

Abstract
Self-renewal capacity is rapidly lost during differentiation of hematopoietic stem cells to lineage-committed progenitors. We demonstrate here that regulated intracellular signaling through the cytokine receptor Mpl induces profound expansion of not only multipotent (ie, lymphomyeloid) but also lymphoid-committed human hematopoietic progenitors. A fusion protein containing the intracellular signaling domain of Mpl and a dimerization domain was constitutively expressed in populations enriched in human lymphomyeloid progenitor/stem cells (CD34+CD38−Lin−CD7−) and multilymphoid progenitors (CD34+CD38−Lin−CD7+). Intracellular dimerization of Mpl in target cells was induced by in vitro or in vivo administration of a diffusible synthetic ligand. In vitro, Mpl dimerization produced divisions of clonogenic, multilineage CD34+ cells able to engraft immunodeficient mice. When dimerization was induced in vivo after transplantation of either lymphomyeloid or multilymphoid progenitors, donor-derived hematopoiesis was sustained for at least 12 weeks and primitive CD34+Lin− progenitors were expanded more than 1000-fold. Lineage potential of progenitors was not altered and differentiation was not prevented by synthetically induced Mpl signaling. These data demonstrate that dimerization of a single cytokine receptor can deliver a profound expansion signal in both uncommitted and lymphoid-committed human hematopoietic progenitors.
Introduction
Studies of the hematopoietic system have formed the foundation for the concept of a stem and progenitor cell hierarchy.1 This paradigm links the potential for proliferation with that of lineage potency, with self-renewal capacity restricted to multipotent stem cells and a progressive loss of proliferative potential occurring at each successive stage of lineage commitment. The stages of differentiation are each regulated by a complex interaction between cell intrinsic factors and extrinsic factors from the microenvironment. Experimental attempts to harness the self-renewal process of stem cells have included ex vivo stimulation with multiple cytokines2 and forced induction of transcriptional pathways thought to be critical for stem cell expansion.3 Although some approaches have appeared to induce self-renewal of murine hematopoietic stem cells,4–8 the human system has been much more challenging, with ex vivo stimulation of stem cells tending to lead rapidly to loss of lineage potency and engraftment potential.
Mpl is a hematopoietic growth factor receptor expressed on hematopoietic stem cells, myeloid precursors, and megakaryocytes.9–11 The ligand for Mpl, thrombopoietin (Tpo), is a key regulator of megakaryocytopoiesis,12 and when used in combination with other cytokines, Tpo induces hematopoietic progenitor expansion in long-term cultures.13 The intracellular signaling domain of Mpl can be expressed in target cells as a fusion protein containing a sequence (F36V) that binds to the diffusible, synthetic ligand AP20187, a chemical inducer of dimerization (CID). This strategy has produced varied results depending on the animal model studied and the type of target cells transduced; previous results with mixed populations of human CD34+ progenitors have produced only myeloid, megakaryocytic, and erythroid differentiation in vitro and in vivo with no evidence of stem or progenitor expansion.14–16
By targeting defined populations of primitive human progenitors, we show that Mpl dimerization induced in vitro can produce a rapid expansion of engraftable, multipotent progenitors in the absence of exogenous cytokines. When Mpl dimerization was induced in vivo after transplantation, sustained and profound progenitor expansion was generated not only from transplanted multilineage (CD34+CD38−Lin−CD7−) progenitors, but also from multilymphoid (CD34+CD38−Lin−CD7+) progenitors. These studies demonstrate that synthetic dimerization of the intracellular component of the Mpl receptor can selectively expand multipotent and lymphoid-restricted human progenitors without loss of lineage potential. The robust in vivo proliferation of lymphoid progenitors and their progeny suggests that this approach might be used to achieve rapid immune reconstitution after bone marrow transplantation.
Methods
Isolation of human progenitor populations
Umbilical cord blood was collected from normal deliveries, according to guidelines approved by the Childrens Hospital Los Angeles Committee on Clinical Investigation (investigational review board). Immunomagnetic enrichment for CD34+ cells was performed as per the manufacturer's instructions using the magnetic-activated cell sorting (MACS) system (Miltenyi Biotec, Auburn, CA) or StemSep human CD34+ selection system (StemCell Technologies, Vancouver, BC).
CD34+ enriched cells were stained with the following anti-human–specific monoclonal antibodies: CD34− PerCP-Cy5.5, CD38− PE-Cy7, CD7-PE, and the FITC-labeled lineage-specific antibodies: CD14, CD15, CD56, CD57 (Becton Dickinson, San Jose, CA), and Gly A (Beckman Coulter/Immunotech, Fullerton, CA), and the lineage APC-labeled antibodies: CD3, and CD19 (all from Becton Dickinson). IgG1 isotype control antibodies (Becton Dickinson) were used to define negative gates. Populations enriched for lymphomyeloid stem/progenitors were defined as CD34+CD38−Lin−CD7− and multilymphoid progenitors as CD34+CD38−Lin−CD7+cells.17 Fluorescence-activated cell-sorting (FACS) analysis and sorting were performed on a FACSVantage (Becton Dickinson).
Lentiviral vector expression of fusion proteins
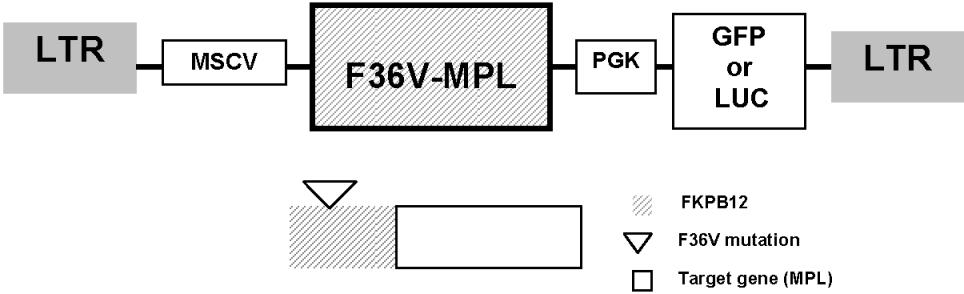
Lentiviral (HIV-1) vectors were constructed that express the fusion protein (F36V-Mpl) expressed under control of the murine stem-cell leukemia virus (MSCV) promoter and a downstream marker gene (green fluorescent protein; GFP for in vitro, and luciferase [LUC] for in vivo studies) expressed under control of the hPGK (human phosphoglycerate kinase) promoter (Figure S1, available on the Blood website; see the Supplemental Materials link at the top of the online article). The F36V-MPL-GFP plasmid did not contain the myristylation domain and was a generous gift of Dr C. A. Blau (University of Washington, Seattle, WA).18 Control vectors expressed F36V and a downstream marker gene (GFP or LUC). Vectors were pseudotyped using vesicular stomatitis virus (VSV) envelope by transient transfection into 293T cells using standard protocols, and were concentrated by ultrafiltration and ultracentrifugation as described before.19–21
Cells were transduced in non–tissue-culture 48-well plates coated with the recombinant fibronectin fragment CH-296 (50 μg/mL; Takara Shuzo Otsu, Japan) in 200 μL serum-free transduction medium X-vivo 15 (BioWhittaker, Walkersville, MD). Viral supernatant (2 × 107 IU/mL) was incubated on CH-296–coated plates for 24 hours prior to addition of human stem/progenitor cells. Medium was then aspirated and discarded prior to adding progenitor populations in 200 μL fresh viral supernatant, X-vivo 15, and cytokines. Fresh viral supernatant (multiplicity of infection [MOI] = 40) was added to the cells twice, after 12 and 24 hours of cytokine stimulation. The following cytokines were added during transduction only (stem cell factor [SCF], 50 ng/mL; Flt 3 ligand [FL[, 5 ng/mL; Tpo, 50 ng/mL; interleukin-3 [IL-3], 20 ng/mL; and IL-6, 20 ng/mL; all from R&D Systems, Minneapolis, MN). IL-3 was not used for transduction in PKH26 experiments; IL-3 and IL-6 were not used for transduction prior to in vivo CID experiments to minimize the proliferation and differentiation stimuli of cells during transduction.
After 36 hours of total exposure to vector, cells were thoroughly washed 5 times with Dulbecco phosphate-buffered saline (DPBS; Mediatech, Herndon, VA) before analysis in in vitro or in vivo assays. Cells were transduced in a common pool and then divided for culture or for transplantation with or without CID.
CID AP20187 formulation and administration
Lyophylized AP20187 (ARIAD Pharmaceuticals [Cambridge, MA] regulation kit) was solubilized in 100% ethanol to produce 1 mM stock solution (for in vitro use) or 3.5 mM (for in vivo use) and stored at −20°C. For in vitro experiments, AP20187 was diluted fresh in the culture medium to a final concentration of 100 nM. Half of the culture medium was changed every 3 days. For in vivo administration, stock solution was diluted fresh on the day of injection. The final solution for injection contained 10% PEG 400 and 1.4% Tween 80.
B- and NK-cell cultures
Cells were plated in bulk or as single cells (by automated cell deposition unit [ACDU] of FACSVantage) in 96-well tissue-culture plates onto established murine marrow MS5 stromal cell line22 in lymphoid medium (RPMI 1640 [Irvine Scientific, Santa Ana, CA], 5% FCS [screened for B-cell cultures], 50 mM 2-ME, 50 U/mL penicillin/50 μg/mL streptomycin, and 200 mM l-glutamine).23
Lymphomyeloid progenitors (CD34+CD38−Lin−CD7−), multilymphoid progenitors (CD34+38−Lin−CD7+), or mixed progenitor/hematopoietic stem cell (HSC) populations (CD34+) were transduced as in “Lentiviral vector expression of fusion proteins” with either F36V-Mpl or F36V control vectors and cultured in the presence or absence of the CID AP 20187 (100 nM) and in the absence of cytokines. In specific experiments, control nontransduced and F36V-MPL–transduced CD34+ or CD34+CD38−Lin−CD7− cells were cultured in the presence of Tpo (50 ng/mL).
Every 7 to 14 days thereafter, depending on the density of the culture, half the cells were recovered for analysis with vigorous pipetting and replaced with fresh medium containing AP20187. Viable cells were counted using trypan blue. Human lineage-specific differentiation was assessed by FACS as described in “Immunophenotypic analysis of human hema-topoietic cells.” Fold increase in the total number of cells compared with the original number of cells plated was calculated allowing for cells lost during serial analysis. The frequency of myeloerythroid progenitors in the cultures was assessed using a colony-forming unit cell (CFU-C) assay by replating equal number of cells in methylcellulose assay using METHOCULT GF+ H4435 (StemCell Technologies) and counting colonies after 14 days.
Immunophenotypic analysis of human hematopoietic cells
FACS analysis was performed on FACSCalibur or FACScan (Becton Dickinson) by direct immunofluorescence staining with human specific monoclonal antibodies and by detection of GFP expression after incubation in 1.2% human intravenous immunoglobulin (IVIG; Cutter, Berkley, CA). Analysis used Cellquest (Becton Dickinson) or FlowJo (Tree Star, Ashland, OR).
Lineage-specific human antigen expression was determined using the following antibodies. Conjugated to APC: CD45, CD10, CD34, CD19, CD38, CD56, and CD4 (all from Becton Dickinson); conjugated to PE: CD45, CD10, CD11b, CD19, CD56, CD57, CD4, CD8 (all from Becton Dickinson), CD7, CD10, Gly A–PE (Beckman Coulter), and CD56; conjugated to FITC: CD3, CD7, CD10, CD19, CD56, CD57, CD14, CD15, CD66B (all from Becton Dickinson), and Gly A; and conjugated to PerCP: CD3, CD4, CD8, CD19, CD34, and CD45 (all from Becton Dickinson).
In vitro tracking of cell proliferation (PKH26 assay)
CD34+ cells were isolated by FACS and transduced with F36V-MPL-GFP or control F36V-GFP vectors, then incubated in 2 mL of diluent with PKH26 (final concentration 2 × 10−6 M; Sigma-Aldrich, St Louis, MO) for 1.5 minutes at room temperature. FCS (10%) was then added to block further adsorption of dye, and the cells were washed 6 times. After 12 hours of culture on CH-296 to allow stabilization of the PKH26 stain, a narrow band of viable PKH26-bright (generation 1 = parent population) cells was isolated, and subsequently cultured in 6-well tissue-culture plates in the lymphoid medium described in “B- and NK-cell cultures” with or without CID and in absence of any cytokines. A small aliquot of the cells was analyzed serially by FACS for PKH26, CD34, and GFP expression and cell count. ModFit LT software (Verity Software House, Topsham, ME) was used to analyze the data. After 7 days in culture, cells which had undergone 3 divisions (generation 4) in the presence or the absence of CID were isolated by FACS and either transplanted into immunodeficient mice or plated as single cells for clonal lineage analysis.
Transplantation assays
Nonobese diabetic/severe combined immunodeficiency (NOD/SCID) β2-microglobulinnull mice, obtained from Jackson Laboratories (Bar Harbor, ME), were used for in vivo experiments according to protocols approved by the Insitutional Animal Care and Use Committee of Childrens Hospital Los Angeles.
Transduced human cells were suspended in DPBS and inoculated (10 μL/animal) into the right tibia of sublethally irradiated (300 cGy 1 hour before transplantation) mice. Engraftment and differentiation of human cells was assessed by in vivo bioluminescence imaging and/or FACS from harvested organs after preincubation in 1.2% human IVIG (Cutter) and purified rat anti–mouse CD16/CD32 (FcγIII/II receptor; Becton Dickinson) to prevent nonspecific antibody binding. The transplantation of relatively low numbers of cells (1 × 104 CD34+CD38−Lin−CD7− and 6 × 103 CD34+CD38−Lin−CD7+ per mouse) was designed to provide minimal baseline engraftment from unstimulated cells. Mice were given the CID AP20187 intraperitoneal (IP) 10 mg/kg (or control vehicle) daily starting 1 day prior to transplantation (day −1) until time of killing.
Human-specific CFU-C assay
Bone marrow cells harvested from mice that received transplants and control animals that did not receive transplants were cultured in 1% METHOCULT H4230 (StemCell Technologies) in recombinant human cytokines which do not cross react with murine cells (20 ng/mL IL-3 and 20 ng/mL granulocyte-macrophage colony-stimulating factor [GM-CSF; R&D Systems]). CFU-Cs were confirmed to be of human origin by Alu polymerase chain reaction (PCR) from DNA isolated from individual colonies (Document S1).
Additional information is provided in Document S1.
Bioluminescence imaging
In vivo bioluminescence imaging was performed under general anesthesia as described.24 Prior to imaging, each mouse was given 125 mg/kg intraperitoneal luciferin (Promega, Madison, WI). In vivo optical imaging was performed with a prototype IVIS 3D bioluminescence/fluorescence optical imaging system (Xenogen, Alameda, CA) at different time points. Regions were manually drawn around the bodies of the mice to assess signal intensity emitted. Ventral and dorsal images were acquired and mean signal was calculated for each time point.
PCR analysis
To analyze lentiviral vector integrants in engrafted human cells, genomic DNA from unfractionated bone marrow of CID treated mice that received transplants of transduced CD34+CD38−Lin−CD7− or CD34+CD38−Lin−CD7+ cells and transduced with F36V-MPL vector was extracted using the DNeasy Tissue kit (Qiagen, Valencia, CA). The genomic-proviral junction sequence was identified using linear amplification–mediated (LAM)–PCR (modified from previous studies19). Linear amplification of target DNA was performed by repeated primer extension with a lentivirus long-term repeat (LTR)–specific 5′ biotinylated primers (LV1, 5′ CCTGGGAGCTCTCTGGCTAACTAGG-3′; and LV2, 5′ CTAGGGAACCCACTGCTTAAGCCTC-3′). To enhance detection from linear amplification, we purified primary PCR products (5′ biotin on primer LV2) and used them as template for the secondary PCR reaction.
Statistical analysis
Data were analyzed using statistical tests as following: Figures 1A and 4A used analysis of variance (ANOVA); Figures 5B and 6B used linear regression; and Tables 3 and 4 used the rank-sum test. Results were considered significant when P was less than or equal to .05.
Figure 1.

In vitro signaling through F36V-Mpl expands the number of transduced CD34+ cells and maintains multilineage potential. (A) Lymphomyeloid (CD34+CD38−Lin−CD7−) progenitors transduced with F36V-MPL-GFP or control (F36V-GFP) vector were cultured on MS5 stroma without growth factors with or without CID. Fold increase (mean, SEM) in total number and CD34+GFP+ cells relative to cells plated on day 0 (D0) is shown (n = 5 experiments, in triplicates); P = .002 and .023, respectively, for F36V-MPL-GFP–transduced cells with or without CID. (B) Immunophenotype of transduced CD34+CD38−Lin−CD7− cells cultured as in panel A, showing generation of CD34+GFP+ (day 25), lymphoid (day 35), and myeloerythroid (day 49) cells. (C) Total number of cells in culture (left panel), number of CD34+ cells in culture (middle panel), frequency of clonogenic cells (mean, SEM percentage of CFU-Cs; right panel), generated from transduced CD34+CD38−Lin−CD7− progenitors cultured with CID or Tpo, and control nontransduced CD34+CD38−Lin−CD7− progenitors cultured with Tpo (n = 3 independent experiments, each in triplicate).
Figure 4.

In vitro stimulation of F36V-Mpl increases lymphoid output from multi-lymphoid progenitors. (A) Multilymphoid progenitors (CD34+CD38−Lin−CD7+) transduced with the F36V-MPL-GFP were cultured on MS5 stroma without growth factors with or without CID. Fold increase (mean, SEM) in total number and CD19+GFP+ cells relative to day 0 is shown (n = 5 experiments, in triplicate); P = .04 and .047, respectively. (B) FACS analysis of cells cultured (D29-35) as in panel A. (C) B-cell progenitors (CD34+CD19+) and B cells (CD34−CD19+) were transduced with F36V-MPL-GFP and cultured with or without CID on MS5 stroma without cytokines. (n = 3 wells/arm; mean ± SE).
Figure 5.

In vivo stimulation of F36V-Mpl signaling in human lymphomyeloid (CD34+CD38−Lin−CD7−) progenitors. Each mouse (NOD/SCID/β2-microglobulin−/−) received transplants of human CD34+CD38−Lin−CD7− progenitors transduced with F36V-MPL-LUC (F36V-MPL) or F36V-LUC (F36V) control vector. Mice were treated with or without CID from day 1 until killing 35 to 49 days after transplantation. (A) In vivo longitudinal imaging of luciferase expression in 2 mice that both received transplants of human CD34+CD38−Lin−CD7− progenitors transduced with F36V-MPL. D indicates dorsal, and and V indicates ventral view of the same mouse. (B) Quantification of signal for all animals, n = 6 mice per arm, 2 independent experiments (mean, SD; P < .001 for difference between F36V-MPL with CID and all other groups). (C) Lineage analysis of human CD45+ cells in bone marrow of representative CID-treated mouse. Boxes below show percentage in each quadrant.
Figure 6.

In vivo stimulation of F36V-Mpl signaling in human multilymphoid progenitors. (A) In vivo longitudinal imaging of luciferase expression in 2 NOD/SCID/β2-microglobulin−/− mice that received transplants with CD34+CD38−Lin−CD7+ cells transduced with F36V-MPL and treated with or without CID from day 1; D indicates dorsal view; V, ventral views. (B) Quantification of signal for all animals, n = 6 mice per arm, 2 independent experiments; mean and SD; P = .001 for difference between F36V-MPL with CID and all the other groups). (C) Lineage analysis of human cells in bone marrow of representative CID-treated mouse.
Table 3.
Engraftment data for human lymphomyeloid progenitor transplants assessed by FACS
| Mouse ID | Vector | CID | Day post BMT | % huCD45+ cells | %CD34+Lin−/huCD45+ | Fold increase CD34+Lin− human cells |
|---|---|---|---|---|---|---|
| Experiment A | ||||||
| 1 | F36V-MPL | + | 35 | 22.4 | 46 | 215.3 |
| 2 | F36V-MPL | + | 49 | 52.8 | 36.1 | 345 |
| 3 | F36V-MPL | − | 35 | 0.71 | 0.1 | 0.02 |
| 4 | F36V-MPL | − | 49 | 0 | 0 | 0 |
| 5 | F36V-MPL | − | 49 | 0.05 | 0 | 0 |
| 6 | Control (no transplant) | 49 | 0 | 0 | 0 | |
| Experiment B | ||||||
| 1 | F36V-MPL | + | 49 | 21.9 | 23.2 | 75.7 |
| 2 | F36V-MPL | + | 49 | 26.1 | 13.1 | 31.8 |
| 3 | F36V-MPL | − | 49 | 0.76 | 20.6 | 0.6 |
| 4 | F36V-MPL | − | 49 | 0.83 | 18.3 | 0.5 |
| 5 | F36V-MPL | − | 49 | 0 | 0 | 0 |
| 6 | F36V | − | 49 | 0.64 | 22.2 | 0.5 |
| 7 | F36V | − | 49 | 0 | 0 | 0 |
| 8 | Control (no transplant) | 49 | 0 | 0 | 0 | |
| Summary | Mean % human CD45+ cells | Mean %CD34+Lin−/huCD45+ cells | Mean fold increase CD34+Lin− human cells | |||
| F36V-MPL | + | 30.8 | 29.6 | 167 | ||
| F36V-MPL | − | 0.4 | 6.5 | 0.2 |
Fold expansion of human CD34+Lin− cells is derived from the percentage of huCD45+CD34+Lin− cells and total cells harvested from bone marrow (femurs, tibias) divided by the number of CD34+CD38−Lin−CD7− human cells transplanted (n = 14 mice, 2 independent experiments). Fold expansion of F36V-MPL–transduced CD34+Lin− cells in CID-treated group is significantly (P = .004) greater compared with all other groups.
Table 4.
Engraftment data for multilymphoid progenitor transplants assessed by FACS
| Mouse ID | Vector | CID | Day post BMT | % huCD45+ cells | %CD34+Lin−/huCD45+ cells | Fold increase CD34+Lin− human cells |
|---|---|---|---|---|---|---|
| Experiment A | ||||||
| 1 | F36V-MPL | + | 49 | 46.7 | 13.8 | 154.7 |
| 2 | F36V-MPL | + | 49 | 19.9 | 20.8 | 114.1 |
| 3 | F36V-MPL | − | 49 | 0.14 | 0 | 0 |
| 4 | F36V-MPL | − | 49 | 0 | 0 | 0 |
| 5 | F36V-MPL | − | 49 | 0.49 | 0 | 0 |
| 6 | Control (no transplant) | 49 | 0 | 0 | 0 | |
| Experiment B | ||||||
| 1 | F36V-MPL | + | 9 | 30.5 | 18.3 | 101 |
| 2 | F36V-MPL | + | 37 | 7.67 | 53.4 | 69 |
| 3 | F36V-MPL | − | 9 | 0.23 | 31.2 | 0.4 |
| 4 | F36V-MPL | − | 37 | 0.086 | 0 | 0 |
| 5 | F36V-MPL | − | 37 | 0.38 | 0 | 0 |
| 6 | Control (no transplant) | − | 37 | 0 | 0 | 0 |
| Summary | Mean % huCD45+ cells | Mean %CD34+Lin−/huCD45+ cells | Mean fold increase CD34+Lin− human cells | |||
| F36V-MPL | + | 26.2 | 26.5 | 109.7 | ||
| F36V-MPL | − | 0.15 | 5.2 | 0.06 |
Fold expansion of human CD34+Lin− cells is derived from the percentage of huCD45+CD34+Lin− cells and total cells harvested from bone marrow (femurs, tibias) at times shown divided by number of cells transplanted (n = 12 mice, 2 independent experiments), fold expansion of CD34+Lin− cells is significantly greater in CID-treated mice (P = .006) compared with all other groups.
Results
Ex vivo dimerization of the F36V-Mpl fusion protein selectively expands human lymphomyeloid progenitors
To explore the effects of synthetically inducing signaling through the thrombopoietin receptor Mpl, purified cells were isolated from umbilical cord blood using the immunophenotype CD34+CD38−Lin−CD7−, a population previously shown to be highly enriched in lymphomyeloid human stem/progenitor cells.17,23 A bicistronic lentiviral vector was used to coexpress the fusion protein F36V-Mpl and the marker gene GFP in target cells (Figure S1). Control CD34+CD38−Lin−CD7− cells were transduced with a similar lentiviral vector that coexpressed the dimerization domain F36V and GFP but lacked the Mpl signaling domain. Transduced progenitors were then cultured long term on a preestablished stromal cell line MS5, with or without the dimerizer AP20187, in the absence of exogenous cytokines.
In the presence of AP20187, the numbers of total cells and of CD34+GFP+ cells generated in long-term culture were significantly higher compared with cells cultured without AP20187 (P = .002 and .023, respectively; Figure 1A). After 3 weeks in culture, most of the F36V-MPL-GFP–transduced cells cultured with AP20187 were viable (viability 92.4% ± 5.7% with CID versus 18.8% ± 16.1% without CID) and coexpressed CD34 and GFP. Myeloid, erythroid, and lymphoid cells were also generated in the presence of CID (Figure 1B). In contrast, transduced CD34+CD38−Lin−CD7− cells cultured without CID did not proliferate, and CD34+ cells were rapidly lost from culture. Consistent with the persistence of CD34+ cells, serial replating of cells from stromal cocultures into methylcellulose revealed that clonogenic cells (CFU-Mix, erythroid burst-forming units [BFU-Es], and CFU-GMs) remained detectable in extended long-term cultures only in the presence of CID-mediated Mpl signaling (total CFU frequency in stromal cocultures at day 128 with CID, 0.9% ± 0.7% CFUs; without CID, CFUs not detectable; n = 3). Thus, AP20187-induced signaling through Mpl allowed long-term expansion of clonogenic CD34+ cells, but did not block either lymphoid or myeloerythroid differentiation.
In contrast, in vitro stimulation of CD34+CD38−Lin−CD7− cells via the natural ligand Tpo led to a progressive loss of total cells, CD34+ cells, and CFUs (Figure 1C). Analysis of the lineage pattern of cultures showed no difference in the proportion of B, natural killer (NK), erythroid, and myeloid cells regardless of whether CD34+CD38−Lin−CD7− cells were stimulated by Tpo or CID.
AP20187-induced Mpl dimerization expands CD34+ cells that retain the ability to engraft
To track the effects of F36V-Mpl signaling on progenitor cell proliferation, transduced CD34+ cells were labeled with the fluorescent membrane marker PKH26 and cultured in the presence or absence of CID. These cultures were performed in the absence of any cytokines or stromal cells. Addition of CID to cultures induced more cells to divide; evidence of a selective effect on transduced cells was seen as a progressive enrichment of GFP+ cells with each generation (Figure 2A). The cells that divided in the presence of CID preferentially coexpressed CD34 and GFP, indicating that Mpl dimerization selectively expanded transduced CD34+ cells (Figure 2B).
Figure 2.

In vitro signaling through F36V-Mpl induces cell division and generation of immunophenotypic and functionally primitive progenitors. (A) CD34+ cells transduced with F36V-MPL-GFP, labeled with PKH26, and analyzed by FACS after 7 days of culture with or without CID. Bar graph shows percentage of GFP+ cells gated from each generation with or without CID at day 7 of culture. (B) FACS analysis of cells cultured with or without CID, after gating on generation 4 (ie, after 3 divisions). (C) Bone marrow engraftment (6 weeks after transplantation) of human cells isolated from generation 4 and transplanted into β2-microglobulin NOD/SCID−/− mice (40 000 cells per mouse) Control indicates mice that did not undergo transplantation. (D) Human-specific Alu PCR of CFU-Cs generated from bone marrow of mice that received transplants of human cells: lanes 1 and 2 show CFUs generated from BM of 2 mice that received transplants of generation 4 of CID-cultured cells; lanes 3 and 4 show duplicate samples of whole bone marrow from mice that received transplants of generation 4 of non-CID–cultured cells; lanes 5 and 6 show duplicate samples of whole bone marrow from control mice that did not undergo transplantation; lane 7 shows +Control (human cord blood); and lane 8 shows −Control (murine cell line).
Equal numbers of cells that had undergone 3 divisions (ie, generation 4) were transplanted into immunodeficient mice. Only cells that had divided in the presence of CID were able to repopulate the mice (1.7%-11.1% human [hu] CD45+; Figure 2C). Bone marrow harvested 6 weeks after transplantation from animals that had engrafted with fourth-generation, CID-expanded cells, contained CD19+ B cells and clonogenic human myeloerythroid progenitors (Figure 2D; Tables 1,2).
Table 1.
Mean number of human CFUs present in murine bone marrow after transplantation
| BM cells plated, 1 × 105 | BFU-E | CFU-mix | CFU-GM |
|---|---|---|---|
| +CID | 4.1 (1-10) | 8.1 (1-20) | 3 (1-9) |
| −CID | 0 | 0 | 0 |
| Control | 0 | 0 | 0 |
N = 6 mice total. Values are no. (range).
Table 2.
Percentage of huCD45+ and huCD19/huCD45 in whole bone marrow from mice that received transplants of generation 4 of cells cultured with and without CID
| Mouse 1: + CID | Mouse 3: + CID | Mouse 4: + CID | Mouse 5: −CID | Mouse 6: control | |
|---|---|---|---|---|---|
| % huCD45+ cells | 3.3 | 1.2 | 11.1 | <0.001 | 0 |
| % CD19+ per huCD45+ cells | 91.2 | 79.1 | 89.4 | <0.001 | 0 |
Control indicates mice that did not undergo transplantaton.
To explore the lineage potential of progenitors expanded through F36V-Mpl signaling at a clonal level, transduced CD34+CD38−Lin−CD7− cells labeled with PKH26 were cultured with CID in cytokine-free cultures. After 4 weeks, single cells that remained CD34+ but had lost all PKH26 fluorescence (indicating at least 7 cell divisions) were isolated by FACS and cloned individually to test lineage potential. Total cloning efficiency of PKH26−CD34+ cells was 2.3%; 33.3% of the clones generated both B and NK cells, and 22.2% of the assayed clones were able to generate myeloerythroid cells. Thus, in vitro dimerization of F36V-Mpl allowed the functional preservation of multilineage, clonogenic progenitors after multiple cell divisions.
In vitro Mpl signaling in multilymphoid (CD34+CD38−Lin−CD7+) progenitors increases lymphoid output
A role for Mpl signaling in lymphopoiesis has not been reported. The CD34+CD38−Lin−CD7+ immunophenotype marks the earliest stage of lymphoid commitment in human umbilical cord blood; in these cells, B-, NK, dendritic,17 and T-cell25 potential are retained, but myeloerythroid potential is absent. As shown in Figure 3, c-MPL was expressed at an RNA level in CD34+CD38−Lin−CD7− cells and CD34+CD38−Lin−CD7+ cells, but was not detectable at later stages of lymphoid differentiation.
Figure 3.

c-MPL expression by RT-PCR in cord blood cell populations. RNA was extracted from equal numbers (1500 cells) of each population. β2-microglobulin indicates microglobulin-positive control for cDNA loading; MLP, multilymphoid progenitors; and BP, B-cell progenitors.
In view of the expression pattern of c-MPL, the response of multilymphoid progenitors to Mpl dimerization was next studied. CD34+CD38−Lin−CD7+ cells were isolated and transduced with the F36V-MPL-GFP lentiviral vector (or the control vector F36V-GFP) and then cultured with or without CID in the absence of any cytokines.
The number of total viable cells and of CD19+GFP+ B cells generated from CD34+CD38−Lin−CD7+ cells ex vivo was significantly higher in the presence of AP20187 (Figure 4A; P = .04 for total cells, P = .047 for CD19+GFP+cells). After 4 weeks in culture, most of the F36V-MPL-GFP–transduced cells cultured in the presence of AP20187 were viable (viability, 86.3% ± 6.7% with CID vs 11.6% ± 4.1% without CID) and expressed GFP (71.3% ± 9.21% GFP+; Figure 4B). CD56+GFP+ NK cells were also generated in cultures (data not shown), but no myeloerythroid cells were generated from CD34+CD38−Lin−CD7+ cells with or without CID (Figure 4B). Thus, F36V-Mpl dimerization significantly and selectively increased lymphoid cell output from primitive lymphoid-restricted progenitors in the absence of other cytokines, but did not affect lineage potential.
To explore whether the cell expansion generated in cultures initiated from CD34+CD38−Lin−CD7+ cells was a result of signaling in the multilymphoid progenitors themselves or in their more mature lymphoid progeny, B-cell progenitors (CD34+CD19+ cells) and B cells (CD34−CD19+ cells) were isolated, transduced with the F36V-MPL-GFP vector, and cultured with or without AP20187. AP20187 had no effect on cell output from either population, and all viable cells were lost from cultures by day 28 (Figure 4C). Thus, F36V-Mpl signaling affects B lymphopoiesis by acting at a multilymphoid CD34+CD38−Lin−CD7+ stage rather than at later stages of B-lymphoid differentiation.
In vivo signaling through F36V-Mpl selectively expands both lymphomyeloid and lymphoid-committed progenitors
Although these approaches to induce Mpl dimerization ex vivo demonstrated significant cell proliferation, prolonged stimulation resulted ultimately in exhaustion of the cultures. We next explored whether expansion of primitive progenitor cells could be more optimally achieved when dimerization through F36V-Mpl was induced in vivo after transplantation. Lymphomyeloid progenitors (CD34+CD38−Lin−CD7−) or multilymphoid progenitors (CD34+CD38−Lin−CD7+) were transplanted into immunodeficient mice immediately after transduction with either F36V-MPL or control F36V vectors. Mice were treated with daily injections of AP20187 starting 1 day prior to transplantation. The vectors used in these experiments carried the marker gene firefly luciferase, thus allowing longitudinal in vivo tracking of engraftment and proliferation of the human cells.
In all animals whether given transplants of CD34+CD38−Lin−CD7− or CD34+CD38−Lin−CD7+ cells, in vivo administration of AP20187 produced prolonged expansion of F36V-Mpl–expressing cells (Figures 5,6; Tables 3,4). In vivo bioluminescent imaging detected luciferase activity in treated mice at increasing levels until time of killing, 5 to 7 weeks after transplantation (Figures 5A,B;6A,B). Those mice that did not receive AP20187 in vivo had no detectable signal after the first day of imaging. Similarly, mice that received transplants of the control vector (F36V-luciferase) and treated with AP20187 produced no detectable bioluminescent signal, demonstrating that the effect on engraftment and proliferation in the mice that received F36V-Mpl mice was mediated specifically by the presence of the intracellular domain of Mpl.
FACS analysis of harvested bone marrow at the time of death confirmed that in vivo administration of AP20187 significantly increased the engraftment in mice that received transplants of human lymphomyeloid progenitors (P = .01 for transplants with CD34+CD38−Lin−CD7− cells with or without CID; Table 3). Animals that received transplants of F36V-MPL–transduced CD34+CD38−Lin−CD7− cells and treated with AP20187 showed a mean of 30.8% human CD45+ cells in the bone marrow (Table 3) and 9.7% in the spleen, compared with 0.4% and 0.18% CD45+ cells, respectively, in animals that received transplants of F36V-MPL that were not treated with AP20187. Generation of all lymphoid and myeloerythroid lineages was noted in animals that received transplants of CD34+CD38−Lin−CD7− cells and treated with AP20187 (Figure 5C). In addition to mature lineage committed cells, CD34+ cells that did not express any lineage differentiation markers (CD34+Lin−) cells were detected at high levels in the bone marrow and spleen in treated animals. Consistent with the CD34+ immunophenotype, clonogenic myeloerythroid cells (CFU-GMs, CFU-Mix, and BFU-Es) were identified only in the bone marrow of CID-treated mice that received transplants of F36V-MPL–expressing CD34+CD38−Lin−CD7− cells. The frequency of the CD34+Lin− cells detected in the marrow by FACS (approximately 10% of all mononuclear cells; Table 3) correlated closely with the frequency of total CFUs (human CFUs comprised approximately 0.5% of all bone marrow cells upon replating the harvested marrow into methylcellulose (Figure S2A), equating to approximately 5% cloning efficiency of CD34+ cells, similar to that of fresh cord blood CD34+ cells.17 In addition, bone marrow harvested from the animals and plated into T lymphoid cultures demonstrated that the T-cell potential of the transplanted progenitors was maintained during in vivo expansion (Figure S2B).
Based on the number of CD34−CD38−Lin−CD7− cells initially transplanted, and the number of human cells recovered from both lower limbs, the number of CD34+Lin− cells was expanded more than 160-fold via in vivo signaling through F36V-Mpl over 5 to 7 weeks. This estimate does not include human cells identified by imaging in the spleen and throughout the skeleton; based on previous estimates that each of the lower limbs contain approximately 7.5% of total marrow,26 overall expansion of progenitors within the marrow only was estimated as more than 1000-fold. In a more limited series of experiments with longer term follow-up, in vivo dimerization of Mpl in neonatal NOD/SCID γcnull mice that received transplants of F36V-MPL–expressing CD34+CD38−Lin−CD7− cells showed high levels of human engraftment for up to 14 weeks (mean, 63%; range, 58%-68% CD45+ cells with CID vs 0.03% CD45+ cells without CID).
Immunophenotypic analysis of bone marrow from mice that received transplants of multilymphoid progenitors also confirmed the profound engraftment effects seen by imaging (Table 4). Animals that received transplants of F36V-MPL–transduced CD34+CD38−Lin−CD7+ cells and treated daily with AP20187 showed a mean of 26% human CD45+ cells in the bone marrow (Table 4) and 3.2% in the spleen, whereas those animals that received transplants that were not administered CID showed little or no engraftment (P = .01 with or without CID). Lymphoid, but no myeloerythroid, potential was seen in the bone marrow or spleen of all animals that received transplants of multilymphoid progenitors by either flow cytometry (Figure 6C), or by replating bone marrow after transplantation to assay human myeloerythroid CFUs. Thus, signaling through F36V-Mpl did not alter the differentiation pattern or lineage potential of these primitive lymphoid-restricted progenitors.
At time of death, the number of CD34+Lin− lymphoid progenitors generated from CD34+CD38−Lin−CD7+ cells had increased by more than 100-fold in response to AP20187 administration (Table 4), based only on cell numbers harvested from the lower limbs, and more than 1000- fold when corrected for total marrow space.
Analysis of the CID-treated bone marrow in engrafted animals showed normal human karyotypes with no evidence of chromosomal abnormalities. LAM-PCR of bone marrow harvested from animals that received transplants of either CD34+CD38−Lin−CD7− or CD34+CD38−Lin−CD7+ cells and treated with CID in vivo showed at least 7 to 11 different integration patterns with few predominant patterns in each of the mice, indicating that in vivo Mpl signaling produced a polyclonal expansion of transduced human cells (data not shown).
Discussion
These studies demonstrate that stimulation via the intracellular signaling domain of Mpl is sufficient to induce profound expansion of lymphomyeloid progenitor cells and, notably, also lymphoid-committed progenitors from human umbilical cord blood. The expansion effect was directly consequent to Mpl signaling rather than a nonspecific effect from the CID itself or indirectly through accessory cells as evidenced by a selective proliferation only of cells that expressed both the dimerizing receptor and its Mpl fusion partner.
These findings differ qualitatively and quantitatively from earlier studies using the Mpl dimerization approach. Previous studies that targeted unfractionated human CD34+ cells produced erythroid and megakaryocytic cell expansion in vitro similar to the effects of stimulation with thrombopoietin, the natural ligand for Mpl. However, these studies failed to show either immunophenotypic or functional evidence of progenitor expansion, and effects on lymphoid cells were not tested.15,16
Similarly, studies with heterogeneous nonhuman populations resulted in expansion of erythroid and megakaryocytic lineages and little or no expansion of transplantable stem/progenitors cells.18,27–31 One exception was a study of canine bone marrow transplantation which demonstrated in vivo expansion of immunophenotypic (CD34+) progenitors and B-lymphoid cells.32 However, the expanded cells were not clonogenic and lacked multilineage potential. It is likely that the markedly different biological effects seen between the current and previous reports are due to critical differences in the cell populations targeted and other aspects of the experimental design. The 2 cell populations studied here (CD34+CD38−Lin−CD7− lymphomyeloid progenitors and CD34+CD38−Lin−CD7+ multilymphoid progenitors) are functionally much more primitive and homogeneous than the unfractionated bone marrow used in murine or nonhuman primate studies and the CD34+ population in previous human studies.17,33,34 These heterogeneous populations contain numerous myeloerythroid progenitors and precursors that rapidly differentiate in response to thrombopoietin stimulation and would likely obscure and modify the effect of Mpl signaling on more rare, primitive populations contained within the graft.
Induction of cell cycling during cytokine stimulation in vitro leads to a rapid and profound reduction in the frequency of repopulating cells.35,36 In previous studies with Mpl dimerization, multiple cytokines were added during ex vivo manipulation, likely promoting differentiation of the cultures. In experiments in which CID was delivered in vivo, its administration was delayed until days to weeks after transplantation,27,29,32 by which time many engrafting progenitors would have entered an irreversible differentiation program. In our study, the dimerization signal was supplied to highly purified primitive progenitors ex vivo in the absence of cytokines, and in vivo from the moment of engraftment.
Thrombopoietin has shown little effect on progenitor expansion as a single agent.13 Consistent with this, in the current study, thrombopoietin alone produced only short-term cell proliferation in vitro without maintenance of progenitors. At least 2 potential reasons exist for the markedly different biological results seen when Mpl signaling is stimulated by its natural ligand and through the dimerization approach. First, the strategy used here involved constitutive expression of the intracellular component of Mpl in targeted cells and their progeny by obtaining stable integration with a lentiviral vector. In contrast, physiologic expression of the wild-type Mpl receptor is rapidly down-regulated during HSC differentiation, and Mpl is largely absent in mature cells other than megakaryocytes.37,38 Thus, down-regulation of Mpl expression, together with internalization of Mpl upon ligand binding, provides an autoregulatory negative feedback.9
The biological differences resulting from AP20187 and thrombopoietin-induced Mpl activation may also be explained by differences in signaling conferred by the presence or absence of components of the full-length receptor.9 The vector used in our studies expressed the entire intracellular domain of murine Mpl, including the Box 2 region that has been shown to be necessary for Mpl-induced cellular proliferation.39 Our construct did not contain any portion of the transmembrane or extracellular domains of Mpl, which have been shown to contain components that inhibit proliferation mediated by the intracellular domain.40 Clinically, mutations affecting the transmembrane and/or Tpo-binding domain of Mpl produce a myeloproliferative disorder.41 However, as the reported inhibition from the extracellular domain is removed upon binding of the natural ligand thrombopoietin,40 it seems that another mechanism may be responsible for the proliferation seen with our approach. Of note, AP20187-induced dimerization did not result in detectable phosphorylation of Stat5, a key component of the normal Mpl signaling pathway.18 In certain intriguing studies, a mutant form of Mpl that lacked the capacity to activate JAK and Stat5 signaling was shown to retain its ability to induce cellular proliferation42 but not differentiation.43 Thus, as yet undescribed and qualitatively different signaling pathways may mediate the profound proliferative effects from direct dimerization of the intracellular Mpl domain seen in our study.
Various approaches have been used in the past to expand hematopoietic stem and progenitor cells either by constitutive expression of the transcriptional regulators4–6,44–46 or activation of key signaling pathways involved in HSC proliferation.7,47–50 These approaches used multiple cytokines, resulting in varying degrees of ex vivo expansion of long-term reconstituting cells.
Optimal expansion was achieved in the current study when the proliferation signal was administered in vivo, presumably because additional survival signals from the complex microenvironment of the bone marrow are not fully recapitulated using current in vitro systems. The dimerization strategy described here has the benefit of being able to target signaling specifically to transplanted progenitors, resulting in regulated expansion of the transduced cells.
The finding that Mpl dimerization induced expansion not only of primitive lymphomyeloid stem/progenitors, but also of progenitors committed to the lymphoid pathway, was particularly novel. The CD34+CD38−Lin−CD7+ multilymphoid progenitors represent the most primitive stage of human lymphopoiesis yet identified.17 Nevertheless, in their unmanipulated state, these CD7+ progenitors possess significantly less proliferative capacity than their more primitive CD7− counterparts. Although ex vivo stimulation of CD7+ progenitors increased output of mainly mature lymphocytes, the level and duration of lymphoid progenitor expansion induced by in vivo Mpl dimerization represents a profound biological modification in these lineage-restricted progenitors and leads us to challenge the notion that clinical approaches toward hematopoietic cell expansion and gene therapy must necessarily be targeted at hematopoietic stem cells.
Supplementary Material
Acknowledgments
We thank the Childrens Hospital Los Angeles (CHLA) Vector Core, the Labor and Delivery Staff of Kaiser Permanente Sunset, and the Guenther Foundation for supporting the Small Animal Imaging Center of The Saban Research Institute of CHLA.
This work was supported by National Institutes of Health grants R01HL077912 (G.M.C.) and PO1HL073104 (D.B.K. and G.M.C.), and by Las Madrinas. H.A.-A. is a recipient of Fellowship awards from The Saban Research Institute of Childrens Hospital Los Angeles and Amgen Oncology Institute. M.R. is a recipient of Fellowship awards from the Radiological Society of North America Research and Education Foundation, and the Fonar Corporation.
Footnotes
An Inside Blood analysis of this article appears at the front of this issue.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
H.A.-A. designed and performed in vivo and in vitro experiments, contributed to design and production of viral vectors, analyzed data, and wrote the paper; Y.Z. performed and contributed to production of viral vectors, in vitro experiments, and tissue harvest for in vivo experiments; R.H. designed and produced viral vectors and performed LAM-PCR; X.W. contributed to technical assistance in methods and data analysis for some in vivo experiments; S.G. contributed to animal care and in vivo imaging and tissue harvest for some in vivo experiments; Q.-L.H. contributed to technical assistance in methods for some in vitro experiments; G.S. collected data of in vivo imaging; D.B.K. designed and used viral vectors, and critically revised the manuscript; M.R. analyzed and presented imaging data for in vivo experiments; and G.M.C. conceived the experimental question, designed in vitro and in vivo experiments, analyzed data, wrote the paper, and supervised the project.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Gay M. Crooks, Childrens Hospital Los Angeles, MS no. 62, 4650 Sunset Blvd, Los Angeles, CA 90027; e-mail: gcrooks@chla.usc.edu.
References
- 1.McCulloch EA, Till JE. Perspectives on the properties of stem cells. Nat Med. 2005;11:1026–1028. doi: 10.1038/nm1005-1026. [DOI] [PubMed] [Google Scholar]
- 2.Nakahata T. Ex vivo expansion of human hematopoietic stem cells. Int J Hematol. 2001;73:6–13. doi: 10.1007/BF02981897. [DOI] [PubMed] [Google Scholar]
- 3.Sorrentino BP. Clinical strategies for expansion of haematopoietic stem cells. Nat Rev Immunol. 2004;4:878–888. doi: 10.1038/nri1487. [DOI] [PubMed] [Google Scholar]
- 4.Krosl J, Austin P, Beslu N, Kroon E, Humphries RK, Sauvageau G. In vitro expansion of hematopoietic stem cells by recombinant TAT-HOXB4 protein. Nat Med. 2003;9:1428–1432. doi: 10.1038/nm951. [DOI] [PubMed] [Google Scholar]
- 5.Antonchuk J, Sauvageau G, Humphries RK. HOXB4-induced expansion of adult hematopoietic stem cells ex vivo. Cell. 2002;109:39–45. doi: 10.1016/s0092-8674(02)00697-9. [DOI] [PubMed] [Google Scholar]
- 6.Krosl J, Beslu N, Mayotte N, Humphries RK, Sauvageau G. The competitive nature of HOXB4-transduced HSC is limited by PBX1: the generation of ultra-competitive stem cells retaining full differentiation potential. Immunity. 2003;18:561–571. doi: 10.1016/s1074-7613(03)00090-6. [DOI] [PubMed] [Google Scholar]
- 7.Reya T, Duncan AW, Ailles L, et al. A role for Wnt signalling in self-renewal of haematopoietic stem cells. Nature. 2003;423:409–414. doi: 10.1038/nature01593. [DOI] [PubMed] [Google Scholar]
- 8.Zhang CC, Lodish HF. Murine hematopoietic stem cells change their surface phenotype during ex vivo expansion. Blood. 2005;105:4314–4320. doi: 10.1182/blood-2004-11-4418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kaushansky K. The molecular mechanisms that control thrombopoiesis. J Clin Invest. 2005;115:3339–3347. doi: 10.1172/JCI26674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McKinstry WJ, Li CL, Rasko JE, Nicola NA, Johnson GR, Metcalf D. Cytokine receptor expression on hematopoietic stem and progenitor cells. Blood. 1997;89:65–71. [PubMed] [Google Scholar]
- 11.Solar GP, Kerr WG, Zeigler FC, et al. Role of c-mpl in early hematopoiesis. Blood. 1998;92:4–10. [PubMed] [Google Scholar]
- 12.Guerriero R, Testa U, Gabbianelli M, et al. Unilineage megakaryocytic proliferation and differentiation of purified hematopoietic progenitors in serum-free liquid culture. Blood. 1995;86:3725–3736. [PubMed] [Google Scholar]
- 13.Piacibello W, Sanavio F, Garetto L, et al. Extensive amplification and self-renewal of human primitive hematopoietic stem cells from cord blood. Blood. 1997;89:2644–2653. [PubMed] [Google Scholar]
- 14.Nagasawa Y, Wood BL, Wang L, et al. Anatomical compartments modify the response of human hematopoietic cells to a mitogenic signal. Stem Cells. 2006;24:908–917. doi: 10.1634/stemcells.2005-0484. [DOI] [PubMed] [Google Scholar]
- 15.Richard RE, Blau CA. Small-molecule-directed mpl signaling can complement growth factors to selectively expand genetically modified cord blood cells. Stem Cells. 2003;21:71–78. doi: 10.1634/stemcells.21-1-71. [DOI] [PubMed] [Google Scholar]
- 16.Richard RE, Wood B, Zeng H, Jin L, Papayannopoulou T, Blau CA. Expansion of genetically modified primary human hemopoietic cells using chemical inducers of dimerization. Blood. 2000;95:430–436. [PubMed] [Google Scholar]
- 17.Hao QL, Zhu J, Price MA, Payne KJ, Barsky LW, Crooks GM. Identification of a novel, human multilymphoid progenitor in cord blood. Blood. 2001;97:3683–3690. doi: 10.1182/blood.v97.12.3683. [DOI] [PubMed] [Google Scholar]
- 18.Otto KG, Broudy VC, Lin NL, et al. Membrane localization is not required for Mpl function in normal hematopoietic cells. Blood. 2001;98:2077–2083. doi: 10.1182/blood.v98.7.2077. [DOI] [PubMed] [Google Scholar]
- 19.Ailles L, Schmidt M, Santoni de Sio FR, et al. Molecular evidence of lentiviral vector-mediated gene transfer into human self-renewing, multi-potent, long-term NOD/SCID repopulating hematopoietic cells. Mol Ther. 2002;6:615–626. [PubMed] [Google Scholar]
- 20.Dull T, Zufferey R, Kelly M, et al. A third-generation lentivirus vector with a conditional packaging system. J Virol. 1998;72:8463–8471. doi: 10.1128/jvi.72.11.8463-8471.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Haas DL, Case SS, Crooks GM, Kohn DB. Critical factors influencing stable transduction of human CD34(+) cells with HIV-1-derived lentiviral vectors. Mol Ther. 2000;2:71–80. doi: 10.1006/mthe.2000.0094. [DOI] [PubMed] [Google Scholar]
- 22.Itoh K, Tezuka H, Sakoda H, et al. Reproducible establishment of hemopoietic supportive stromal cell lines from murine bone marrow. Exp Hematol. 1989;17:145–153. [PubMed] [Google Scholar]
- 23.Hao QL, Smogorzewska EM, Barsky LW, Crooks GM. In vitro identification of single CD34+CD38- cells with both lymphoid and myeloid potential. Blood. 1998;91:4145–4151. [PubMed] [Google Scholar]
- 24.Wang X, Rosol M, Ge S, et al. Dynamic tracking of human hematopoietic stem cell engraftment using in vivo bioluminescence imaging. Blood. 2003;102:3478–3482. doi: 10.1182/blood-2003-05-1432. [DOI] [PubMed] [Google Scholar]
- 25.Hoebeke I, De Smedt M, Stolz F, et al. T-, B- and NK-lymphoid, but not myeloid cells arise from human CD34(+)CD38(-)CD7(+) common lymphoid progenitors expressing lymphoid-specific genes. Leukemia. 2007;21:311–319. doi: 10.1038/sj.leu.2404488. [DOI] [PubMed] [Google Scholar]
- 26.Papayannopoulou T, Finch CA. On the in vivo action of erythropoietin: a quantitative analysis. J Clin Invest. 1972;51:1179–1185. doi: 10.1172/JCI106911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Richard RE, De Claro RA, Yan J, et al. Differences in F36VMpl-based in vivo selection among large animal models. Mol Ther. 2004;10:730–740. doi: 10.1016/j.ymthe.2004.07.005. [DOI] [PubMed] [Google Scholar]
- 28.Zeng H, Masuko M, Jin L, Neff T, Otto KG, Blau CA. Receptor specificity in the self-renewal and differentiation of primary multipotential hemopoietic cells. Blood. 2001;98:328–334. doi: 10.1182/blood.v98.2.328. [DOI] [PubMed] [Google Scholar]
- 29.Jin L, Zeng H, Chien S, et al. In vivo selection using a cell-growth switch. Nat Genet. 2000;26:64–66. doi: 10.1038/79194. [DOI] [PubMed] [Google Scholar]
- 30.Jin L, Siritanaratkul N, Emery DW, et al. Targeted expansion of genetically modified bone marrow cells. Proc Natl Acad Sci U S A. 1998;95:8093–8097. doi: 10.1073/pnas.95.14.8093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Blau CA, Peterson KR, Drachman JG, Spencer DM. A proliferation switch for genetically modified cells. Proc Natl Acad Sci U S A. 1997;94:3076–3081. doi: 10.1073/pnas.94.7.3076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Neff T, Horn PA, Valli VE, et al. Pharmacologically regulated in vivo selection in a large animal. Blood. 2002;100:2026–2031. doi: 10.1182/blood-2002-03-0792. [DOI] [PubMed] [Google Scholar]
- 33.Hao QL, Thiemann FT, Petersen D, Smogorzewska EM, Crooks GM. Extended long-term culture reveals a highly quiescent and primitive human hematopoietic progenitor population. Blood. 1996;88:3306–3313. [PubMed] [Google Scholar]
- 34.Larochelle A, Vormoor J, Hanenberg H, et al. Identification of primitive human hematopoietic cells capable of repopulating NOD/SCID mouse bone marrow: implications for gene therapy. Nat Med. 1996;2:1329–1337. doi: 10.1038/nm1296-1329. [DOI] [PubMed] [Google Scholar]
- 35.Traycoff CM, Kosak ST, Grigsby S, Srour EF. Evaluation of ex vivo expansion potential of cord blood and bone marrow hematopoietic progenitor cells using cell tracking and limiting dilution analysis. Blood. 1995;85:2059–2068. [PubMed] [Google Scholar]
- 36.Glimm H, Oh IH, Eaves CJ. Human hematopoietic stem cells stimulated to proliferate in vitro lose engraftment potential during their S/G(2)/M transit and do not reenter G(0). Blood. 2000;96:4185–4193. [PubMed] [Google Scholar]
- 37.Ninos JM, Jefferies LC, Cogle CR, Kerr WG. The thrombopoietin receptor, c-Mpl, is a selective surface marker for human hematopoietic stem cells. J Transl Med. 2006;4:9. doi: 10.1186/1479-5876-4-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Edvardsson L, Dykes J, Olofsson T. Isolation and characterization of human myeloid progenitor populations: TpoR as discriminator between common myeloid and megakaryocyte/erythroid progenitors. Exp Hematol. 2006;34:599–609. doi: 10.1016/j.exphem.2006.01.017. [DOI] [PubMed] [Google Scholar]
- 39.Drachman JG, Miyakawa Y, Luthi JN, et al. Studies with chimeric Mpl/JAK2 receptors indicate that both JAK2 and the membrane-proximal domain of Mpl are required for cellular proliferation. J Biol Chem. 2002;277:23544–23553. doi: 10.1074/jbc.M201120200. [DOI] [PubMed] [Google Scholar]
- 40.Sabath DF, Kaushansky K, Broudy VC. Deletion of the extracellular membrane-distal cytokine receptor homology module of Mpl results in constitutive cell growth and loss of thrombopoietin binding. Blood. 1999;94:365–367. [PubMed] [Google Scholar]
- 41.Ding J, Komatsu H, Wakita A, et al. Familial essential thrombocythemia associated with a dominant-positive activating mutation of the c-MPL gene, which encodes for the receptor for thrombopoietin. Blood. 2004;103:4198–4200. doi: 10.1182/blood-2003-10-3471. [DOI] [PubMed] [Google Scholar]
- 42.Dorsch M, Fan PD, Danial NN, Rothman PB, Goff SP. The thrombopoietin receptor can mediate proliferation without activation of the Jak-STAT pathway. J Exp Med. 1997;186:1947–1955. doi: 10.1084/jem.186.12.1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dorsch M, Danial NN, Rothman PB, Goff SP. A thrombopoietin receptor mutant deficient in Jak-STAT activation mediates proliferation but not differentiation in UT-7 cells. Blood. 1999;94:2676–2685. [PubMed] [Google Scholar]
- 44.Buske C, Feuring-Buske M, Abramovich C, et al. Deregulated expression of HOXB4 enhances the primitive growth activity of human hematopoietic cells. Blood. 2002;100:862–868. doi: 10.1182/blood-2002-01-0220. [DOI] [PubMed] [Google Scholar]
- 45.Amsellem S, Pflumio F, Bardinet D, et al. Ex vivo expansion of human hematopoietic stem cells by direct delivery of the HOXB4 homeoprotein. Nat Med. 2003;9:1423–1427. doi: 10.1038/nm953. [DOI] [PubMed] [Google Scholar]
- 46.Thorsteinsdottir U, Mamo A, Kroon E, et al. Overexpression of the myeloid leukemia-associated Hoxa9 gene in bone marrow cells induces stem cell expansion. Blood. 2002;99:121–129. doi: 10.1182/blood.v99.1.121. [DOI] [PubMed] [Google Scholar]
- 47.Austin TW, Solar GP, Ziegler FC, Liem L, Matthews W. A role for the Wnt gene family in hematopoiesis: expansion of multilineage progenitor cells. Blood. 1997;89:3624–3635. [PubMed] [Google Scholar]
- 48.Van Den Berg DJ, Sharma AK, Bruno E, Hoffman R. Role of members of the Wnt gene family in human hematopoiesis. Blood. 1998;92:3189–3202. [PubMed] [Google Scholar]
- 49.Trowbridge JJ, Xenocostas A, Moon RT, Bhatia M. Glycogen synthase kinase-3 is an in vivo regulator of hematopoietic stem cell repopulation. Nat Med. 2006;12:89–98. doi: 10.1038/nm1339. [DOI] [PubMed] [Google Scholar]
- 50.Varnum-Finney B, Brashem-Stein C, Bernstein ID. Combined effects of Notch signaling and cytokines induce a multiple log increase in precursors with lymphoid and myeloid reconstituting ability. Blood. 2003;101:1784–1789. doi: 10.1182/blood-2002-06-1862. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}