

Abstract
Asbestosis is a form of interstitial lung disease caused by the inhalation of asbestos fibers, leading to inflammation and pulmonary fibrosis. Inflammation and oxidant/antioxidant imbalances are known to contribute to the disease pathogenesis. Extracellular superoxide dismutase (EC-SOD) is an antioxidant enzyme that has been shown to protect the lung from oxidant-mediated damage, inflammation, and interstitial fibrosis. Extracellular matrix (ECM) components, such as collagen and glycosaminoglycans, are known to be sensitive to oxidative fragmentation. Heparan sulfate, a glycosaminoglycan, is highly abundant in the ECM and tightly binds EC-SOD. We investigated the protective role of EC-SOD by evaluating the interaction of EC-SOD with heparan sulfate in the presence of reactive oxygen species (ROS). We found that ROS-induced heparin and heparan sulfate fragments induced neutrophil chemotaxis across a modified Boyden chamber, which was inhibited by the presence of EC-SOD by scavenging oxygen radicals. Chemotaxis in response to oxidatively fragmented heparin was mediated by Toll-like receptor-4. In vivo, bronchoalveolar lavage fluid from EC-SOD knockout mice at 1, 14, and 28 days after asbestos exposure showed increased heparan sulfate shedding from the lung parenchyma. We demonstrate that one mechanism through which EC-SOD inhibits lung inflammation and fibrosis in asbestosis is by protecting heparin/heparan sulfate from oxidative fragmentation.
Introduction
Asbestosis, a form of pneumoconiosis, is the prototype of interstitial lung disease caused by the inhalation of mineral fibers. This disease causes significant morbidity and mortality in patients and also increases the risk of lung cancer (12). Between 1940 and 1970, an estimated 27 million workers were exposed to asbestos (14) and currently no treatment is available. Inflammation and oxidant/antioxidant imbalances are known to contribute to the pathogenesis of this disease. Extracellular superoxide dismutase (EC-SOD) is an antioxidant enzyme that has been shown to protect the lung from oxidant-mediated damage, inflammation, and interstitial fibrosis (8). However, the mechanisms through which EC-SOD inhibits pulmonary fibrosis and inflammation remain unclear. EC-SOD is highly expressed in the lung and is known to tightly bind and localize to the glycosaminoglycan, heparan sulfate (HS) (7, 16, 24). Glycosaminoglycans, like heparan sulfate, are known to be sensitive to oxidative fragmentation (1). Furthermore, heparan sulfate has been previously shown to be involved in inflammatory (26) and fibrotic responses (23). EC-SOD is known to bind heparan sulfate; we have hypothesized that one mechanism through which EC-SOD protects the lung from oxidant-induced damage, inflammation, and fibrosis is by preventing oxidative fragmentation of heparin/heparan sulfate in the extracellular matrix (ECM).
EC-SOD is one of three SOD antioxidant enzyme isoforms that function to scavenge superoxide radicals and prevent the formation of peroxynitrite and the highly potent hydroxyl radical that can damage tissue components (5, 16). EC-SOD has been implicated in the pathogenesis of pulmonary diseases involving oxidative stress (2, 6, 7). It plays a role in several models of pulmonary fibrosis including bleomycin-induced and asbestos-induced fibrosis, both of which produce reactive oxygen species (ROS) in the tissue. The mechanisms by which EC-SOD inhibits inflammation and fibrosis require additional investigation.
The ECM of the lung is intricately linked to the processes of interstitial injury. Evidence supports the role of collagen, hyaluronic acid, and heparan sulfate in profibrotic processes of many tissues (11, 18, 20). Fragmentation of collagen and heparin by ROS has also been established (18, 19). Heparan sulfate is a key ECM component and is the most common cell-surface proteoglycan (1). Heparin is a structurally related endogenous glycosaminoglycan, differing primarily in its disaccharide sulfation and lack of a core protein. Within the ECM microenvironment, heparan sulfate binds and localizes many important factors, such as EC-SOD, basic fibroblast growth factor, platelet-derived growth factor, hepatic growth factor, antithrombin III, interferon-γ, and lipoprotein lipase (1, 20). EC-SOD binds to negatively charged heparin/heparan sulfate through a positively charged C-terminal heparin-binding domain (7, 10). The localizing function of heparan sulfate makes it a unique ECM component and one that can alter the microenvironment during injury and disease. Heparan sulfate may also play a role in inflammatory processes and leukocyte trafficking. Lack of heparan sulfate in the endothelium results in decreased neutrophil adhesion and infiltration in inflammatory models (24).
Heparan sulfate is highly abundant in the ECM and tightly binds EC-SOD. After bleomycin exposure, syndecan 1, a heparan sulfate, is shed from the lung parenchyma (11). Because EC-SOD has a high affinity for heparan sulfate, modifications in heparan sulfate integrity may contribute to the loss of EC-SOD from the matrix that occurs during injuries leading to pulmonary fibrosis (3, 16–19). Furthermore, the loss of HS and EC-SOD from the ECM may increase the susceptibility of the lung to oxidative stress during asbestos-induced injury. EC-SOD may protect heparan sulfate from fragmentation induced by asbestos-generated oxidative species within the lung, as depicted in Fig. 1. Because heparan sulfate is thought to be involved in inflammation (17, 24) and inflammation contributes to the pathogenesis of asbestosis (21), we investigated the role of heparan sulfate fragments and EC-SOD in the development of lung inflammation and fibrosis in a mouse model of asbestosis, by evaluating neutrophil chemotaxis and the effects of a lack of EC-SOD in the lung on heparan sulfate integrity. We have hypothesized that one mechanism through which EC-SOD protects the lung from oxidant-induced damage, inflammation, and fibrosis is by preventing oxidative fragmentation of heparin/heparan sulfate (ECM).
FIG. 1. Proposed interactions of EC-SOD, ROS, and heparan sulfate.

(1) Reactive oxygen species (ROS) are generated near the lung epithelium by asbestos fibers; (2) heparan sulfate side chains are fragmented and released into the airspace; (3) EC-SOD has a high affinity for heparan sulfate and binds via a heparin-binding domain. EC-SOD is localized at the cell surface and may prevent heparan sulfate fragmentation by scavenging ROS generated by asbestos. (4) If EC-SOD is insufficient, HS will be cleaved by attacking ROS while the remaining EC-SOD, as well as other bound growth factors and cytokines, will be released from the epithelium. Loss of HS and EC-SOD could increase tissue susceptibility to oxidative injury from asbestos fibers, resulting in inflammation and fibrosis.
Materials and Methods
Materials
Heparin (5,000 USP U/ml) was purchased from Elkin-Sinns, Cherry Hill, NJ. Heparan sulfate sodium salt and titanium dioxide particles were purchased from Sigma, St. Louis, MO. Crocidolite asbestos was received from National Institute of Environmental Health Sciences (Research Triangle Park, NC). WST-1 Proliferation reagent was purchased from Roche (Indianapolis, IN). Heparan sulfate antibody, MAB2040, was purchased from Calbiochem (San Diego, CA). Anti–Toll-like receptor 4 polyclonal blocking antibody, 2246, was purchased from Cell Signaling (Danvers, MA).
Neutrophil chemotaxis
A neutrophil chemotaxis assay with a modified Boyden chamber was used to evaluate the migration of purified human primary neutrophils (PMNs) across a transwell membrane in response to heparin/HS treated with ROS in the absence or presence of human EC-SOD. Human extracellular superoxide dismutase (EC-SOD) was isolated from human aorta, and enzyme activity was determined, as previously described (15). Human polymorphic neutrophils (PMNs) were isolated as previously described (13) from 50 ml of blood drawn from volunteers, as approved by the Institutional Review Board at the University of Pittsburgh. The PMN pellet was resuspended in sterile Dulbecco's modification of Eagle's medium (DMEM) (Fisher, Pittsburgh, PA) for cell counting. PMNs were used at a final concentration of 2.5 × 107 cells/ml. PMNs (5 × 106) were added to the upper chamber of transwell inserts, 5 μm pore size (Corning Inc., Corning, NY) in a 24-well plate, creating the modified Boyden chamber (25). The lower chambers contained 0.6 ml of DMEM with 15 units of heparin or heparan sulfate (10 μg/ml) and various doses of reactive oxygen species (ROS) with or without the presence of EC-SOD (50–100 U/well). ROS were generated by using a CuSO4/ H2O2 system as previously described (18). ROS are generated by adding 0.1 M H2O2 to CuSO4 solution (0.1 M NaH2PO4/50 μM CuSO4; pH 7.4). Various doses of ROS were evaluated at final Cu(II) concentrations of 0.8 μM, 1.04, 1.25, 1.45, and 1.875 μM, and H2O2 at 1.1 mM, 1.3, 1.5, 1.75, and 2.0 mM, respectively. Superoxide produced by this CuSO4/H2O2 system at 1.25 μM CuSO4 (the concentration used in shown chemotaxis experiments) was determined to be 0.068 nmol/min/ml, as assessed by acetylated cytochrome-c reduction analysis, as previously described (4, 21). If EC-SOD was present, it was added to wells before ROS addition. Control wells include media (DMEM) alone (0.6 ml), positive control lipopolysaccharide (2 μg/ml), and heparin (15 U)/HS (10 μg/ml) with no ROS (copper solution without H2O2). The wells were incubated for 2 h at 37°C/5% CO2. Lower-chamber supernatants are collected on ice, and cell counts are performed on a Beckmann Coulter Counter (Beckmann Coulter, Fullerton, CA).
Superoxide production
Superoxide production was measured with a colorometric assay by using WST-1 Proliferation Reagent (Roche, Indianapolis, IN). The sulfonated tetrazolium salt, WST-1, is efficiently reduced by superoxide to a stable water-soluble formazan, yielding a quantitative assessment of superoxide production. Experimental conditions were prepared as for the chemotaxis experiments. In brief, heparin (15 U) or heparan sulfate sodium salt (10 μg/ml) was incubated in DMEM with ROS (H2O2 and CuSO4), no ROS (CuSO4 only), or both EC-SOD and ROS at a total volume of 0.6 ml. WST-1 (50 μl) was added to each well and allowed to incubate for 2.5 h at 37°C/5% CO2 with the reaction. Each experimental condition was completed in triplicate, and each sample was read in triplicate on a spectrophotometer at 450 nm.
Toll-like receptor 4 blocking assay
To evaluate the role of toll-like receptor 4 (TLR-4) in the activation of neutrophils by heparin fragments, a TLR-4–specific rabbit polyclonal blocking antibody, #2246 (Cell Signaling, Inc., Danvers, MA) at 1 and 0.5 μg/ml was added to upper chamber neutrophils for 15 min before heparin-fragment exposure. Chemotaxis experiments were then carried out as described earlier.
Animals
All animal experiments were reviewed and approved by the University of Pittsburgh Institutional Animal Care and Use Committee. Eight- to 10-week-old male C57BL/6 wild-type mice (Taconic, Germantown, NY) and EC-SOD knockout mice (congenic with C57BL/6) were used (n = 4 per time point and genotype). The knockout animals do not express EC-SOD (3). Crocidolite asbestos fibers from the National Institute of Environmental Health & Sciences, or titanium dioxide (inert control particle) at 0.1 mg in sterile saline was intratracheally injected into the lungs of wild-type C57BL/6 or EC-SOD knockout mice. After 1, 14, and 28 days, mice were killed, and bronchoalveolar lavage fluid (BALF) was recovered by using 0.8-ml sterile saline, as previously described (5).
Western blot analysis of bronchoalveolar lavage fluid
BALF was collected from animals and subjected to SDS-PAGE on 5–15% gradient polyacrylamide gels, under reducing conditions, and electrophoretically transferred onto a polyvinylidene fluoride (PVDF) membrane (Millipore, Bedford, MA) and probed with mouse heparan sulfate antibody, MAB2040 (Calbiochem) and then a horseradish peroxidase (HRP)-conjugated anti-mouse secondary antibody, as previously described (6). Enhanced chemiluminescence detection reagents (Amersham Biosciences, Amersham, England) were used for visualization by a Gel Logic 2200 and Kodak MI system (Kodak, Rochester, NY). Band density was reported as described earlier. Ponceau red membrane staining was used for protein-loading normalization.
Statistical analysis
Densitometry and all other quantitative data were assessed for significance by using the Student's t test (two groups) or analysis of variance (ANOVA; for three or more groups) followed by Bonferroni's posttest. Graphpad Prism statistics program was used for analyses. Significance was achieved by a p value < 0.05.
Results
Extracellular superoxide dismutase (EC-SOD) inhibits neutrophil chemotaxis induced by preventing oxidative heparin/heparan sulfate fragmentation
A modified Boyden chamber was used to evaluate the effect of EC-SOD on the migration of primary human neutrophils (PMNs) across a transwell membrane in response to heparin and heparan sulfate treated with ROS. To confirm that ROS are produced in our model system, a colorometric assay was used to confirm superoxide production. Superoxide was significantly higher in reactions containing both H2O2 and CuSO4 in the heparin system (Fig. 2; p < 0.001). Identical ROS production was found in the heparan sulfate system (data not illustrated). We confirmed by gel electrophoresis that both heparin and heparan sulfate are fragmented by ROS in our model, which has been shown previously for heparan sulfate in a xanthine oxidase/xanthine system (19). ROS-induced heparin fragmentation led to an increase in PMN chemotaxis across the transwell membrane compared with heparin without ROS (Fig. 3a; p < 0.001), and EC-SOD inhibited this response (p < 0.001 vs. heparin with ROS). EC-SOD was capable of inhibiting PMN chemotaxis at 50 U (not illustrated) and 100 U (see Fig. 3) per lower-well chamber by inhibiting the formation of ROS and preventing heparin fragmentation and subsequent PMN chemotaxis. No significant difference was found between the control heparin without ROS and heparin with both ROS and EC-SOD. Unfragmented heparin in the absence of ROS did not stimulate chemotaxis (Fig. 3a). Similar chemotaxis results were seen with heparan sulfate fragmented by ROS (Fig. 3b). EC-SOD inhibited PMN chemotaxis due to ROS-induced heparan sulfate fragmentation (p < 0.05 vs. heparan sulfate with no ROS). In addition, no chemotaxis was observed with ROS in the absence of heparin or heparan sulfate (Fig. 3b). Various doses of Cu(II) and H2O2 (producing ROS) caused PMN chemotaxis, specifically across a range of Cu(II) from 0.8 to 1.875 μM and H2O2 at 1.1–2.0 mM, respectively. PMN chemotaxis displayed a bell-shaped curve with increasing chemotaxis with higher concentrations of ROS, followed by a decrease with in the highest concentrations of ROS (dose–response data not illustrated). DMEM (0.6 ml), ROS [1.25 μM Cu (II) and 1.5 mM H2O2], and EC-SOD (100 U) alone do not stimulate chemotaxis. All data presented are for concentrations of ROS, for which a maximal chemotactic response was noted.
FIG. 2. Superoxide is produced in a Fenton-like chemical reaction, and EC-SOD inhibits this production.

WST-1 reagent was used in a colorimetric assay to determine the relative amount of superoxide produced in our ROS reaction system. Absorbance was measured at 450 nm, and the optical density (O.D.) reported is proportional to superoxide production. Similar ROS production was seen with heparan sulfate as well.
FIG. 3. EC-SOD inhibits neutrophil chemotaxis induced by preventing oxidative fragmentation of heparin and heparan sulfate.


Chemotaxis was assessed in a modified Boyden chamber, and ROS, from Fenton-type reactions. EC-SOD significantly inhibited neutrophil chemotaxis induced by ROS-fragmented (A) heparin (*p < 0.001) and (B) heparan sulfate (t, p < 0.05 vs. heparan sulfate without ROS). ROS was used at a Cu(II) concentration of 1.25 μM and 1.5 mM H2O2. Heparin or heparan sulfate without ROS contained only CuSO4 solution and no H2O2. 100 Units of EC-SOD is depicted and functions by scavenging superoxide radicals and preventing further hydroxyl radical formation. Statistical analysis completed by ANOVA and Bonferonni's posttest.
Toll-like receptor-4 is involved in chemotaxis driven by oxidant-produced heparin fragments
Studies suggest that heparin and heparan sulfate signal through TLR4 (9), which is present on inflammatory cells. In our chemotaxis model, anti-TLR4 prevented heparin fragment–induced PMN chemotaxis (Fig. 4). LPS induction of chemotaxis via TLR-4 was used as a positive control. This provides further support for our hypothesis that prevention of oxidant-induced fragmentation of heparin/heparan sulfate is one mechanism through which EC-SOD inhibits inflammation and injury.
FIG. 4. Toll-like receptor 4 mediates heparin fragment-induced PMN chemotaxis.

Anti-TLR4 antibody inhibits PMN chemotaxis induced by ROS-fragmented heparin. *p < 0.05. Data analysis used ANOVA and Bonferonni's posttest.
Native EC-SOD protects against heparan sulfate shedding from the lung extracellular matrix during asbestos-induced injury
Further to elucidate the role of EC-SOD in the protection of the extracellular matrix (ECM), we assessed the effects of asbestos exposure on heparan sulfate integrity in the lungs of wild-type and EC-SOD null mice. Western blot analysis of heparan sulfate in the BALF of asbestos-treated wild-type mice showed an increase in shed heparan sulfate (HS) at 1 day (Fig. 5a), 14 days (Fig. 5b), and 28 days (Fig. 5c) after asbestos exposure. Notably, Fig. 5 also shows that in EC-SOD null mice, asbestos injury led to a more dramatic increase in heparan sulfate shedding at 1 day (HS2), 14 days (HS1 and 2), and 28 days (HS1 and 3). Several types of heparan sulfate were found to increase, noted as HS1, 2, and 3. Based on size, these HS species are in the syndecan family, with HS1 being syndecan-1; HS2, syndecan-2; and HS3, syndecan-4. EC-SOD knockout mice treated with intratracheal asbestos display an increase in PMNs and protein in the BALF and increased total lung fibrosis, as previously reported by our laboratory with this injury model (21). Our laboratory also reported increased EC-SOD in the BALF, with a corresponding decrease in lung EC-SOD in wild-type mice exposed to asbestos (21).
FIG. 5. EC-SOD prevents HS shedding from the ECM after asbestos-induced injury.
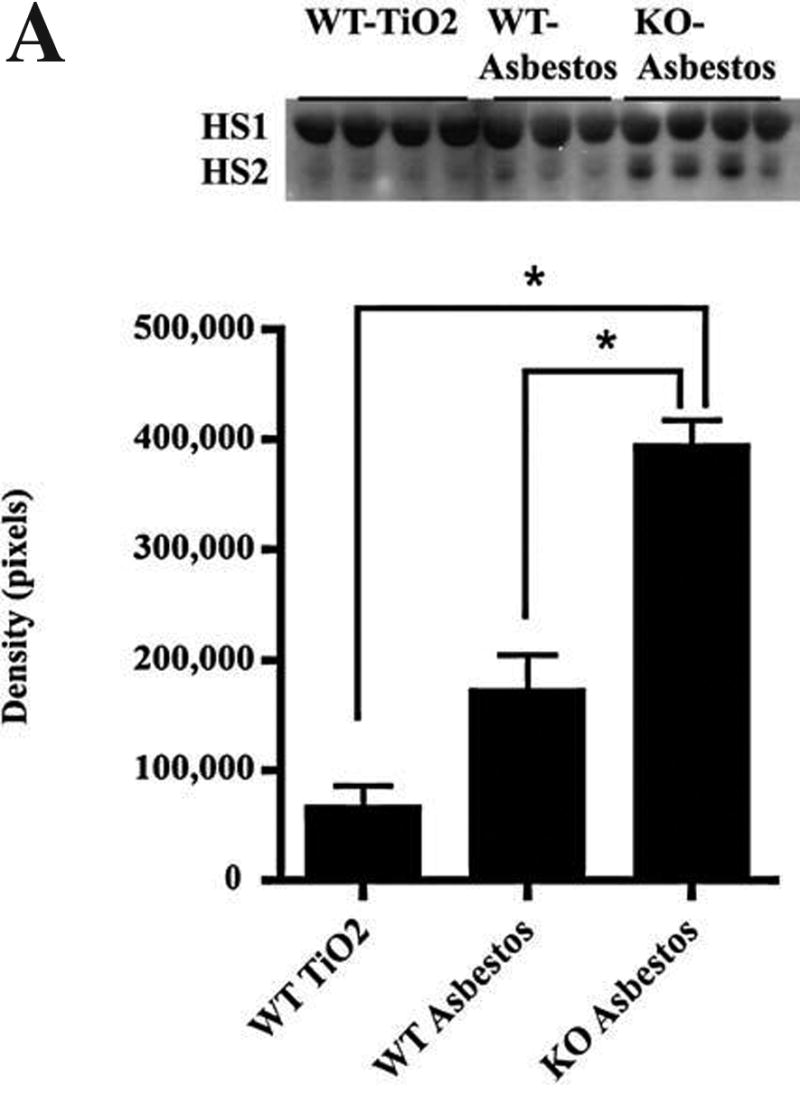


HS shedding increases in the BALF in EC-SOD knockout mice at (A) 1 day (*p < 0.001), (B) 14 days, and (C) 28 days after asbestos exposure compared with wild-type mice. Ten micrograms of protein was loaded for each sample. Significant increases were seen in three HS species (HS1, HS2, and HS3), detected by Western blot with antibody MAB2040 (normalized to protein loading by Ponceau red staining of the membrane). HS1, 2, and 3 are predicted to be syndecans by molecular mass, 35–80 kDa.
Discussion
In the present study, we showed that EC-SOD can protect heparin/heparan sulfate from oxidant-mediated degradation and inhibit associated inflammatory responses. These findings support our hypothesis that one mechanism by which EC-SOD protects the lung from oxidant-induced damage, inflammation, and fibrosis in asbestosis is by preventing heparin/heparan sulfate (HS) fragmentation in the ECM.
EC-SOD is an antioxidant enzyme found in high abundance in the lung compared with other tissues. HS binds to and localizes many factors to the ECM, one of which is EC-SOD (7, 15). Fragmentation and shedding of HS would result in the release of bound factors. EC-SOD is known to protect the lung from oxidant-mediated damage, inflammation, and interstitial fibrosis (3, 21, 26). One mechanism through which EC-SOD may prevent inflammatory cell chemotaxis within the lung is by protecting glycosaminoglycans from oxidative fragmentation. Our study supports this by showing that EC-SOD inhibits neutrophil chemotaxis by preventing the formation of reactive oxygen species (ROS) and heparin/heparan sulfate fragments (see Fig. 3). This chemotactic response at least partially involves TLR-4 on the neutrophils (see Fig. 4). This finding is significant, given that in lung-injury models of asbestosis, substantial neutrophil influx is seen (5, 21).
We also show that in vivo, EC-SOD protects against the shedding of HS from the lung parenchyma. In EC-SOD–null mice treated with intratracheal asbestos, enhanced shedding of heparan sulfate is seen at 1, 14, and 28 days after treatment (see Fig. 5). The type of HSs shed into the BALF changes over the course of injury. HS1 is the most abundant throughout the injury course, whereas HS3 increases later near 28 days after exposure (see Fig. 5). Based on molecular-weight analysis of the shed HSs, HS1 is likely to be syndecan 1. Notably, matrix metalloproteinase 7 is known to contribute to the pathogenesis of pulmonary fibrosis (11, 26), and MMP7-induced shedding of syndecan-1 from the lung parenchyma is believed to be a central mechanism contributing to fibrosis after bleomycin injury (11). Because the pathogenesis of asbestos-induced fibrosis is thought to involve oxidative stress (12), and we have found that HSs are shed during asbestos injury, it is important to determine whether oxidant/antioxidant imbalances can also contribute to HS shedding in pulmonary fibrosis. Because of its close interaction with EC-SOD, HS is a key ECM component to evaluate in the context of fibrosis and oxidants.
Previous studies have shown that after asbestos injury, EC-SOD decreases in the lung parenchyma, with a corresponding increase in the alveolar lining fluid (21, 22). Because HS binds EC-SOD in the lung (7, 10), HS shedding could provide one mechanism for the decrease in EC-SOD distribution in the lung after asbestos exposure. HS shedding with subsequent loss of EC-SOD from the lung parenchyma could therefore lead to an increase in lung susceptibility to oxidative stress, injury, and inflammation (see Fig. 1). Additional investigation is needed to determine the direct interactions between EC-SOD and HS in vivo.
Through this study, the discovery of oxidative fragmentation of heparin/heparan sulfate and protection by EC-SOD in the lung is a novel finding in the context of asbestosis and advances the understanding of key interactions between oxidant/antioxidant imbalances and inflammation after asbestos exposure. EC-SOD is an important enzyme involved in the pathogenesis of pulmonary fibrosis, and further investigation into its interactions with the extracellular matrix will advance the current knowledge of many interstitial lung diseases. These studies may provide targets for the discovery of therapeutic agents for asbestosis and other interstitial pulmonary injuries.
Acknowledgments
This work was supported by funding from NIH, HL63700 and an American Heart Association Established Investigator award.
Abbreviations
- DMEM
Dulbecco's modified Eagle's medium
- EC-SOD
extracellular superoxide dismutase
- HS
heparan sulfate
- IPF
idiopathic pulmonary fibrosis
- LPS
lipopolysaccharide
- ROS
reactive oxygen species
- TLR4
Toll-like receptor 4
References
- 1.Bernfield M, Gotte M, Park PW, Reizes O, Fitzgerald ML, Lincecum J, Zako M. Functions of cell surface heparan sulfate proteoglycans. Annu Rev Biochem. 1999;68:729–777. doi: 10.1146/annurev.biochem.68.1.729. [DOI] [PubMed] [Google Scholar]
- 2.Bowler RP, Crapo JD. Oxidative stress in airways: Is there a role for extracellular superoxide dismutase? Am J Respir Crit Care Med. 2002;166:S38–S43. doi: 10.1164/rccm.2206014. [DOI] [PubMed] [Google Scholar]
- 3.Carlsson LM, Jonsson J, Edlund T, Marklund SL. Mice lacking extracellular superoxide dismutase are more sensitive to hyperoxia. Proc Natl Acad Sci U S A. 1995;92:6264–6268. doi: 10.1073/pnas.92.14.6264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cohen HJ, Chovaniec ME. Superoxide generation by digitonin-stimulated guinea pig granulocytes: a basis for a continuous assay for monitoring superoxide production and for the study of the activation of the generating system. J Clin Invest. 1978;61:1081–1087. doi: 10.1172/JCI109007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fattman CL, Chu CT, Kulich SM, Enghild JJ, Oury TD. Altered expression of extracellular superoxide dismutase in mouse lung after bleomycin treatment. Free Radic Biol Med. 2001;31:1198–207. doi: 10.1016/s0891-5849(01)00699-2. [DOI] [PubMed] [Google Scholar]
- 6.Fattman CL, Enghild JJ, Crapo JD, Schaefer LM, Valnickova Z, Oury TD. Purification and characterization of extracellular superoxide dismutase in mouse lung. Biochem Biophys Res Commun. 2000;275:542–548. doi: 10.1006/bbrc.2000.3327. [DOI] [PubMed] [Google Scholar]
- 7.Fattman CL, Schaefer LM, Oury TD. Extracellular superoxide dismutase in biology and medicine. Free Radic Biol Med. 2003;35:236–256. doi: 10.1016/s0891-5849(03)00275-2. [DOI] [PubMed] [Google Scholar]
- 8.Fattman CL, Tan RJ, Tobolewski JM, Oury TD. Increased sensitivity to asbestos-induced lung injury in mice lacking extracellular superoxide dismutase. Free Radic Biol Med. 2006;40:601–607. doi: 10.1016/j.freeradbiomed.2005.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Johnson GB, Brunn GJ, Platt JL. Cutting edge: an endogenous pathway to systemic inflammatory response syndrome (SIRS)-like reactions through toll-like receptor 4. J Immunol. 2004;172:20–24. doi: 10.4049/jimmunol.172.1.20. [DOI] [PubMed] [Google Scholar]
- 10.Karlsson K, Lindahl U, Marklund SL. Binding of human extracellular superoxide dismutase C to sulphated glycosaminoglycans. Biochem J. 1988;256:29–33. doi: 10.1042/bj2560029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li Q, Park PW, Wilson CL, Parks WC. Matrilysin shedding of syndecan-1 regulates chemokine mobilization and transepithelial efflux of neutrophils in acute lung injury. Cell. 2002;111:635–646. doi: 10.1016/s0092-8674(02)01079-6. [DOI] [PubMed] [Google Scholar]
- 12.Manning CB, Vallyathan V, Mossman BT. Diseases caused by asbestos: mechanisms of injury and disease development. Int Immunopharmacol. 2002;2:191–200. doi: 10.1016/s1567-5769(01)00172-2. [DOI] [PubMed] [Google Scholar]
- 13.Monboisse JC, Bellon G, Randoux A, Dufer J, Borel JP. Activation of human neutrophils by type I collagen: requirement of two different sequences. Biochem J. 1990;270:459–462. doi: 10.1042/bj2700459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Murphy RL., Jr The diagnosis of nonmalignant diseases related to asbestos. Am Rev Respir Dis. 1987;136:1516–1517. doi: 10.1164/ajrccm/136.6.1516. [DOI] [PubMed] [Google Scholar]
- 15.Oury TD, Crapo JD, Valnickova Z, Enghild JJ. Human extracellular superoxide dismutase is a tetramer composed of two disulphide-linked dimers: a simplified, high-yield purification of extracellular superoxide dismutase. Biochem J. 1996;317:51–57. doi: 10.1042/bj3170051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Oury TD, Day BJ, Crapo JD. Extracellular superoxide dismutase: a regulator of nitric oxide bioavailability. Lab Invest. 1996;75:617–636. [PubMed] [Google Scholar]
- 17.Parish CR. The role of heparan sulphate in inflammation. Nat Rev Immunol. 2006;6:633–643. doi: 10.1038/nri1918. [DOI] [PubMed] [Google Scholar]
- 18.Petersen SV, Oury TD, Ostergaard L, Valnickova Z, Wegrzyn J, Thogersen IB, Jacobsen C, Bowler RP, Fattman CL, Crapo JD, Enghild JJ. Extracellular superoxide dismutase (EC-SOD) binds to type i collagen and protects against oxidative fragmentation. J Biol Chem. 2004;279:13705–13710. doi: 10.1074/jbc.M310217200. [DOI] [PubMed] [Google Scholar]
- 19.Raats CJ, Bakker MA, van den Born J, Berden JH. Hydroxyl radicals depolymerize glomerular heparan sulfate in vitro and in experimental nephrotic syndrome. J Biol Chem. 1997;272:26734–26741. doi: 10.1074/jbc.272.42.26734. [DOI] [PubMed] [Google Scholar]
- 20.Raats CJ, Van Den Born J, Berden JH. Glomerular heparan sulfate alterations: mechanisms and relevance for proteinuria. Kidney Int. 2000;57:385–400. doi: 10.1046/j.1523-1755.2000.00858.x. [DOI] [PubMed] [Google Scholar]
- 21.Tan RJ, Fattman CL, Watkins SC, Oury TD. Redistribution of pulmonary EC-SOD after exposure to asbestos. J Appl Physiol. 2004;97:2006–2013. doi: 10.1152/japplphysiol.00480.2004. [DOI] [PubMed] [Google Scholar]
- 22.Tan RJ, Lee JS, Manni ML, Fattman CL, Tobolewski JN, Zheng M, Kolls JK, Martin TR, Oury TD. Inflammatory cells as a source of airspace extracellular superoxide dismutase after pulmonary injury. Am J Respir Cell Mol Biol. 2006;34:226–232. doi: 10.1165/rcmb.2005-0212OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Venkatesan N, Roughley PJ, Ludwig MS. Proteoglycan expression in bleomycin lung fibroblasts: role of transforming growth factor-beta(1) and interferon-gamma. Am J Physiol Lung Cell Mol Physiol. 2002;283:L806–L814. doi: 10.1152/ajplung.00061.2002. [DOI] [PubMed] [Google Scholar]
- 24.Wang L, Fuster M, Sriramarao P, Esko JD. Endothelial heparan sulfate deficiency impairs L-selectin- and chemokine-mediated neutrophil trafficking during inflammatory responses. Nat Immunol. 2005;6:902–910. doi: 10.1038/ni1233. [DOI] [PubMed] [Google Scholar]
- 25.Zen K, Reaves TA, Soto I, Liu Y. Response to genistein: assaying the activation status and chemotaxis efficacy of isolated neutrophils. J Immunol Methods. 2006;309:86–98. doi: 10.1016/j.jim.2005.11.014. [DOI] [PubMed] [Google Scholar]
- 26.Zuo F, Kaminski N, Eugui E, Allard J, Yakhini Z, Ben-Dor A, Lollini L, Morris D, Kim Y, DeLustro B, Sheppard D, Pardo A, Selman M, Heller RA. Gene expression analysis reveals matrilysin as a key regulator of pulmonary fibrosis in mice and humans. Proc Natl Acad Sci U S A. 2002;99:6292–6297. doi: 10.1073/pnas.092134099. [DOI] [PMC free article] [PubMed] [Google Scholar]