol (0.125 μM) and 250 μmol (50 μM) with a correlation coefficient (r2) of 0.9995.
Hydrogen Peroxide Assay
The concentration of a stock H2O2 solution was determined using an molar absortivity of 43.6 M−1 cm−1 at 240 nm. From this stock solution, standards ranging from 0.56 to 100 μM were prepared, and unknown H2O2 concentrations in lipid or amine oxidation reactions were determined using these standards in the FOX2 assay (50). This is a colorimetric assay that measures peroxide by its ability to convert Fe(II) to Fe(III), which in turn forms a purple complex with xylenol orange at low pH.
RESULTS
Prooxidant Activity of Aβ42
Aβ42 has been shown to generate H2O2 and reduce Cu(II) ions while promoting the oxidation of various biochemical substrates (27-29, 51). To test whether a polyunsaturated lipid may serve as an oxidizable substrate in such reactions, SAPC and H2O2 concentrations were measured over time in various mixtures. DMPC was included in these mixtures as an internal standard because it is not susceptible to oxidation. When Aβ42 and metal ions were both present in an experiment, they were premixed and incubated for 30 min prior to the addition of other reactants so that any redox or folding reactions that occur upon metal binding to Aβ42 may proceed without interference from other components in the mixture. Although several experiments were nominally free of Cu(II) ions, no special procedures were employed to remove trace amounts of Cu(II) in our water except where noted.
SAPC concentrations remained stable at 10 μM over 120 min in the presence of 5 μM Aβ42 and 0.5 μM Cu(II) (Figure 1A). Over the same time interval, SAPC concentration decreased by 15% in the presence of 5 μM Aβ42 and 50 μM ascorbate and by 40% in the presence of 0.5 μM Cu(II) and 50 μM ascorbate. Adding Aβ42 to this last mixture accelerated SAPC loss, causing 100% loss at 120 min.
Figure 1.
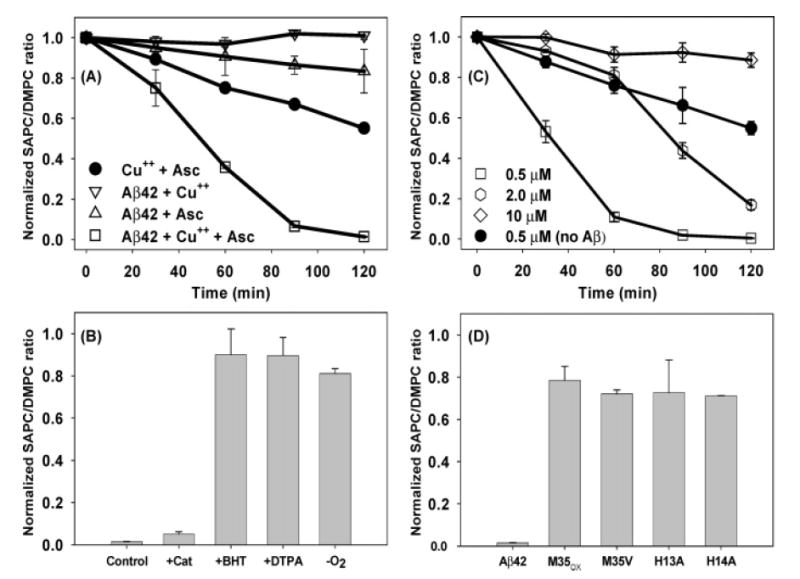
Prooxidant activity of Aβ42. (A) Phospholipid loss in the presence of Aβ42 (0.5 μM, American Peptide, open symbols), Cu(II) (0.5 μM), and/or ascorbate (50 μM). (B) Effects of catalase (1000 units/mL), BHT (7.5 μM), DTPA (10 μM), and anaerobic conditions on Aβ42-mediated lipid loss at 120 min. (C) Effects of preincubating Aβ42 (rPeptide) with increasing copper concentrations (0.5, 2, 10 μM) on lipid loss. (D) Effects of Aβ variants on lipid loss (M35OX–methionine sulfoxide, M35V, H13A, and H14A Aβ42 with single residue changes). Note that two of the experiments in panels A and C represent identical conditions but different commercial sources of Aβ42.
Various control experiments confirm that SAPC loss is due to oxidative damage. Using a negative ion mode mass spectrometric technique capable of detecting 0.1 μM of each fatty acid, no arachidonate or stearate could be detected during the 120 min oxidation reaction, indicating that SAPC was not being lost to simple hydrolysis. SAPC loss was largely prevented by an antioxidant (7.5 μM BHT), by metal chelation (10 μM DTPA), and by removal of oxygen from the solvents. Oxidative damage to SAPC was unaffected by 1000 units/mL catalase (Figure 1B), suggesting that damage was not mediated by H2O2. SAPC loss was not accelerated by Aβ42 variants in which Ala residues replaced His at positions 13 or 14 or in which the Met residue at position 35 was oxidized (M35OX) or replaced by Val (M35V), suggesting that the ability of Aβ42 to bind copper ions via the His residues or to undergo an alternative oxidation reaction (at the Met residue) was necessary to promote lipid oxidation (Figure 1D). Although the foregoing experiments showed that Cu(II) is required for Aβ42 to accelerate lipid oxidation, increasing Cu(II) ion concentrations from 0.5 to 2 and 10 μM reduced the oxidative loss of SAPC (Figure 1C).
Physical State of Aβ42
When 5 μM Aβ42 solutions were incubated for 30 min with and without 0.5 μM Cu(II) and then filtered through a 10 kDa cutoff filter, no protein appeared in the filtrate, indicating that the protein had aggregated. The same solutions, examined by internal reflection infrared spectroscopy in a D2O-based buffer, yield a broad, relatively featureless amide I band that is maximal at 1625 cm−1 (Figure 2). This spectrum resembles previously published spectra of Aβ proteins freshly adsorbed onto oxidatively damaged membranes. Note that the amide I spectrum of the Aβ42 used in these experiments is quite distinct from that of unfibrillized protein (maximal at ~1657 cm−1) and from fully formed amyloid fibrils (maximal at ~1623 cm−1; see Figure 2 of ref 7). We conclude that Aβ42 is aggregated but nonfibrillar under the conditions in which we observe accelerated lipid oxidation.
Figure 2.
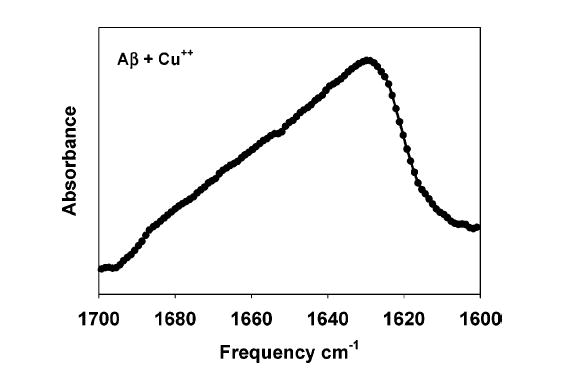
Secondary structure of prooxidant Aβ42. Amide I region of an infrared spectrum of 5 μM Aβ42 following 30 min incubation with 0.5 mM Cu(II) in 5 mM D2O–HEPES, pD 7.4.
H2O2 Production
When SAPC was treated with ascorbate and Cu(II) in the absence of Aβ42, H2O2 concentrations quickly reached levels in excess of 25 μM (Figure 3A). Catalase (1000 units/mL) markedly reduced the H2O2 concentration (Figure 3B), indicating that the assay is truly measuring H2O2 and that failure of catalase to prevent loss of SAPC in the experiments described above was not due to its ineffectiveness. The addition of Aβ42 markedly depressed H2O2 production under these conditions, although increasing Cu(II) concentrations to 2 and 10 μM partially reversed this depression (Figure 3A). The combination of Aβ42 and catalase was no more effective at reducing H2O2 levels than either protein alone (Figure 3B). We conclude that the high levels of H2O2 produced by ascorbate and Cu(II) are not a direct cause of lipid oxidation.
Figure 3.
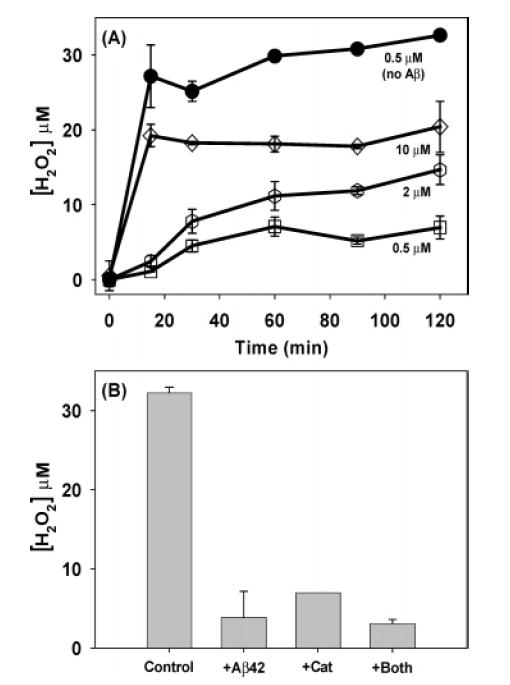
H2O2 production. (A) Effect of copper concentration (0.5, 2, 10 μM) on H2O2 production in the presence (open symbols)and absence (filled symbols) of Aβ42. (B) Effect of Aβ42 andcatalase on H2O2 production at 120 min. The Aβ42 concentrationwas 5 μM (when present) and the ascorbate concentration was 50 μM for all of the data in both panels.
Antioxidant Activity of Aβ42
Cu(II) and H2O2, without ascorbate, are often used to induce lipid oxidation under laboratory conditions. To investigate the effect of Aβ42 on lipid oxidation via this reaction, it was necessary to use somewhat higher concentrations of Cu(II) and H2O2 than in the previous experiments so that oxidation proceeded at a comparable rate. Cu(II) (5 μM) and 200 μM H2O2 completely oxidized 10 μM SAPC in 90 min (Figure 4A). When 1 μM Aβ42 was present, 35% of the lipid remained after 120 min, and when 5 μM Aβ42 was present, 85% of the lipid remained. The latter conditions represented a 1:1 mole ratio of Aβ42 and Cu(II). Aβ1–11, Aβ1–28, and Aβ22–35 present at a 1:1 mole ratio also protected SAPC from oxidation, but α-MSH and neurotensin did not (Figure 4B). When DA was used as the oxidizable substrate instead of SAPC, it was also oxidized extensively by Cu(II) and H2O2 over 120 min (Figure 4C). Aβ42, Aβ1–28, and Aβ22–35 inhibited DA oxidation but not the control peptides α-MSH and neurotensin. We conclude that Aβ peptides specifically inhibit oxidation of substrates such as SAPC and DA under these conditions.
Figure 4.
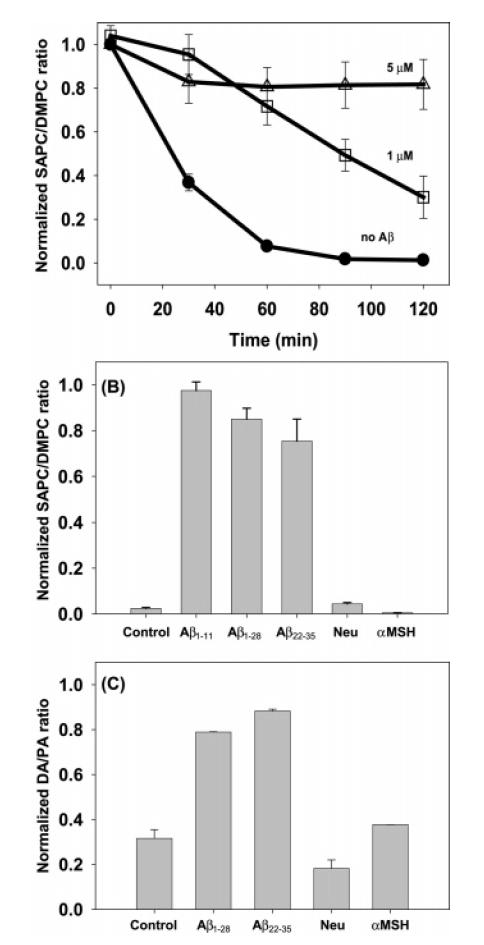
Antioxidant activity of Aβ42. (A) Lipid oxidation by Cu(II) (2.0 μM) and H2O2 (200 μM) in the absence (filled symbols) of Aβ42 and in the presence of 1 or 5 μM Aβ42 (open symbols). (B) Lipid and (C) dopamine concentrations at 120 min of oxidation in the presence of various Aβ and control peptides (5 μM) [Neu = neurotensin, α-MSH]. Aβ was not preincubated with copper in these experiments; Cu(II) (2 μM) and H2O2 (200 μM) were added to a mixture of Aβ and lipid to initiate the experiment.
DISCUSSION
These results demonstrate that Aβ42 promotes oxidative lipid damage in the presence of copper. This is a key finding for several reasons. First, oxidative lipid damage is a consistent finding in Alzheimer’s disease, and nonenzymatic oxidative damage of lipids containing ω-6 fatty acids (such as SAPC) yields products that are promising biomarkers of Alzheimer’s disease (52). Second, oxidatively damaged lipid membranes may initiate the pathological process by misfolding Aβ proteins (6, 7). Third, the oxidative lipid damage that is promoted by Aβ42 only requires 50 μM ascorbate, which is well within the range considered physiologically normal for human brain tissue (53), and substoichiometric amounts of copper relative to Aβ42. Copper is highly protein bound in vivo, and these conditions are mimicked in our experiments wherein 0.5 μM copper is likely to be completely bound by 5 μM Aβ42 (54).
The reaction between ascorbate (AscH−) and Cu(II) produces ascorbyl radical (Asc•−) and Cu(I). Asc•− is relatively unreactive among radical species and most likely undergoes disproportionation with itself to regenerate a molecule of AscH− and a molecule of dehydroascorbate (55). Cu(I) readily oxidizes to produce a variety of reactive oxygen species:
Given an initial ascorbate concentration of 50 μM and dissolved oxygen at approximately 250 μM (55), these two reactions readily account for most of the ~30 μM H2O2 that is produced in the absence of Aβ42 (Figure 3). However, this concentration of H2O2 is relatively ineffective at oxidizing lipid (Figure 4A). Moreover, the extent of lipid oxidation in the presence of Aβ42 is inversely related to both Cu(II) and H2O2 concentration (compare Figures 1C and 3A). Therefore, the species most likely responsible for lipid oxidation is the hydroxyl radical, OH•. This radical may be produced by either of two classic reactions:
The first reaction is the copper-catalyzed Haber–Weiss reaction, while the second is the copper analogue of the Fenton reaction. The observation that H2O2 levels decline as lipid oxidation increases is consistent with either reaction, but the tendency for increased Cu(II) concentrations to inhibit lipid oxidation suggests that Cu(II) decreases OH• production by driving the second reaction in reverse. Polyunsaturated lipids react with OH• and dissolved oxygen via well-known mechanisms to produce lipid hydroperoxides (LOOH) which, in turn, are decomposed by Cu(I) and Cu(II) into chain-reacting radical lipid species, e.g.
Because Aβ42 stabilizes reduced copper (26) and exhibits an affinity for lipid membranes, the most likely mechanism by which it promotes lipid oxidation is by facilitating redox activity while associated with membranes. This mechanism is supported by our observation that variant forms of Aβ42 lacking residues implicated in copper binding (His13 and His14) do not accelerate lipid oxidation. In addition, it hasbeen suggested that Aβ42 traps O2 in a microenvironmentthat facilitates its reduction by electron transfer from metals (27). By facilitating redox reactions involving copper and O2 in the vicinity of a lipid membrane, the H2O2 that isproduced may partition into the membranes, react with Aβ42-bound copper, and produce OH• in close proximity topolyunsaturated lipid acyl chains. Partitioning of H2O2 intomembranes would explain the inability of catalase to inhibit the oxidation. However, the M35V variant has even greater membrane affinity and binds copper (21), yet it inhibits rather than promotes lipid oxidation. This suggests that residue Met35 is involved in facilitating the exchange of electrons between copper and lipid hydroperoxides, as the copper undergoes redox cycling. Met35 has been previously implicated in free radical formation and neurotoxicity (10, 56). Note that simple oxidation of Met35 by H2O2 does not explain our findings, since this produces M35OX which inhibits rather than promotes lipid oxidation (Figure 1D). However, other free radical forms of Met35 may well be involved (31, 33, 57).
The prooxidant activity of Aβ42 described in this paper and that proposed by Opazo et al. (28) represent different chemical reactions but may involve related chemical mechanisms. In both cases, copper ions bound to Aβ42 undergo redox cycling, an electron-donating substrate is required, and H2O2 is produced. However, Aβ42–metal complexes canproduce H2O2 even when there is no apparent source of electrons and when trapping reagents are unable to detect superoxide intermediates (27, 28). In our experiments, it is unlikely that lipids were the electron-donating substrates for H2O2 production according to the mechanism of Opazo etal. because the extent of lipid oxidation was inversely related to H2O2 concentration. Lipid oxidation required ascorbate (Figure 1A), suggesting instead that ascorbate is the principal electron donor for H2O2 production. A specific coreductant (ascorbate) was not required for cholesterol oxidation by Aβ42 and Cu(II) (29). However, it is difficult to compare our results with the chemistry of lipid oxidation in that system because the reaction mixture in the cholesterol experiments was considerably more complex, employing detergent, a different buffer, and a 40:1 molar ratio of Cu(II) to Aβ42.
Previous studies from this laboratory have demonstrated that Aβ42 does indeed have a particular affinity for oxidatively damaged lipid membranes and that on such membranes it can seed and promote fibril formation by Aβ40 (6, 7). Moreover, infrared amide I spectra of Aβ42 in these experiments (Figure 2) are virtually identical to spectra of Aβ40 on Aβ42-seeded membranes (7), while being distinct from those of unaggregated Aβ42 and Aβ fibrils. This suggests that the Aβ proteins promoting oxidative damage share some form of structural similarity with the Aβ proteins that accumulate on oxidatively damaged membranes. They may in fact correspond to the toxic intermediates or soluble oligomers that are suggested to be the most pathological form of Aβ proteins (58-60). Technical difficulties currently preclude verifying this identity by collecting an infrared spectrum of these intermediates or soluble oligomers. It should be noted that prior studies describing metal ion dependencies for Aβ protein association with membranes were performed with lipids that are relatively resistant to oxidative damage (51).
In contrast to its prooxidant properties, Aβ42 inhibits oxidation in the absence of ascorbate and in the presence of exogenously added H2O2. Under these circumstances, increased Cu(II) concentrations reverse the inhibition. Our observations parallel those made in an earlier study of AD-afflicted brains in which immunohistochemical estimates of Aβ were inversely correlated to a biochemical marker of oxidative damage (42). The same study also found that copper significantly increased the neurotoxic effects of Aβ42. For several reasons, however, it would be overly simplistic to conclude from that prior study or our current study that Aβ42 is a neuroprotective antioxidant. One reason is that our in vitro demonstration of Aβ42 antioxidant activityrequired relatively high concentrations of copper and H2O2 while its prooxidant activity was evident at much lower concentrations. Another reason is that irrespective of any negative correlation, Aβ protein and oxidative markers are both elevated in AD-afflicted brain (4, 61).
The inhibitory effect of Aβ42 in our experiments may involve peroxide-mediated oxidation of Met35 which, as noted above, abolishes its ability to accelerate lipid oxidation. However, this explanation does not account for the equally potent inhibitory effects of Aβ1–11 and Aβ1–28, both of which lack Met35. Likewise, sequestration of copper does not explain the inhibition of oxidation by Aβ42 because copper is bound by M35OX and is redox active (20). Furthermore, several of the other variants examined lack two or more of the His residues implicated in high-affinity copper binding (62), and they also inhibit oxidation. Inhibition of oxidation is not due to nonspecific effects of polypeptides, since neurotensin and αMSH did not inhibit oxidation. Similar results were obtained using DA as an oxidizable substrate, demonstrating that these results were not particular to the presence of a lipid phase. Therefore, the antioxidant mechanism of Aβ42 remains unexplained.
In summary, Aβ42 accelerates the oxidative lipid damage caused by ascorbate and Cu(II) ions. The mechanism most likely involves the adsorption of Aβ42 onto lipid membranes, the concentration of bound redox-active metal ion and molecular oxygen in the vicinity of the membrane, and residue Met35. Aβ42 is neither unfolded nor fibrillar in this capacity, and it may be a protofibril, a soluble oligomer, or another intermediate form that is on-path to fibril formation. The ability of Aβ42 to promote lipid oxidation is significant because oxidatively damaged membranes induce misfolding and fibrillization of Aβ42. Insights into the behavior of Aβ proteins in more complex mixtures and multiple phases, such as these involving a mixed lipid phase, bring us closer to understanding of in vivo pathogenesis.
Acknowledgments
The authors are grateful to John Hevko, Jeff Miller, and Bob Bausman from Applied Biosystems, Thomas Waeghe from Agilent, and Kevin Barnham and Carlos Opazo for advice and discussions at early stages of this investigation. Ashley Bush, Trevor Penning, and Harry Ishiropoulos provided helpful comments on the manuscript. Kevin Barnham generously supplied variant forms of Aβ42 for this work.
Footnotes
This work was supported by grants from the Alzheimer’s Disease Research program of the American Health Assistance Foundation (A2004-248 to I.V.J.M.), the National Institute on Aging (AG20238 to P.H.A.), and the Alzheimer’s Association (IIRG-02-3874 to P.H.A.).
Abbreviations: Aβ, amyloid β; SAPC, 1-stearoyl-2-arachidonyl-sn-glycero-3-phosphocholine; DMPC, 1,2-dimyristoyl-sn-glycero-3-phosphocholine; PA, phenylethylamine; DA, dopamine; BHT, 2,6-di-tert-butyl-4-hydroxytoluene; DTPA, diethylenetriaminepentaacetic acid; ESI, electrospray ionization; LC/MS, liquid chromatography/mass spectrometry; AD, Alzheimer’s disease.
References
- 1.Markesbery WR. Oxidative stress hypothesis in Alzheimer’s disease. Free Radical Biol Med. 1997;23:134–147. doi: 10.1016/s0891-5849(96)00629-6. [DOI] [PubMed] [Google Scholar]
- 2.Markesbery WR, Carney JM. Oxidative alterations in Alzheimer’s disease. Brain Pathol. 1999;9:133–146. doi: 10.1111/j.1750-3639.1999.tb00215.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pratico D, Delanty N. Oxidative injury in diseases of the central nervous system: Focus on Alzheimer’s disease. Am J Med. 2000;109:577–585. doi: 10.1016/s0002-9343(00)00547-7. [DOI] [PubMed] [Google Scholar]
- 4.Butterfield DA, Lauderback CM. Lipid peroxidation and protein oxidation in Alzheimer’s disease brain: Potential causes and consequences involving amyloid beta-peptide-associated free radical oxidative stress. Free Radical Biol Med. 2002;32:1050–1060. doi: 10.1016/s0891-5849(02)00794-3. [DOI] [PubMed] [Google Scholar]
- 5.Drake J, Link CD, Butterfield DA. Oxidative stress precedes fibrillar deposition of Alzheimer’s disease amyloid beta-peptide (1–42) in a transgenic Caenorhabditis elegans model. Neurobiol Aging. 2003;24:415–420. doi: 10.1016/s0197-4580(02)00225-7. [DOI] [PubMed] [Google Scholar]
- 6.Koppaka V, Axelsen PH. Accelerated accumulation of amyloid beta proteins on oxidatively damaged lipid membranes. Biochemistry. 2000;39:10011–10016. doi: 10.1021/bi000619d. [DOI] [PubMed] [Google Scholar]
- 7.Koppaka V, Paul C, Murray IVJ, Axelsen PH. Early synergy between A beta 42 and oxidatively damaged membranes in promoting amyloid fibril formation by A beta 40. J Biol Chem. 2003;278:36277–36284. doi: 10.1074/jbc.M301334200. [DOI] [PubMed] [Google Scholar]
- 8.McIntosh LJ, Trush MA, Troncoso JC. Increased susceptibility of Alzheimer’s disease temporal cortex to oxygen free radical-mediated processes. Free Radical Biol Med. 1997;23:183–190. doi: 10.1016/s0891-5849(96)00573-4. [DOI] [PubMed] [Google Scholar]
- 9.Marcus DL, Thomas C, Rodriguez C, Simberkoff K, Tsai JS, Strafaci JA, Freedman ML. Increased peroxidation and reduced antioxidant enzyme activity in Alzheimer’s disease. Exp Neurol. 1998;150:40–44. doi: 10.1006/exnr.1997.6750. [DOI] [PubMed] [Google Scholar]
- 10.Varadarajan S, Yatin S, Aksenova M, Butterfield DA. Review: Alzheimer’s amyloid beta-peptide-associated free radical oxidative stress and neurotoxicity. J Struct Biol. 2000;130:184–208. doi: 10.1006/jsbi.2000.4274. [DOI] [PubMed] [Google Scholar]
- 11.Prasad MR, Lovell MA, Yatin M, Dhillon H, Markesbery WR. Regional membrane phospholipid alterations in Alzheimer’s disease. Neurochem Res. 1998;23:81–88. doi: 10.1023/a:1022457605436. [DOI] [PubMed] [Google Scholar]
- 12.Pettegrew JW, Panchalingam K, Hamilton RL, McClure RJ. Brain membrane phospholipid alterations in Alzheimer’s disease. Neurochem Res. 2001;26:771–782. doi: 10.1023/a:1011603916962. [DOI] [PubMed] [Google Scholar]
- 13.Han XL, Holtzman DM, McKeel DW, Kelley J, Morris JC. Substantial sulfatide deficiency and ceramide elevation in very early Alzheimer’s disease: potential role in disease pathogenesis. J Neurochem. 2002;82:809–818. doi: 10.1046/j.1471-4159.2002.00997.x. [DOI] [PubMed] [Google Scholar]
- 14.Mulder C, Wahlund LO, Teerlink T, Blomberg M, Veerhuis R, van Kamp GJ, Scheltens P, Scheffer PG. Decreased lysophosphatidylcholine/phosphatidylcholine ratio in cerebrospinal fluid in Alzheimer’s disease. J Neural Transm. 2003;110:949–955. doi: 10.1007/s00702-003-0007-9. [DOI] [PubMed] [Google Scholar]
- 15.Lovell MA, Ehmann WD, Butler SM, Markesbery WR. Elevated thiobarbituric acid-reactive substances and antioxidant enzyme-activity in the brain in Alzheimers-disease. Neurology. 1995;45:1594–1601. doi: 10.1212/wnl.45.8.1594. [DOI] [PubMed] [Google Scholar]
- 16.Lovell MA, Ehmann WD, Mattson MP, Markesbery WR. Elevated 4-hydroxynonenal in ventricular fluid in Alzheimer’s disease. Neurobiol Aging. 1997;18:457–461. doi: 10.1016/s0197-4580(97)00108-5. [DOI] [PubMed] [Google Scholar]
- 17.Mattson MP. Central role of oxyradicals in the mechanism of amyloid b-peptide cytotoxicity. Alzheimer’s Dis Rev. 1997;2:1–14. [Google Scholar]
- 18.Montine TJ, Markesbery WR, Morrow JD, Roberts LJ. Cerebrospinal fluid F-2-isoprostane levels are increased in Alzheimer’s disease. Ann Neurol. 1998;44:410–413. doi: 10.1002/ana.410440322. [DOI] [PubMed] [Google Scholar]
- 19.Pratico D, Lee VMY, Trojanowski JQ, Rokach J, FitzGerald GA. Increased F-2-isoprostanes in Alzheimer’s disease: evidence for enhanced lipid peroxidation in vivo. FASEB J. 1998;12:1777–1783. doi: 10.1096/fasebj.12.15.1777. [DOI] [PubMed] [Google Scholar]
- 20.Barnham KJ, Ciccotosto GD, Tickler AK, Ali FE, Smith DG, Williamson NA, Lam YH, Carrington D, Tew D, Kocak G, Volitakis I, Separovic F, Barrow CJ, Wade JD, Masters CL, Cherny RA, Curtain CC, Bush AI, Cappai R. Neurotoxic, redox-competent Alzheimer’s beta-amyloid is released from lipid membrane by methionine oxidation. J Biol Chem. 2003;278:42959–42965. doi: 10.1074/jbc.M305494200. [DOI] [PubMed] [Google Scholar]
- 21.Ciccotosto GD, Tew D, Curtain CC, Smith D, Carrington D, Masters CL, Bush AI, Cherny RA, Cappai R, Barnham KJ. Enhanced toxicity and cellular binding of a modified amyloid beta peptide with a methionine to valine substitution. J Biol Chem. 2004;279:28425–42538. doi: 10.1074/jbc.M406465200. [DOI] [PubMed] [Google Scholar]
- 22.Tickler AK, Smith DG, Ciccotosto GD, Tew DJ, Curtain CC, Carrington D, Masters CL, Bush AI, Cherny RA, Cappai R, Wade JD, Barnham KJ. Methylation of the imidazole side chains of the Alzheimer disease amyloid-{beta} peptide results in abolition of superoxide dismutase-like structures and inhibition of neurotoxicity. J Biol Chem. 2005;280:13355–13363. doi: 10.1074/jbc.M414178200. [DOI] [PubMed] [Google Scholar]
- 23.Wang SSS, Rymer DL, Good TA. Reduction in cholesterol and sialic acid content protects cells from the toxic effects of beta-amyloid peptides. J Biol Chem. 2001;276:42027–42034. doi: 10.1074/jbc.M102834200. [DOI] [PubMed] [Google Scholar]
- 24.Waschuk SA, Elton EA, Darabie AA, Fraser PE, McLaurin J. Cellular membrane composition defines Abeta–lipid interactions. J Biol Chem. 2001;276:33561–33568. doi: 10.1074/jbc.M103598200. [DOI] [PubMed] [Google Scholar]
- 25.Strausak D, Mercer JFB, Dieter HH, Stremmel W, Multhaup G. Copper in disorders with neurological symptoms: Alzheimer’s, Menkes, and Wilson diseases. Brain Res Bull. 2001;55:175–185. doi: 10.1016/s0361-9230(01)00454-3. [DOI] [PubMed] [Google Scholar]
- 26.Huang XD, Cuajungco MP, Atwood CS, Hartshorn MA, Tyndall JDA, Hanson GR, Stokes KC, Leopold M, Multhaup G, Goldstein LE, Scarpa RC, Saunders AJ, Lim J, Moir RD, Glabe C, Bowden EF, Masters CL, Fairlie DP, Tanzi RE, Bush AI. Cu(II) potentiation of Alzheimer A beta neurotoxicitys—Correlation with cell-free hydrogen peroxide production and metal reduction. J Biol Chem. 1999;274:37111–37116. doi: 10.1074/jbc.274.52.37111. [DOI] [PubMed] [Google Scholar]
- 27.Huang XD, Atwood CS, Hartshorn MA, Multhaup G, Goldstein LE, Scarpa RC, Cuajungco MP, Gray DN, Lim J, Moir RD, Tanzi RE, Bush AI. The A beta peptide of Alzheimer’s disease directly produces hydrogen peroxide through metal ion reduction. Biochemistry. 1999;38:7609–7616. doi: 10.1021/bi990438f. [DOI] [PubMed] [Google Scholar]
- 28.Opazo C, Huang XD, Cherny RA, Moir RD, Roher AE, White AR, Cappai R, Masters CL, Tanzi RE, Inestrosa NC, Bush AI. Metalloenzyme-like activity of Alzheimer’s disease beta-amyloid—Cu-dependent catalytic conversion of dopamine, cholesterol, and biological reducing agents to neurotoxic H2O2. J Biol Chem. 2002;277:40302–40308. doi: 10.1074/jbc.M206428200. [DOI] [PubMed] [Google Scholar]
- 29.Nelson TJ, Alkon DL. Oxidation of cholesterol by amyloid precursor protein and beta-amyloid peptide. J Biol Chem. 2005;280:7377–7387. doi: 10.1074/jbc.M409071200. [DOI] [PubMed] [Google Scholar]
- 30.Barnham KJ, Haeffner F, Ciccotosto GD, Curtain CC, Tew D, Beyreuther K, Carrington D, Masters CL, Cherny RA, Cappai R, Bush AI. Tyrosine gated electron transfer is key to the toxic mechanism of Alzheimer’s disease β-amyloid. FASEB J. 2004;18:1427–1429. doi: 10.1096/fj.04-1890fje. [DOI] [PubMed] [Google Scholar]
- 31.Butterfield DA, Kanski J. Methionine residue 35 is critical for the oxidative stress and neurotoxic properties of Alzheimer’s amyloid beta-peptide 1–42. Peptides. 2002;23:1299–1309. doi: 10.1016/s0196-9781(02)00066-9. [DOI] [PubMed] [Google Scholar]
- 32.Butterfield DA. Amyloid beta-peptide (1–42)-induced oxidative stress and neurotoxicity: Implications for neurodegeneration in Alzheimer’s disease brain. A review. Free Radical Res. 2002;36:1307–1313. doi: 10.1080/1071576021000049890. [DOI] [PubMed] [Google Scholar]
- 33.Butterfield DA, Bush AI. Alzheimer’s amyloid beta-peptide (1–42): involvement of methionine residue 35 in the oxidative stress and neurotoxicity properties of this peptide. Neurobiol Aging. 2004;25:563–568. doi: 10.1016/j.neurobiolaging.2003.12.027. [DOI] [PubMed] [Google Scholar]
- 34.Berthon G. Does human beta A4 exert a protective function against oxidative stress in Alzheimer’s disease? Med Hypotheses. 2000;54:672–677. doi: 10.1054/mehy.1999.0924. [DOI] [PubMed] [Google Scholar]
- 35.Sayre LM, Perry G, Harris PLR, Liu YH, Schubert KA, Smith MA. In situ oxidative catalysis by neurofibrillary tangles and senile plaques in Alzheimer’s disease: A central role for bound transition metals. J Neurochem. 2000;74:270–279. doi: 10.1046/j.1471-4159.2000.0740270.x. [DOI] [PubMed] [Google Scholar]
- 36.Kontush A, Donarski N, Beisiegel U. Resistance of human cerebrospinal fluid to in vitro oxidation is directly related to its amyloid-beta content. Free Radical Res. 2001;35:507–517. doi: 10.1080/10715760100301521. [DOI] [PubMed] [Google Scholar]
- 37.Kontush A, Berndt C, Weber W, Akopyan V, Arlt S, Schippling S, Beisiegel U. Amyloid-β is an anti-oxidant for lipoproteins in cerebrospinal fluid and plasma. Free Radical Biol Med. 2001;30:119–128. doi: 10.1016/s0891-5849(00)00458-5. [DOI] [PubMed] [Google Scholar]
- 38.Kontush A. Amyloid-[beta]: an antioxidant that becomes a pro-oxidant and critically contributes to Alzheimer’s disease. Free Radical Biol Med. 2001;31:1120–1131. doi: 10.1016/s0891-5849(01)00688-8. [DOI] [PubMed] [Google Scholar]
- 39.Arlt S, Beisiegel U, Kontush A. Lipid peroxidation in neurodegeneration: new insights into Alzheimer’s disease. Curr Opin Lipidol. 2002;13:289–294. doi: 10.1097/00041433-200206000-00009. [DOI] [PubMed] [Google Scholar]
- 40.Smith MA, Casadesus G, Joseph JA, Perry G. Amyloid-beta and tau serve antioxidant functions in the aging and Alzheimer brain. Free Radical Biol Med. 2002;33:1194–1199. doi: 10.1016/s0891-5849(02)01021-3. [DOI] [PubMed] [Google Scholar]
- 41.Atwood CS, Obrenovich ME, Liu TB, Chan H, Perry G, Smith MA, Martins RN. Amyloid-beta: a chameleon walking in two worlds: a review of the trophic and toxic properties of amyloid-beta. Brain Res Rev. 2003;43:1–16. doi: 10.1016/s0165-0173(03)00174-7. [DOI] [PubMed] [Google Scholar]
- 42.Cuajungco MP, Goldstein LE, Nunomura A, Smith MA, Lim JT, Atwood CS, Huang X, Farrag YW, Perry G, Bush AI. Evidence that the beta-amyloid plaques of Alzheimer’s disease represent the redox-silencing and entombment of Abeta by zinc. J Biol Chem. 2000;275:19439–19442. doi: 10.1074/jbc.C000165200. [DOI] [PubMed] [Google Scholar]
- 43.Nunomura A, Perry G, Pappolla MA, Friedland RP, Hirai K, Chiba S, Smith MA. Neuronal oxidative stress precedes amyloid-beta deposition in Down syndrome. J Neuropathol Exp Neurol. 2000;59:1011–1017. doi: 10.1093/jnen/59.11.1011. [DOI] [PubMed] [Google Scholar]
- 44.Zou K, Gong JS, Yanagisawa K, Michikawa M. A novel function of monomeric amyloid beta-protein serving as an antioxidant molecule against metal-induced oxidative damage. J Neurosci. 2002;22:4833–4841. doi: 10.1523/JNEUROSCI.22-12-04833.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.White DA. The Phospholipid Composition of Mammalian Tissue. Elsevier; New York: 1973. [Google Scholar]
- 46.Bartlett GR. Phosphorus assay in column chromatography. J Biol Chem. 1959;234:466–468. [PubMed] [Google Scholar]
- 47.Kushnir MM, Urry FM, Frank EL, Roberts WL, Shushan B. Analysis of catecholamines in urine by positive-ion electrospray tandem mass spectrometry. Clin Chem. 2002;48:323–331. [PubMed] [Google Scholar]
- 48.Li WL, Rossi DT, Fountain ST. Development and validation of a semi-automated method for L-dopa and dopamine in rat plasma using electrospray LC/MS/MS. J Pharm Biomed Anal. 2000;24:325–333. doi: 10.1016/s0731-7085(00)00422-2. [DOI] [PubMed] [Google Scholar]
- 49.Pulfer M, Murphy RC. Electrospray mass spectrometry of phospholipids. Mass Spectrom Rev. 2003;22:332–364. doi: 10.1002/mas.10061. [DOI] [PubMed] [Google Scholar]
- 50.Nourooz-Zadeh J. Ferrous ion oxidation in the presence of xylenol orange for detection of lipid hydroperoxides in plasma. Methods Enzymol. 1999;300:58–62. doi: 10.1016/s0076-6879(99)00113-5. [DOI] [PubMed] [Google Scholar]
- 51.Curtain CC, Ali FE, Smith DG, Bush AI, Masters CL, Barnham KJ. Metal ions, pH, and cholesterol regulate the interactions of Alzheimer’s disease amyloid-beta peptide with membrane lipid. J Biol Chem. 2003;278:2977–2982. doi: 10.1074/jbc.M205455200. [DOI] [PubMed] [Google Scholar]
- 52.Pratico D. Peripheral biomarkers of oxidative damage in Alzheimer’s disease: the road ahead. Neurobiol Aging. 2005;26:581–583. doi: 10.1016/j.neurobiolaging.2004.09.020. [DOI] [PubMed] [Google Scholar]
- 53.Rice ME. Ascorbate regulation and its neuroprotective role in the brain. Trends Neurosci. 2000;23:209–216. doi: 10.1016/s0166-2236(99)01543-x. [DOI] [PubMed] [Google Scholar]
- 54.Atwood CS, Scarpa RC, Huang XD, Moir RD, Jones WD, Fairlie DP, Tanzi RE, Bush AI. Characterization of copper interactions with Alzheimer amyloid beta peptides: Identification of an attomolar-affinity copper binding site on amyloid beta 1–42. J Neurochem. 2000;75:1219–1233. doi: 10.1046/j.1471-4159.2000.0751219.x. [DOI] [PubMed] [Google Scholar]
- 55.Halliwell B, Gutteridge JMC. Free Radicals in Biology and Medicine. Oxford University Press; Oxford: 1999. [Google Scholar]
- 56.Yatin SM, Varadarajan S, Link CD, Butterfield DA. In vitro and in vivo oxidative stress associated with Alzheimer’s amyloid beta-peptide (1–42) Neurobiol Aging. 1999;20:325–330. doi: 10.1016/s0197-4580(99)00056-1. [DOI] [PubMed] [Google Scholar]
- 57.Schoneich C, Pogocki D, Hug GL, Bobrowski K. Free radical reactions of methionine in peptides: Mechanisms relevant to beta-amyloid oxidation and Alzheimer’s disease. J Am Chem Soc. 2003;125:13700–13713. doi: 10.1021/ja036733b. [DOI] [PubMed] [Google Scholar]
- 58.Caughey B, Lansbury PT. Protofibrils, pores, fibrils, and neurodegeneration: Separating the responsible protein aggregates from the innocent bystanders. Annu Rev Neurosci. 2003;26:267–298. doi: 10.1146/annurev.neuro.26.010302.081142. [DOI] [PubMed] [Google Scholar]
- 59.Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. Naturally secreted oligomers of amyloid beta protein potently inhibit hippo-campal long-term potentiation in vivo. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- 60.Walsh DM, Klyubin I, Fadeeva M, Rowan MJ, Selkoe DJ. Amyloid-beta oligomers: their production, toxicity and therapeutic inhibition. Biochem Soc Trans. 2005;30:552–557. doi: 10.1042/bst0300552. [DOI] [PubMed] [Google Scholar]
- 61.Nunomura A, Perry G, Pappolla MA, Wade R, Hirai K, Chiba S, Smith MA. RNA oxidation is a prominent feature of vulnerable neurons in Alzheimer’s disease. J Neurosci. 1999;19:1959–1964. doi: 10.1523/JNEUROSCI.19-06-01959.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Syme CD, Nadal RC, Rigby SEJ, Viles JH. Copper binding to the amyloid-beta (A beta) peptide associated with Alzheimer’s diseases—Folding, coordination geometry, pH dependence, stoichiometry, and affinity of A beta-(1–28): Insights from a range of complementary spectroscopic techniques. J Biol Chem. 2004;279:18169–18177. doi: 10.1074/jbc.M313572200. [DOI] [PubMed] [Google Scholar]