

Abstract
Fishes have been recently recognized as a suitable model organism to study vertebrate physiological processes, in particular skeletal development and tissue mineralization. However, there is a lack of well characterized in vitro cell systems derived from fish calcified tissues. We describe here a protocol that was successfully used to develop the first calcified tissue-derived cell cultures of fish origin. Vertebra and branchial arches collected from young gilthead seabreams were fragmented then submitted to the combined action of collagenase and trypsin to efficiently release cells embedded in the collagenous extracellular matrix. Primary cultures were maintained under standard conditions and spontaneously transformed to form continuous cell lines suitable for studying mechanisms of tissue mineralization in seabream. This simple and inexpensive protocol is also applicable to other calcified tissues and species by adjusting parameters to each particular case.
Keywords: Calcified tissues, Gilthead seabream (Sparus aurata L.), In vitro cell system, Mineralization, Cell culture protocol
Introduction
The relative abundance and extensive biochemical and genetic characterization of in vitro cell systems in mammals contrasts singularly with the information available for other vertebrate organisms and particularly fish, the most diverse and oldest class of vertebrates (Bolis et al. 2001). Fish, by sharing with mammals a large number of important characteristics (e.g. similar organ systems, developmental organization and physiological/biochemical mechanisms) and by presenting various technical advantages (Patton and Zon 2001; Berghmans et al. 2005; McGonnell and Fowkes 2006), has become a promising vertebrate model for most biological studies and a suitable alternative to mammalian systems (Hightower and Renfro 1988; Kelly et al. 1998; Braga et al. 2006). Fish, in particular zebrafish, has emerged as an important organism for studies on skeletal development in vertebrates, e.g. successful identification of the gene responsible for the chihuahua mutation, collagen I(α1), where severe skeletal deformities are present, accurately modeling human osteogenesis imperfecta (Fisher et al. 2003). More recently, a new screen for craniofacial mutants identified gene mutations related to two human syndromes (Nissen et al. 2006), and highlighted the fact that many more can probably be uncovered due to high degree of evolutionary conservation in gene organization and function between human and fish. As a result of the growing interest for fish models (e.g. zebrafish, but also medaka, fugu, killifish, green-spotted pufferfish, goldfish, etc.), a number of tools and techniques have been developed in the last decade, including in vitro cell systems such as cell lines. However, although various fish cell lines are available, few of them have been characterized (most of them were developed for virus susceptibility testing) and important biological processes, such as tissue mineralization, are still lacking cell lines suitable for their study in fish. Only two fish cell lines have been successfully developed, and described, from calcified tissues, both in our laboratory (Pombinho et al. 2004) and both from the gilthead seabream (Sparus aurata L.), a marine teleost extensively cultured in most Mediterranean countries (Bejar et al. 1997). The reduced availability of fish cell lines suitable to study molecular and cellular mechanisms of bone formation and skeletal development is hampering the development of fish as a model animal to study vertebrate mineralization, probably as a result of the lack of any published protocol describing the preparation of primary cell culture from fish calcified tissues. We present here a successful, simple and inexpensive method that can be used to obtain primary cell cultures and cell lines from fish calcified tissues.
Materials
Fish: juvenile gilthead seabream (S. aurata L.), obtained from natural spawned eggs, were bred at 16–20 °C in 100-L seawater tanks with a 12 h light-dark photoperiod, aeration of 100 mL min−1, and renewal flow of 1 tank per day. Fish up to 50 days after hatching were fed with artemia and/or rotifers and older fish with artificial food (Sorgal, Porto, Portugal).
Cell culture apparatus: CO2 incubator MCO-18AIC and cooled incubator MIR-152 from SANYO Electric (Osaka, Japan); biological safety cabinet class II BBV4 from B. BRAUN (Melsungen, Germany).
Cell culture medium: Dulbecco’s modified Eagle medium (DMEM) and Leibovitz’s medium (L15), from Invitrogen (Carlsbad, CA).
Medium supplements: Fetal bovine serum (FBS), l-glutamine, penicillin-streptomycin and fungizone, all from Invitrogen.
Solutions: phosphate-buffered saline (PBS: 137 mM NaCl, 2.7 mM KCl, 15.8 mM Na2HPO4, 1.23 mM KH2PO4), digestion solution (0.125% collagenase in PBS) from Boehringer (Ingelheim, Germany), trypsin-EDTA solution (in PBS; 1.1 mM EDTA, 0.2% trypsin from Invitrogen).
Plasticware: cell culture dishes and serologic pipettes from Sarstedt (Nümbrecht, Germany).
Cell cryopreservation: Cryogenic tubes and cell freezing device (Mr. Frosty) from Nalgene (Roskilde, Denmark), liquid nitrogen storage container Locator Jr. Plus from Thermolyne (Dubuque, IA).
All other chemicals were purchased from Sigma-Aldrich (St. Louis, MO), unless otherwise stated.
Methods
Juvenile and healthy seabreams were rapidly anesthetized with 2-phenoxyethanol diluted 1:10,000 in seawater.
Fish were sacrificed by decapitation and quickly washed with diluted bleach (1:100) then 70% ethanol to remove most surface contaminants.
Calcified tissues (e.g. vertebrae, branchial arches) were collected using sterile instruments and cleaned from adherent tissues using a scalpel, then washed 5 × 5 min with PBS supplemented with 5% (v/v) penicillin/streptomycin and 1% (v/v) fungizone to prevent contamination of primary cell cultures.
Tissues were minced manually to small fragments of about 8 mm3 using sterile instruments.
Fragments were first digested with collagenase for various periods of time (1, 5, 10 and 20 h) then with trypsin for 5 min. All digestions were performed at room temperature (approximately 22–24 °C) and with agitation.
Digestion solution was removed and fragments were washed 3 × 5 min with serum-free media (L15 or DMEM) supplemented with 5% (v/v) antibiotics and 1% (v/v) fungizone.
Fragments were then placed into a 24-well plate containing 250 μL of supplemented media (15% FBS, 1% fungizone and 1% antibiotics) and incubated, according to the medium used, in a standard incubator (L15) or a CO2 incubator (DMEM) at various temperatures (ranging from 22 to 33 °C).
Cells were allowed to migrate from fragments and attach to the plate for approximately 2–4 weeks with medium renewed twice a week.
Cells from confluent cultures were collected using trypsin-EDTA solution and seeded into a 6-well plate containing 2 mL of appropriate medium then, when confluent again, transferred using a similar methodology, but this time into a 100-mm plate containing 8 mL of medium.
Cell cultures were routinely sub-cultured (1:2) at confluence by trypsinization and maintained in continuous culture up to passage 130.
At appropriate passages, cells from healthy, pre-confluent cultures were trypsinized and harvested by centrifugation (6 min at 2,000g). Cell pellet, containing 3.5 × 106 cells, was gently resuspended in ice-cold culture medium containing 10% DMSO by pipetting slowly up and down. Cell suspension was then aliquoted into 2-mL cryogenic tubes subsequently cooled down to −80 °C using Mr. Frosty device to achieve a constant rate of freezing. Cryotubes were finally transferred into a cell container and immersed in liquid nitrogen for long-term storage.
To thaw the cells, cryotubes were removed from liquid nitrogen and quickly thawed at 25 °C (using a water bath for rapid thawing). As soon as the medium containing the cells was sufficiently thawed to be released from the walls of the cryotube, the vial was quickly immersed into 70% ethanol, then cleaned and opened, and cells transferred into a 100-mm dish containing fresh medium. Cells were gently dispersed, then incubated for 6–8 h to allow the cells to adhere. Medium was then renewed to remove DMSO.
Results and discussion
We present here a successful, simple and inexpensive method that can be used to obtain primary cell cultures and cell lines from fish calcified tissues. The efficient release of cells embedded in the collagenous extracellular matrix is crucial for the successful development of in vitro cell cultures from hard tissues. This critical step has been particularly improved by the fragmentation of the calcified tissue in small pieces then the successive action of two different proteases: first, collagenase to degrade collagen fibers that hold animal tissues, followed by a trypsin digestion to break down structural proteins such as those involved in cell-matrix interaction. Digestion time and temperature have been optimized for S. aurata calcified tissues but may need to be adjusted to each particular tissue and species.
Using the protocol described here, primary cultures have been developed from S. aurata vertebrae (Pombinho et al. 2004) and branchial arches (Fig. 1). After extended time in culture, some cells of the primary cultures transformed spontaneously (in absence of any exogenous agent). These cells grow rapidly (doubling time: 36–48 h) and do not exhibit passage-related senescence, at least up to 130 passages, behaving as if they were immortalized. Spontaneous transformation of primary cell culture into a continuous cell line is common in some species (Crane 1999), such as fish (Wolf and Mann 1980; Fernandez et al. 1993). The successful cryopreservation (using dimethyl sulphoxide—DMSO—as cryoprotectant) and thawing of the three cell lines, named VSa13, VSa16 and ABSa15 (from vertebrae and branchial arches, respectively), was also demonstrated to be achieved with survival rates >70%. Furthermore, efficient DNA transfer, crucial for analysis of gene function, was obtained using polyethylenimine (PEI) (Braga et al. 2006).
Fig. 1.
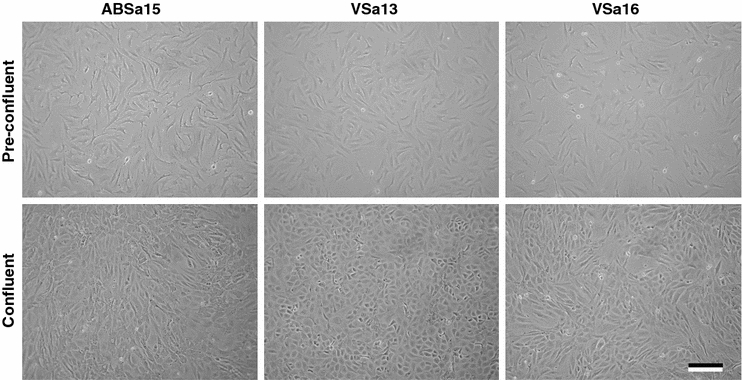
Phase-contrast micrographs of S. aurata ABSa15, VSa13 and VSa16 pre-confluent and confluent cultures in DMEM supplemented with 10% FBS. Micrographs were obtained using an Axiovert 25 inverted light microscope (Zeiss, Göttingen, Germany) equipped with phase-contrast and linked to a C-3030 digital camera (Olympus, Hamburg, Germany). Bar is 200 μm
The three different cell lines exhibit a homogeneous and stable phenotype, which is polygonal when cultured in DMEM (Fig. 1) and are able to mineralize their extracellular matrix (ECM), when cultured under mineralization conditions (Fig. 2) demonstrating their suitability to analyze mineralization-related mechanisms (Pombinho et al. 2004). The expression of bone and cartilage-related genes (e.g. osteocalcin, osteonectin, osteopontin, matrix Gla protein, bone morphogenetic protein 2, alkaline phosphatase, etc.) and their regulation during in vitro mineralization were also demonstrated, further confirming their suitability to identify molecular determinants of tissue calcification (Laizé et al. 2005; Rafael et al. 2006; Fonseca et al. 2007). Finally, these cells are well adapted to grow in standard media (DMEM or L15) in the presence of mammalian serum (10% fetal bovine serum), seriously decreasing costs associated to cell maintenance (fish sera are available, but expensive) and allowing direct comparison with studies using mammalian bone-derived cells (Kellermann et al. 1990; Fournier and Price 1991; Stanford et al. 1995; Costa and Fernandes 2000).
Fig. 2.
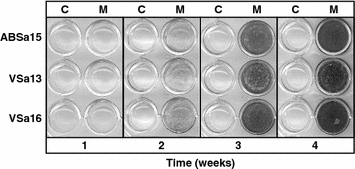
Micrographs of von Kossa-stained ABSa15, VSa13 and VSa16 cells cultured under control (C) and mineralization conditions (M). Extracellular matrix mineralization was induced in cells grown in normal culture medium (DMEM with 10% FBS) supplemented with 50 μg mL−1 L-ascorbic acid, 10 mM β-glycerophosphate and 4 mM CaCl2. Medium was renewed twice a week. After 4 weeks, mineral deposition was revealed by von Kossa staining. Cells were washed 3 times with PBS, fixed with 10% (v/v) formaldehyde (in PBS) for 30 min at 4 °C, washed 3 times with distilled water then incubated with 5% silver nitrate for 10 min under ultraviolet light. Black areas indicate presence of silver-stained mineral nodules. Cell passages were 35, 45 and 36, respectively
The protocol described here has been successfully applied to the development of the first calcified tissue-derived cell lines of fish origin. Its simplicity and low cost should permit the development of additional fish cell lines from other species and suitable to study mechanisms of tissue mineralization, providing that some parameters (e.g. digestion time, temperature of cell growth) are adjusted to each particular tissue and species. Primary cell cultures derived from Adriatic sturgeon (Acipenseriformes, Acipenser naccarii) and Atlantic salmon (Salmoniformes, Salmo salar) mineralized tissues have already been developed and successfully sub-cultured several times using this method (our unpublished results), further demonstrating its robustness and efficiency.
Acknowledgements
This work was partially supported by grants POCTI/BCI/48748/2002 from the Portuguese Science and Technology Foundation (FCT) and GOCE-CT-2004-505403 (Marine Genomics Europe) from the European Commission under the 6th Framework Program. C. L. Marques and M. S. Rafael were the recipients of a CCMAR/Instituto de Emprego e Formação Profissional and a FCT doctoral (SFRH/BD/22695/2005) fellowships, respectively.
Abbreviations
- DMSO
Dimethyl sulphoxide
- PEI
Polyethylenimine
- ECM
Extracellular matrix
References
- Bejar J, Borrego JJ, Alvarez MC (1997) A continuous cell line from the cultured marine fish gilt-head seabream (Sparus aurata L.). Aquaculture 150:143–153 [DOI]
- Berghmans S, Jette C, Langenau D, Hsu K, Stewart R, Look T, Kanki JP (2005) Making waves in cancer research: new models in the zebrafish. Biotechniques 39:227–237 [DOI] [PubMed]
- Bolis CL, Piccolella M, Dalla Valle AZ et al (2001) Fish as model in pharmacological and biological research. Pharmacol Res 44:265–280 [DOI] [PubMed]
- Braga D, Laizé V, Tiago DM, Cancela ML (2006) Enhanced DNA transfer into fish bone cells using polyethylenimine. Mol Biotechnol 34:51–54 [DOI] [PubMed]
- Costa MA, Fernandes MH (2000) Long-term effects of parathyroid hormone, 1,25-dihydroxyvitamin d(3), and dexamethasone on the cell growth and functional activity of human osteogenic alveolar bone cell cultures. Pharmacol Res 42:345–353 [DOI] [PubMed]
- Crane MS (1999) Mutagensis and cell transformation in cell culture. Methods Cell Sci 21:245–253 [DOI] [PubMed]
- Fernandez RD, Yoshimizu M, Kimura T, Ezura Y (1993) Establishment and characterization of seven continuous cell lines from freshwater fish. J Aquat Anim Health 5:137–147 [DOI]
- Fisher S, Jagadeeswaran P, Halpern ME (2003) Radiographic analysis of zebrafish skeletal defects. Dev Biol 264:64–76 [DOI] [PubMed]
- Fonseca VG, Laizé V, Valente MS, Cancela ML (2007) Identification of an osteopontin-like protein in fish associated with mineral formation. FEBS J 274:4428–4439 [DOI] [PubMed]
- Fournier B, Price PA (1991) Characterization of a new human osteosarcoma cell line OHS-4. J Cell Biol 114:577–583 [DOI] [PMC free article] [PubMed]
- Hightower LE, Renfro JL (1988) Recent applications of fish cell culture to biomedical research. J Exp Zool 248:290–302 [DOI] [PubMed]
- Kellermann O, Buc-Caron MH, Marie PJ, Lamblin D, Jacob F (1990) An immortalized osteogenic cell line derived from mouse teratocarcinoma is able to mineralize in vivo and in vitro. J Cell Biol 110:123–132 [DOI] [PMC free article] [PubMed]
- Kelly KA, Havrilla CM, Brady TC, Abramo KH, Levin ED (1998) Oxidative stress in toxicology: established mammalian and emerging piscine model systems. Environ Health Perspect 106:375–384 [DOI] [PMC free article] [PubMed]
- Laizé V, Pombinho AR, Cancela ML (2005) Characterization of Sparus aurata osteonectin cDNA and in silico analysis of protein conserved features: evidence for more than one osteonectin in Salmonidae. Biochimie 87:411–420 [DOI] [PubMed]
- McGonnell IM, Fowkes RC (2006) Fishing for gene function—endocrine modelling in the zebrafish. J Endocrinol 189:425–439 [DOI] [PubMed]
- Nissen RM, Amsterdam A, Hopkins N (2006) A zebrafish screen for craniofacial mutants identifies wdr68 as a highly conserved gene required for endothelin-1 expression. BMC Dev Biol 6:28 [DOI] [PMC free article] [PubMed]
- Patton EE, Zon LI (2001) The art and design of genetic screens: zebrafish. Nat Rev Genet 2:956–966 [DOI] [PubMed]
- Pombinho AR, Laizé V, Molha DM, Marques SM, Cancela ML (2004) Development of two bone-derived cell lines from the marine teleost Sparus aurata; Evidence for extracellular matrix mineralization and cell-type-specific expression of matrix Gla protein and osteocalcin. Cell Tissue Res 315:393–406 [DOI] [PubMed]
- Rafael MS, Laizé V, Cancela ML (2006) Identification of Sparus aurata bone morphogenetic protein 2: molecular cloning, gene expression and in silico analysis of protein conserved features in vertebrates. Bone 39:1373–1381 [DOI] [PubMed]
- Stanford CM, Jacobson PA, Eanes ED, Lembke LA, Midura RJ (1995) Rapidly forming apatitic mineral in an osteoblastic cell line. J Biol Chem 270:9420–9428 [DOI] [PubMed]
- Wolf K, Mann JA (1980) Poikilotherm vertebrate cell lines and viruses: a current listing for fishes. In Vitro 16:168–179 [DOI] [PubMed]