

Abstract
Patients with the rare genetic disorders, xeroderma pigmentosum (XP), trichothiodystrophy (TTD) and Cockayne syndrome (CS) have defects in DNA nucleotide excision repair (NER). The NER pathway involves at least 28 genes. Three NER genes are also part of the basal transcription factor, TFIIH. Mutations in 11 NER genes have been associated with clinical diseases with at least 8 overlapping phenotypes. The clinical features of these patients have some similarities and but also have marked differences. NER is involved in protection against sunlight induced DNA damage. While XP patients have 1000-fold increase in susceptibility to skin cancer, TTD and CS patients have normal skin cancer risk. Several of the genes involved in NER also affect somatic growth and development. Some patients have short stature and immature sexual development. TTD patients have sulfur deficient brittle hair. Progressive sensorineural deafness is an early feature of XP and CS. Many of these clinical diseases are associated with developmental delay and progressive neurological degeneration. The main neuropathology of XP is a primary neuronal degeneration. In contrast, CS and TTD patients have reduced myelination of the brain. These complex neurological abnormalities are not related to sunlight exposure but may be caused by developmental defects as well as faulty repair of DNA damage to neuronal cells induced by oxidative metabolism or other endogenous processes.
Keywords: cancer, neurodegeneration, DNA repair, genetic disease, neurocutaneous diseases
INTRODUCTION
Xeroderma pigmentosum (XP), trichothiodystrophy (TTD) and Cockayne syndrome (CS) are rare, inherited neurocutaneous disorders (Table 1) 1,8,9,11,19,23. Affected patients have skin hypersensitivity to sun exposure in association with abnormalities in the nervous system. These autosomal recessive diseases are caused by defects in nucleotide excision repair (NER). The NER pathway serves to repair DNA that is damaged by ultraviolet radiation in sunlight as well as by other agents that induce bulky DNA distorting lesions. Thus NER recognizes UV radiation induced photoproducts such as cyclobutane pyrimidine dimers, cigarette smoke induced benzo-a-pyrene DNA adducts and chemical carcinogen induced 4-nitroquinoline oxide DNA adducts. NER also has a role in normal human development including the nervous system. This article will describe major clinical features of XP, TTD, CS, and the XP/CS complex and provide a summary of the NER pathway. Finally, the complex relationship between the clinical features and the molecular defects in NER will be outlined.
Table 1.
COMPARISON OF FEATURES OF XERODERMA PIGMENTOSUM (XP), TRICHOTHIODYSTROPHY (TTD) AND COCKAYNE SYNDROME (CS)
| FEATURE | XERODERMA PIGMENTOSUM(XP) | XP WITH NEUROLGICAL DISEASE | TTD | CS | XP/CS COMPLEX |
|---|---|---|---|---|---|
| SKIN | |||||
| Skin sun sensitivity | yes | severe | yes/no | yes | yes |
| Increased skin pigmentation | yes | yes | no | no | yes |
| Sunlight -induced skin cancer | yes | yes | no | no | yes |
| EYES | |||||
| Photophobia | yes | yes | yes/no | yes | yes |
| Conjunctival growths | yes | yes | no | no | yes |
| Cancer (anterior eye/lids) | yes | yes | no | no | not reported |
| Congenital cataracts | no | no | yes | yes | no |
| Pigmentary retinal degeneration | no | no | no | yes | yes |
| SOMATIC | |||||
| Short stature | no | no/yes | yes | yes | yes |
| Immature sexual development | no | no | no/yes | yes | yes |
| NERVOUS SYSTEM | |||||
| Sensorineural deafness | no | yes | no | yes | yes |
| Developmental delay | no | yes | yes | yes | yes |
| Progressive neurological degeneration | no | yes | unknown | yes | yes |
| Primary neuronal degeneration | no | yes | no | no | no |
| Dysmyelination | no | no | yes | yes | yes |
| Cerebral atrophy | no | yes | no/yes | yes | yes |
| Cerebellar atrophy | no | yes | no | yes | yes |
| Calcifiation (basal ganglia) | no | no | no/yes | yes | yes |
| DISEASE MECHANISM | |||||
| Nucleotide excision repair defect | yes | yes | yes | yes | yes |
| Reaction to exogenous or endogenous damaging agents | yes - severe | yes - severe | no | yes | yes |
| Developmental defect | no | yes | yes - severe | yes -severe | yes - severe |
XERODERMA PIGMENTOSUM
XP patients have marked skin sun sensitivity (Table 1). About half of the patients give a history of acute burning on minimal sun exposure. Typically the burn will appear about one day after the sun exposure. XP patients may develop blistering and severe redness that persists for days. The patients with the more severe burns tend to be those that later develop neurological abnormalities. Other XP patients do not experience sun burning but tan normally. However, following sun exposure all XP patients develop increased freckle-like pigmentation on exposed skin (Figure 1 A, B and C). The presence of these pigmentary abnormalities in children under age 2 years is a clinical marker of XP patients.
Figure 1.

Skin and eye involvement in XP. A. Patient XP6BE, 17 year old female with marked poikilodermatous changes of her face with areas of increased freckle-like pigmentation, areas of decreased pigment and atrophy. The lips show cheilitis and there is absence of eye lashes on both lower lids. She had multiple surgeries for removal of skin cancers including basal cell carcinoma and melanoma. B. Back of patient XP5BE, age 26 years, sister of XP6BE. Her skin shows marked poikilodermatous changes with a sharp cutoff at her sun protected buttocks. These sisters had XP neurological disease. They inherited a defect in the XPD (ERCC2) DNA repair gene. Detailed information is reported in 22. C. Right calf of patient XP1BE at age 46 years. She had multiple large pigmented lesions. Lesion 15 was a large melanoma that measured 0.55 mm in depth when excised. D. Right eye of patient XP1BE at age 29 years. There is clouding of the cornea, prominent vascular growth on the conjunctiva approaching the limbus and loss of lashes on the lower lid. She is wearing a contact lens. She subsequently had a corneal transplant and an ocular squamous cell carcinoma that required enucleation. This patient inherited a defect in the XPC DNA repair gene. Detailed information is reported in 21,22
With continued sun exposure the skin and the eyes become damaged. The skin of children with XP can develop pre-cancerous lesions such as actinic keratoses. XP patients develop skin cancers at a mean age of less than 10 years. This is a 50 year reduction in age of onset of skin cancer in comparison to the U.S. general population and thus XP represents a form of premature aging. Similarly, the eyes may have chronic UV induced conjunctivitis and keratitis. (Figure 1C). These may progress to inflammatory lesions such as pingueculae (conjunctival growths limited to the bulbar conjunctiva) and ptergyia (conjunctival growths that extend onto the corneal surface). In the general population, these lesions are rarely seen in children but have a fairly strong association with UV exposure in normal individuals such as farmers and sailors. The XP patients have 1000-fold increase in cancer of the skin and eyes 11,12. There is evidence that early diagnosis and rigorous sun protection will prevent more of the serious skin cancers in XP patients and prolong their life expectancy.
XP WITH NEUROLOGICAL DISEASE
About 30% of XP patients have a progressive neurological degeneration in addition to the skin abnormalities (Table 1). In contrast to CS, patients with XP neurological disease usually have normal sexual development. The course of the neurological degeneration is variable. The earliest clinical signs of the presence of XP neurological disease are diminished or absent deep tendon reflexes and high frequency hearing loss. Progression of sensorineural deafness may necessitate use of a hearing aid. Other changes may develop including abnormal gait, and difficulty walking eventually leading to the use of a wheelchair (Figure 2 A, B, and C) or to quadraparesis. During childhood, development may progress, with intellectual capacity initially increasing, however, this may be followed by deterioration over the course of years to decades. Swallowing difficulties may become severe leading to aspiration of food. At this late stage patients are often fed with an implanted gastric tube.
Figure 2.

Progressive cachexia in patient XP12BE. At age 4 years she had prominent pigmentary changes on the sun exposed portions of her body (face, arms) with sparing of her chest. By age 17 years she had had many basal cell and squamous cell carcinomas excised. Her main neurological abnormalities were loss of deep tendon reflexes and bilateral sensorineural hearing loss. By age 37 years she had become cachectic, was unable to swallow her food and required assistance to walk. Details of her neurological status up to age 22 years are described in 20. This patient inherited a defect in the XPA DNA repair gene 3.
The MRI shows atrophy of the cerebrum and cerebellum with sparing of the white matter (Table 1). Enlarged ventricles may be seen early in childhood. This process may progress leading to greatly enlarged ventricles. The primary histological finding is neuronal degeneration without inflammation or abnormal depositions.
TRICHOTHIODYSTROPHY
TTD patients have sulfur deficient brittle hair with characteristic alternating dark and light banding appearance (“tiger tail”) with use of a light microscope with polarizing filters (Figure 3 A, B, C and D) 7,14,15,18. The clinical features in different patients are remarkably varied ranging from only hair involvement to severe neurological and somatic developmental abnormalities. Many TTD patients were born prematurely or were small for gestational dates. Some patients may have a form of TTD called PIBIDS (for Photosensitivity, Ichthyosis, Brittle hair, Infertility, Decreased intelligence and Short stature). There is also a high frequency of congenital cataracts and of multiple infections. They may have skeletal abnormalities including peripheral osteopenia and central osteosclerosis. TTD patients, while sun sensitive, do not develop the pigmentary abnormalities of XP and do not have an increased frequency of skin cancer.
Figure 3.
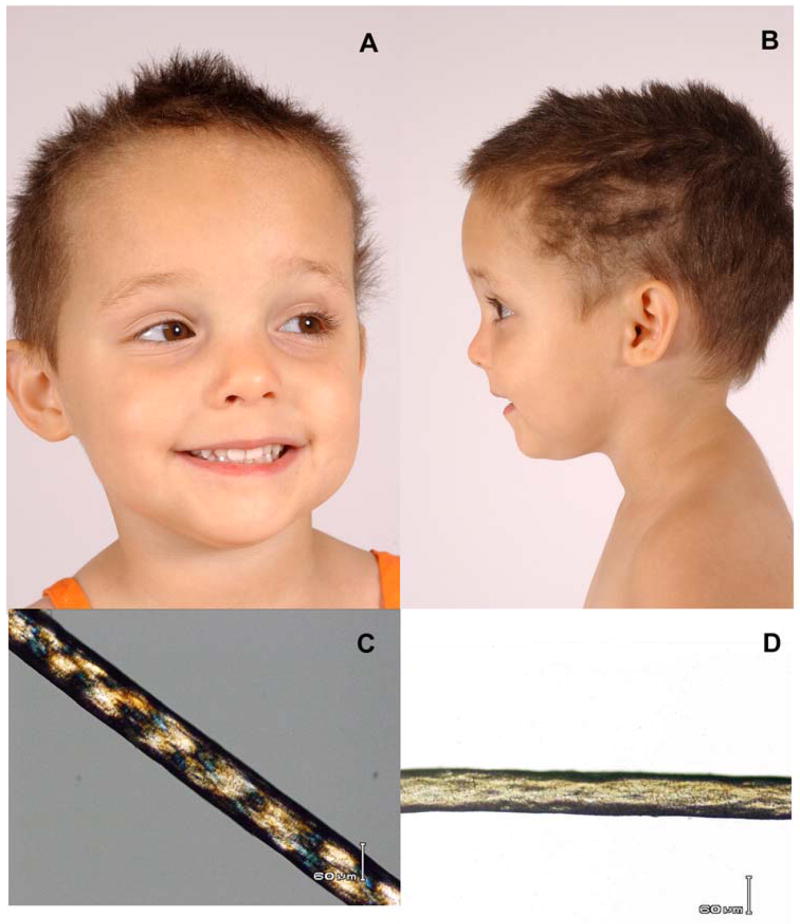
Clinical appearance of TTD. A, B) Three year old female with short, brittle hair which is sparse and broken off at different lengths. She rarely has haircuts except to trim uneven areas. C) Tiger-tail appearance of hair with polarizing microscopy. D) Irregular, undulating hair shaft with light microscopy. From 14
TTD patients may have developmental defects in the nervous system including microcephaly. While TTD patients may have intellectual impairment, they usually are very social and have an outgoing, engaging, friendly personality. Typically, standard intelligence tests appear to underestimate their capability for social interactions. The patients may have ataxia. The MRI shows predominately hypomyelination of the white matter of the cerebrum. Atrophy of the brain is not a major feature. Some patients may have calcification of the basal ganglia. At present the clinical course of TTD patients is not known.
COCKAYNE SYNDROME
CS patients are often normal at birth but experience postnatal failure of brain growth. 17,19. They have a characteristic facies with deep set eyes, prominent ears and a wizened facial appearance. CS patients have extremely short stature and immature sexual development. They are sun sensitive but do not have the pigmentary changes or increased skin cancer frequencies seen in XP (Table 1). Unlike XP, where diminished deep tendon reflexes may be an early sign of neurologic involvement, deep tendon reflexes may be increased in CS. CS patients have a progressive sensorineural deafness beginning with high frequency hearing loss. The eye shows progressive visual loss with pigmentary retinal degeneration (“salt and pepper retinopathy”) and involvement of the lens and cornea 4. CS patients develop profound cachexia and often are fed with a gastric tube. Their clinical course typifies premature aging and usually results in early death.
Like TTD patients, CS patients are very sociable with an outgoing affect. They develop progressive ataxia and loss of ability to walk and care for themselves. The brain MRI shows absence of myelin. Cerebral atrophy may be present. CT scan may reveal prominent calcification of the basal ganglia and other areas in the brain (Figure 4[BPB1] A and B). The primary histological finding in CS is a demyelinating neuropathy rather than the neuronal degeneration seen in XP.
Figure 4.

Calcification of brain in CS. A. Axial brain CT scan of a 3 year old girl showing bilateral and symmetric speckled calcifications of the putamen. Tomographic sections at higher levels (not shown) revealed mild enlargement of the lateral ventricles, decreased attenuation in the white matter due to leukodystrophy and thick skull. She had CS with skin sun sensitivity, short stature and progressive neurological degeneration. B. Axial brain CT scan of a 16 year old girl showing extensive bilateral and symmetric calcifications in the putamen, the globus pallidus and the head of the caudate nucleus. Prominent calcifications are also identified in both cerebellar hemispheres. Less prominent calcifications were present at the gray-white matter junction of the frontal and the parietal lobes which were appreciated in higher tomographic sections. Furthermore in the higher sections there was evident leukodystrophy in the white matter of the cerebral hemispheres and increased skull thickness. She had skin sun sensitivity, short statue and progressive neurological degeneration. Modified from 9.
XERODERMA PIGMENTOSUM/COCKAYNE SYNDROME COMPLEX
Patients with the XP/CS complex have skin and eye disease of XP and the somatic and neurological abnormalities of CS (Table 1) 19. Thus their skin is hypersensitive to sunlight and they develop the freckling and other pigmentary changes of XP. They have the short stature and immature sexual development as well as the retinal degeneration as in CS. The disease course is one of progressive neurological degeneration (Figure 5 A-G). Less than two dozen patients with the XP/CS complex have been reported in the literature.
Figure 5.

Patient XP20BE with XP/CS complex. A. Patient at age 4 months after he developed severe sunburn in March, the first sign of illness. Note the sharp change in redness on his lower legs where his socks blocked some sunlight. B. Normal fullness of the face at age 1 year. C. Age 1.5 years. Characteristic skin lesions of XP: freckling and hypopigmented macules of skin and lips. A benign pedunculated lesion is circled. D. Age 6 years. Typical appearance of CS with deep set eyes, prominent ears, and profound cachexia with posturing of the hand. E. Age 6 years. Pigmentary changes and wrinkling of the dorsum of the hand showing signs of premature aging. F. Age 6 years. Signs of advanced CS with profound weakness of dorsiflexion of the hand. Note small size in comparison to the mother who is holding him. G. Profound wasting and contractures of the legs with distal cyanosis and permanently upgoing toes. From 16
NUCLEOTIDE EXCISION REPAIR PATHWAY
Patients with XP, TTD, CS and the XP/CS complex have defects in the NER pathway (Figure 6) 23. This pathway serves to remove DNA damage caused by exposure to ultraviolet radiation and some other exogenous and endogenous agents such as some bulky lesions caused by chemical carcinogens or by oxidative damage. There are at least 28 proteins in the NER pathway. They serve to recognize DNA lesions, unwind the surrounding DNA, excise the lesion and then fill in the resulting gap. Three of the proteins are also part of the 10 protein complex of the basal transcription factor, TFIIH. Thus the NER pathway is closely linked to transcription.
Figure 6.
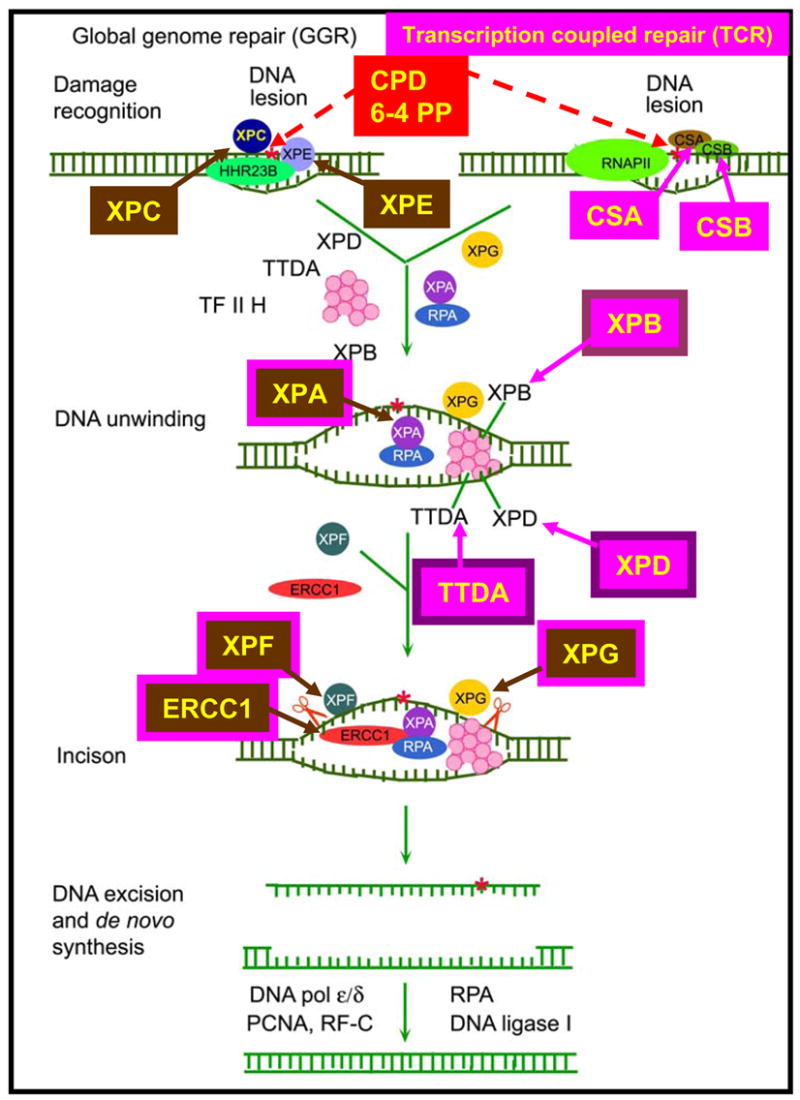
Nucleotide excision repair pathway. Transcription coupled repair (TCR) removes damage from actively transcribing genes while global genome repair (GGR) removes damage from the remainder of the genome. In GGR damage such as ultraviolet induced cyclobutane pyrimidine dimers (CPD) or 6–4 photoproducts (6–4 PP) are recognized by proteins including the XPE (DDB2) and XPC gene products. In TCR, the lesion appears to block the progress of RNA polymerase II in a process involving the CSA and CSB gene products. Following initial damage recognition the pathways converge. The XPB (ERCC3) and XPD (ERCC2) helicases unwind the region surrounding the lesion along with the XPA and XPG (ERCC5) gene products, and replication protein A (RPA). The XPF and XPG (ERCC5) endonucleases perform incisions to remove the lesion in a fragment of about 30 nucleotides. The resulting gap is filled in by de novo DNA synthesis. This system is coordinated so that if one part of the pathway is mutated the entire pathway fails to function normally. Mutations in the genes in rectangles have been associated with clinical disease. This diagram is modified from 23.
Repair of actively transcribing genes (transcription coupled repair) proceeds more rapidly than DNA repair of damage located in the remainder of the genome. DNA damage in these actively transcribing genes is signaled by CSA and CSB proteins along with other proteins. The XPC and XPE (DDB2) proteins are involved in recognition of DNA damage in the remainder of the genome (global genome repair). The XPB, XPD and TTDA proteins are part of the basal transcription factor TFIIH. The XPB and XPD helicases unwind the DNA. The XPF and XPG proteins are endonucleases that make asymmetric single strand breaks on either side of the damage about 30 nucleotides apart.
RELATIONSHIP OF CLINICAL DISEASE TO NER MUTATIONS
There is a complex relationship between the clinical diseases and the molecular defects in NER (Figure 7). Patients with one of clinical disease may have inherited a defect in one of several different NER genes. Since the NER pathway functions in sequence, a defect in one portion of the pathway impairs the function of the subsequent steps. Thus patients with XP can have defects in XPA, XPB(ERCC3), XPC, XPD(ERCC2), XPE(DDB2), XPF(ERCC4) or XPG(ERCC5) genes. Conversely, different defects in one gene may lead to different clinical diseases. Thus different mutations in the XPD (ERCC2) gene may lead to one of 6 different clinical disorders: XP, XP neurological disease, TTD, the XP/CS complex, XP/TTD complex or a severe form of CS known as COFS (cerebral, ocular, facial, skeletal syndrome).
Figure 7.
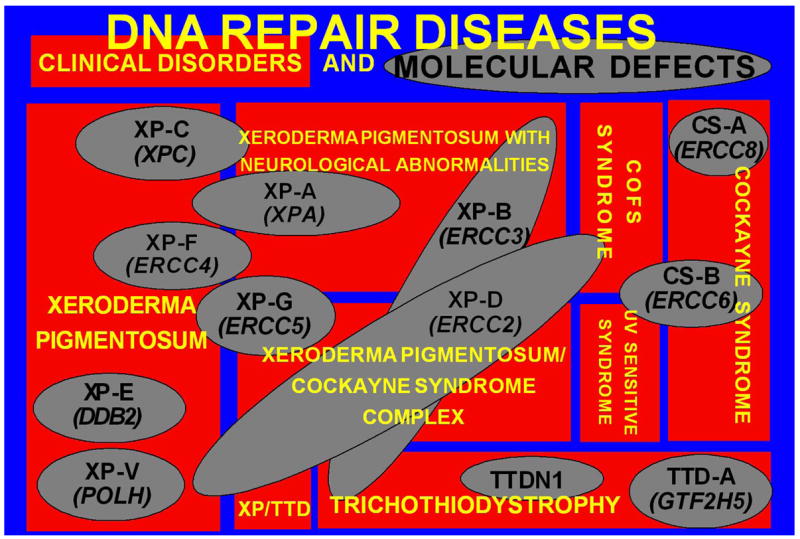
DNA repair diseases - relationship of clinical disorders (red rectangles) to molecular defects (gray ovals) in DNA Repair diseases. Eight clinical diseases are represented. One disease may be caused by mutations in several different genes. Conversely, different mutations in one gene may result in several different clinical diseases. Modified from 10.
THEORETICAL MECHANISMS OF DISEASE
The skin and eye disease in XP and the 1000-fold increased frequency of neoplasms in these sun exposed organs is related to faulty repair of ultraviolet damage to DNA. This results in increased cell killing and increased frequency of mutations in many of the genes in the surviving cells. Accumulation of mutations that activate oncogenes or inhibit tumor suppressor genes in dividing cells eventually leads to cancer.
In the XP patients who develop progressive neurological degeneration the brain is not exposed to ultraviolet radiation. The progressive nature of the degeneration suggests that there might be ongoing damage that is not repaired. Since neurons do not divide, unrepaired DNA damage could result in impaired cellular function and eventually to cell death. An effect of the damage might be to alter the process of transcription so that mutations might be introduced into newly synthesized RNA. This could impair functioning of essential proteins. The NER pathway is present in all cells in the body including those in the nervous system. Recent studies from several laboratories have reported the discovery of some bulky DNA lesions produced by oxidative damage that are substrates for the NER pathway. Unrepaired lesions such as cyclo-deoxyadenine or cyclo-deoxyguanine might accumulate in the DNA of neurons of patients with defective NER and lead to the progressive neurological degeneration 2. There are many unanswered questions relating to this theory including why only some of the XP patients develop neurological degeneration and why certain parts of the brain are typically affected more than others.
Patients with TTD have neurological defects that appear to be mainly related to impaired development and maturation of the nervous system. They have decreased to absent myelin in the cerebrum. While not proven definitively, it seems that the myelin in these patients never formed properly (dysmyelination) rather than sustaining a loss of normally formed myelin (demyelination). This suggests the presence of a developmental defect as is also seen in the congenital cataracts in the eye, the problems during in utero growth, and the short stature. In addition TTD patients do not have an increased frequency of skin cancer. TTD patients have defects in XPD, XPB or TTDA genes. These genes are part of the basal transcription factor, TFIIH as well as in NER. It has been proposed that the specific defects in these genes in TTD patients have a greater effect on the transcription activities of these genes than on their repair activities 5. A striking example is the finding that mutations in the XPD component of TFIIH result in beta-thalassemia in TTD patients without mutations in hemoglobin genes24. At present we have no evidence of progressive neurological degeneration in TTD patients.
CS patients have both developmental defects and progressive neurological degeneration. Like TTD patients there is primary involvement of the myelin in the brain. The CSA and CSB proteins have a role in transcription coupled repair and may also be involved in transcription of undamaged genes. CSA and CSB proteins are involved in regulation of recruitment of chromatin remodeling and repair factors to stalled RNA polymerase II after UV damage6. The transcriptional response after oxidative damage is defective in cells from patients with mutations in the CSB gene13. Thus the progressive neurological degeneration in CS may involve faulty repair of oxidative damage or of bulky DNA damage as in XP patients. However major questions remain such as the differences in the type of involvement – for example retinal degeneration is present in CS but not in XP - and the differences in cancer susceptibility.
Acknowledgments
This research was supported by the Intramural Research Program of the NIH and the Center for Cancer Research of the National Cancer Institute, the Clinical Center, the National Institute of Neurological Diseases and Stroke, and the National Eye Institute.
Abbreviations
- XP
xeroderma pigmentosum
- TTD
trichothiodystrophy
- CS
Cockayne syndrome
- NER
nucleotide excision repair
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bootsma D, Kraemer KH, Cleaver JE, Hoeijmakers JHJ. Nucleotide excision repair syndromes: xeroderma pigmentosum, Cockayne syndrome, and trichothiodystrophy. In: Vogelstein B, Kinzler KW, editors. The Genetic Basis of Human Cancer. 2. McGraw-Hill; New York: 2002. pp. 211–237. [Google Scholar]
- 2.Brooks PJ, Wise DS, Berry DA, Kosmoski JV, Smerdon MJ, Somers RL, Mackie H, Spoonde AY, Ackerman EJ, Coleman K, Tarone RE, Robbins JH. The oxidative DNA lesion 8,5′-(S)-cyclo-2′-deoxyadenosine is repaired by the nucleotide excision repair pathway and blocks gene expression in mammalian cells. J Biol Chem. 2000;275:22355–22362. doi: 10.1074/jbc.M002259200. [DOI] [PubMed] [Google Scholar]
- 3.Cleaver JE, Thompson LH, Richardson AS, States JC. A summary of mutations in the UV-sensitive disorders: xeroderma pigmentosum, Cockayne syndrome, and trichothiodystrophy. Hum Mutat. 1999;14:9–22. doi: 10.1002/(SICI)1098-1004(1999)14:1<9::AID-HUMU2>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 4.Dollfus H, Porto F, Caussade P, Speeg-Schatz C, Sahel J, Grosshans E, Flament J, Sarasin A. Ocular manifestations in the inherited DNA repair disorders. Surv Ophthalmol. 2003;48:107–122. doi: 10.1016/s0039-6257(02)00400-9. [DOI] [PubMed] [Google Scholar]
- 5.Dubaele S, Proietti d S, Bienstock RJ, Keriel A, Stefanini M, Van Houten B, Egly JM. Basal transcription defect discriminates between xeroderma pigmentosum and trichothiodystrophy in XPD patients. Mol Cell. 2003;11:1635–1646. doi: 10.1016/s1097-2765(03)00182-5. [DOI] [PubMed] [Google Scholar]
- 6.Fousteri M, Vermeulen W, van Zeeland AA, Mullenders LH. Cockayne syndrome A and B proteins differentially regulate recruitment of chromatin remodeling and repair factors to stalled RNA polymerase II in vivo. Mol Cell. 2006;23:471–482. doi: 10.1016/j.molcel.2006.06.029. [DOI] [PubMed] [Google Scholar]
- 7.Itin PH, Sarasin A, Pittelkow MR. Trichothiodystrophy: update on the sulfur-deficient brittle hair syndromes. J Am Acad Dermatol. 2001;44:891–920. doi: 10.1067/mjd.2001.114294. [DOI] [PubMed] [Google Scholar]
- 8.Kraemer KH. Cellular hypersensitivity and DNA repair. In: Freedberg IM, Eisen AZ, Wolff K, Austen KF, Goldsmith LA, Katz SI, editors. Fitzpatrick’s Dermatology in General Medicine. McGraw Hill; New York: 2003. pp. 351–358. [Google Scholar]
- 9.Kraemer KH. Heritable diseases with increased sensitivity to cellular injury. In: Freedberg IM, Eisen AZ, Wolff K, Austen KF, Goldsmith LA, Katz SI, editors. Fitzpatrick’s Dermatology in General Medicine. McGraw-Hill; New York: 2003. pp. 1508–1521. [Google Scholar]
- 10.Kraemer KH. From proteomics to disease. Nat Genet. 2004;36:677–678. doi: 10.1038/ng0704-677. [DOI] [PubMed] [Google Scholar]
- 11.Kraemer KH, Lee MM, Scotto J. Xeroderma pigmentosum. Cutaneous, ocular, and neurologic abnormalities in 830 published cases. Arch Dermatol. 1987;123:241–250. doi: 10.1001/archderm.123.2.241. [DOI] [PubMed] [Google Scholar]
- 12.Kraemer KH, Lee MM, Andrews AD, Lambert WC. The role of sunlight and DNA repair in melanoma and nonmelanoma skin cancer: The xeroderma pigmentosum paradigm. Arch Dermatol. 1994;130:1018–1021. [PubMed] [Google Scholar]
- 13.Kyng KJ, May A, Brosh RM, Jr, Cheng WH, Chen C, Becker KG, Bohr VA. The transcriptional response after oxidative stress is defective in Cockayne syndrome group B cells. Oncogene. 2003;22:1135–1149. doi: 10.1038/sj.onc.1206187. [DOI] [PubMed] [Google Scholar]
- 14.Liang C, Kraemer KH, Morris A, Schiffmann R, Price VH, Menefee E, DiGiovanna JJ. Characterization of tiger tail banding and hair shaft abnormalities in trichothiodystrophy. J Am Acad Dermatol. 2005;52:224–232. doi: 10.1016/j.jaad.2004.09.013. [DOI] [PubMed] [Google Scholar]
- 15.Liang C, Morris A, Schlucker S, Imoto K, Price VH, Menefee E, Wincovitch SM, Levin IW, Tamura D, Strehle KR, Kraemer KH, DiGiovanna JJ. Structural and molecular hair abnormalities in trichothiodystrophy. J Invest Dermatol. 2006;126:2210–2216. doi: 10.1038/sj.jid.5700384. [DOI] [PubMed] [Google Scholar]
- 16.Lindenbaum Y, Dickson D, Rosenbaum P, Kraemer K, Robbins I, Rapin I. Xeroderma pigmentosum/cockayne syndrome complex: first neuropathological study and review of eight other cases. Eur J Paediatr Neurol. 2001;5:225–242. doi: 10.1053/ejpn.2001.0523. [DOI] [PubMed] [Google Scholar]
- 17.Nance MA, Berry SA. Cockayne syndrome: Review of 140 cases. Am J Med Genet. 1992;42:68–84. doi: 10.1002/ajmg.1320420115. [DOI] [PubMed] [Google Scholar]
- 18.Price VH, Odom RB, Ward WH, Jones FT. Trichothiodystrophy: sulfur-deficient brittle hair as a marker for a neuroectodermal symptom complex. Arch Dermatol. 1980;116:1375–1384. doi: 10.1001/archderm.116.12.1375. [DOI] [PubMed] [Google Scholar]
- 19.Rapin I, Lindenbaum Y, Dickson DW, Kraemer KH, Robbins JH. Cockayne syndrome and xeroderma pigmentosum. Neurology. 2000;55:1442–1449. doi: 10.1212/wnl.55.10.1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Robbins JH, Brumback RA, Mendiones M, Barrett SF, Carl JR, Cho S, Denckla MB, Ganges MB, Gerber LH, Guthrie RA. Neurological disease in xeroderma pigmentosum. Documentation of a late onset type of the juvenile onset form. Brain. 1991;114:1335–1361. doi: 10.1093/brain/114.3.1335. [DOI] [PubMed] [Google Scholar]
- 21.Robbins JH, Brumback RA, Moshell AN. Clinically asymptomatic xeroderma pigmentosum neurological disease in an adult: Evidence for a neurodegeneration in later life caused by defective DNA repair. Eur Neurol. 1993;33:188–190. doi: 10.1159/000116932. [DOI] [PubMed] [Google Scholar]
- 22.Robbins JH, Kraemer KH, Lutzner MA, Festoff BW, Coon HG. Xeroderma pigmentosum. An inherited disease with sun sensitivity, multiple cutaneous neoplasms, and abnormal DNA repair. Ann Intern Med. 1974;80:221–248. doi: 10.7326/0003-4819-80-2-221. [DOI] [PubMed] [Google Scholar]
- 23.Van Steeg H, Kraemer KH. Xeroderma pigmentosum and the role of UV-induced DNA damage in skin cancer. Mol Med Today. 1999;5:86–94. doi: 10.1016/s1357-4310(98)01394-x. [DOI] [PubMed] [Google Scholar]
- 24.Viprakasit V, Gibbons RJ, Broughton BC, Tolmie JL, Brown D, Lunt P, Winter RM, Marinoni S, Stefanini M, Brueton L, Lehmann AR, Higgs DR. Mutations in the general transcription factor TFIIH result in beta-thalassaemia in individuals with trichothiodystrophy. Hum Mol Genet. 2001;10:2797–2802. doi: 10.1093/hmg/10.24.2797. [DOI] [PubMed] [Google Scholar]