

SUMMARY
F425-B4e8 (B4e8) is a monoclonal antibody isolated from a human immunodeficiency virus type 1 (HIV-1)-infected individual that recognizes the V3 variable loop on the gp120 subunit of the viral envelope spike. B4e8 neutralizes a subset of HIV-1 primary isolates from subtypes B, C and D, which places this antibody among the very few human anti-V3 antibodies with notable cross-neutralizing activity. Here, the crystal structure of the B4e8 Fab’ fragment in complex with a 24-mer V3-peptide (RP142) at 2.8 Å resolution is described. The complex structure reveals that the antibody recognizes a novel V3 loop conformation, featuring a 5-residue α-turn around the conserved GPGRA apex of the β-hairpin loop. In agreement with previous mutagenesis analyses, the Fab’ interacts primarily with V3 through side-chain contacts with just two residues, IleP309 and ArgP315, while the remaining contacts are to the main chain. The structure helps explain how B4e8 can tolerate a certain degree of sequence variation within V3 and, hence, is able to neutralize an appreciable number of different HIV-1 isolates.
Keywords: HIV-1, neutralizing antibody, V3, gp120, X-ray crystallography
INTRODUCTION
The envelope spike of the human immunodeficiency virus type 1 (HIV-1) is responsible for viral infectivity by binding to cell surface receptors, thus mediating cell entry1. The viral spike is a membrane-anchored, trimeric assembly formed by the non-covalently associated glycoproteins gp120 and gp41. All HIV-1 neutralizing antibodies are directed against these envelope glycoproteins.
Recent crystal structures of broadly neutralizing anti-HIV antibodies have provided insight into the epitope targets of these antibodies and, consequently, strategies to neutralize this constantly evolving virus. One of these epitopes is the membrane proximal region on gp41, which is recognized by human monoclonal antibodies (mAbs) 4E10, the most broadly anti-HIV-1 neutralizing antibody known2,3, and 2F54. Another suitable epitope for broad neutralization by an antibody is the recessed, but conserved, CD4 binding site on gp120, which is recognized by human mAb b125,6. Structural studies of anti-HIV antibodies have also revealed unique Fab configurations, such as the domain-swapped architecture of mAb 2G12 Fab, that forms a multivalent paratope, specific for oligomannose clusters on gp1207. In contrast to these unique broadly neutralizing antibodies, mAbs to the third hypervariable loop (V3) on gp120 neutralize a much smaller range of primary isolates and many tend to be isolate specific8-11.
The V3 region of gp120 is critical for viral infectivity. V3 mediates the binding to coreceptor molecules on target cells and its sequence determines coreceptor tropism12-14. It has been suggested, based on mutagenesis experiments and structural data, that the relatively conserved V3 tip interacts with the extracellular loops of CCR5, thus participating in the gp120 chemokine receptor interaction15,16. During cell entry, gp120 undergoes several structural changes, during which V3 may change its conformation, disposition and accessibility. Conformational flexibility in gp120 is supported in part by the crystal structure of unliganded simian immunodeficiency virus (SIV) gp120 core17,18, which shows significant structural changes in comparison to CD4-bound HIV gp12019,20 and by an engineered gp120 in complex with mAb b126.
A recent crystal structure of HIV-gp120 bound to CD4 and neutralizing antibody X5 revealed the first structural insights into the conformation and relative orientation of the intact V3 loop15. The V3 loop comprises approximately 35 residues, linked by a disulfide bond at its base (Cys296-Cys331, HXB2 numbering21) and can be subdivided into three parts: the base (residues 296-300 and 326-331), the stem (residues 301-305 and 321-325) and the tip or crown (residues 306-320). In the gp120 structure, the stem region is quite flexible, whereas the base and tip region consist of two antiparallel β-strands that form a β-hairpin loop15. The apex of this loop (residues 312-315) is conserved with a GPGR motif in many subtype B viruses and a GPGQ in nearly all non subtype B viruses.
A number of crystal structures of anti-V3 Fabs in complex with V3 peptides have been determined22-28. The Fab interactions are mostly centered around the tip region of V3 with its β-turn, with an extended β-structure on its N terminal side; these substructures align well with their corresponding sections in the gp120 structure. V3 peptides have been complexed with murine antibodies 50.122,23, 59.124,25, 58.223, 83.126, and human mAbs 221927 and 447-52D28. All of these V3-structures contain a type II β-turn around the conserved GPGR motif, with the exception of 83.1, that adopts a type I β-turn. In agreement with these crystal structures, the NMR structures of isolated V3-peptides show a high turn propensity around the apex of the hairpin loop29.
Despite a large repertoire of V3-specific antibodies studied, only a few exhibit neutralization that is not isolate specific. Of these, the two human mAbs 447-52D and 2219 are the best characterized. Human mAb 447-52D utilizes its extended CDR H3 loop to form a mixed β-sheet with the V3-hairpin loop and enlists mainly main-chain interactions to achieve its high affinity and broad specificity28. However, it also interacts extensively with the conserved GPGR-crown region, which limits its reactivity to viruses with a GPGR sequence at the V3 tip. Human mAb 2219, on the other hand, binds to the conserved β-structure in the β-hairpin, namely residues P303 to P309, with the crown region, in this case, exposed to solvent. This mode of interaction allows mAb 2219 to recognize V3 peptides with unusual crown sequences27.
B4e8 is an anti-V3 antibody that initially was thought to recognize an epitope at the base of the V3 loop30. However, a recent study31 re-assessed the epitope designation. It was shown that B4e8 binds to the tip of V3 and that the presence of IleP309, ArgP315 and PheP317 are important for high-affinity binding. In this later study, B4e8 also could neutralize a subset of primary isolates from subtypes B, C and D. In order to investigate the structural basis of this cross-reactivity, we have now determined the crystal structure of B4e8 in complex with the V3 peptide RP142 at 2.8 Å resolution.
RESULTS
Fab’ structure
Fab’ B4e8 (human, IgG2, κ) was crystallized in complex with the 24-mer,V3-peptide RP142 (YP301NKRKRIHIP309GP312PGRAFYTTKNIIGCP326) and the structure was determined to 2.8 Å resolution. The heavy and light chains are numbered by the standard Kabat and Wu numbering system32, whereas the peptide is numbered according to the HXB2 reference sequence21. B4e8 is the first human IgG2 that has been structurally characterized (Fig. 1). As expected, the IgG2 CH1 domain is structurally very similar to human IgG1 CH1 (rmsd of 0.7 Å for all CH1 Cα atoms of B4e8 and human IgG1 HIL [PDB ID: 8FAB]); only 6 residues, corresponding to positions 135, 136, 197, 198, 211 and 222, are different.
Figure 1.

Crystal structure of the B4e8-RP142 complex. The peptide is colored in yellow and the Fab’ heavy and light chains in dark and light gray, respectively; the Fab’ CDR loops are colored in orange for L1, purple for L2, red for L3, cyan for H1, salmon for H2 and light green for H3. The sugar and AsnH28, to which it is linked, are shown in stick representation and colored in dark green. Images were produced with Pymol (http://pymol.sourceforge.net/).
The CDR loops L1, L2, H1 and H2 fall into canonical classes33,34 κ2, 1, 1 and 3A, respectively. B4e8 CDR loop L3 shows an insertion after residue 95 which designates it among CDR L3 loops of canonical class κ5. However, the inserted residue is an Asp and not a Pro, as reported for other members of that class35 and the overall conformation is different from all previously described classes. Thus, the CDR L3 in B4e8 represents a new canonical class for κ-light chains (Fig. 2). The CDR H3 loop is of moderate length (14 residues) compared to other human anti-HIV-1 antibodies with very long H3 loops, such as 2F5 (22), 447-52D (20) or b12 (18). It has a kinked base, as predicted from its sequence36.
Figure 2.

New canonical class for CDR L3. (a) Comparison of the CDR L3 loop of B4e8 (red) to members of canonical class κ-1 (PDB-code: 1REI, lightblue), κ-2 (2FBJ, yellow), κ-3 (1YQV, aquamarine), κ-4 (1DFB, salmon) and κ-5 (palegreen, 1BAF). (b) The B4e8 kappa chain CDR L3 has an Asp instead of the usual Pro at position 95a and represents a new canonical structure.
Interestingly, the Fab’ is glycosylated at AsnH28, which is part of framework region (FR) 1, with interpretable electron density for the core trisaccharide, (GlcNAc)2Man. A single point mutation in the VH gene segment creates a glycosylation site by mutation of the conserved ThrH28 to AsnH28. However, the sugar does not contact the antigen and, therefore, does not participate in Fab’-peptide interaction. Computer modeling of the antibody structure onto the V3-containing HIV-1 gp120 structure15 also suggests no likely interaction of the sugar with CD4-bound gp120. Glycosylation of variable antibody domains has been observed previously37 and at least one other Fab contains a glycosylation site on a conserved framework region38.
Peptide structure and interactions
The 24-mer V3-peptide RP142 exhibits clear and interpretable electron density in the Fab’-peptide complex for residues P304-P320, except for the ArgP304 and LysP305 side chains (Fig. 3). A total of 96 contacts, including four hydrogen bonds and one salt bridge, are formed between B4e8 and RP142 (Table 2). The buried molecular surface area on the Fab’ is 447 Å2 and is contributed slightly unequally by the heavy (57%) and light chain (43%), using CDR loops L1 (7%), L3 (36%), H1 (8%), H2 (28%) and H3 (22%). No contacts are made by CDR L2, as frequently observed for haptens and non-protein ligands39. The corresponding area on the peptide is 411 Å2 and involves all residues from IleP307 to ThrP319 with the largest contribution from ArgP315 (24%) and IleP309 (21%) (Table 2).
Figure 3.

Electron density (2Fobs-Fcalc) for the RP142-peptide in complex with B4e8, contoured at 1.0σ. All residues, except the side chains of LysP305 and ArgP304, are well defined.
Table 2.
Total hydrogen bond and van der Waals contacts of peptide with B4e8 and buried surface areas of the V3 peptidea
| Peptide residue | Mainb | Sideb | Surface (Å2) |
|---|---|---|---|
| IP307 | 0 / 0 | 0 / 0 | 8.6 |
| HP308 | 0 / 0 | 0 / 0 | 7.4 |
| IP309 | 0 / 2 | 0 / 13 | 87.9 |
| GP312 | 0 / 1 | 0 / 0 | 9.8 |
| PP313 | 1 / 5 | 0 / 4 | 41.9 |
| GP314 | 0 / 6 | 0 / 0 | 19.5 |
| RP315 | 1 / 5 | 1 / 29 | 96.3 |
| AP316 | 0 / 6 | 0 / 2 | 34.1 |
| FP317 | 1 / 9 | 0 / 3 | 45.7 |
| YP318 | 0 / 0 | 0 / 7 | 51.7 |
| TP319 | 0 / 0 | 0 / 0 | 10.2 |
Residues with no buried surface area are not listed.
Shown as: hydrogen bonds / van der Waals contacts
The interaction between the Fab’ with the V3 peptide is primarily mediated by limited peptide side-chain, as well as some main-chain, contacts. The side-chain interactions are focussed on only two residues, IleP309 and ArgP315. Together with main-chain contacts along the peptide backbone, these residues participate in 83% of the van der Waals contacts, all hydrogen bonds and the only salt bridge (Fig. 4). Two main-chain hydrogen bonds are formed by AsnH108 with ProP313 and ArgP315 from the apex of the β-hairpin (Fig. 5). Another main-chain hydrogen bond between the Fab’ and the V3-peptide is formed between LeuL94 and PheP317, which further stabilizes their interaction.
Figure 4.

Specific binding interactions between B4e8 and RP142. Atomic coloring is used for the atoms with the carbon atoms of the peptide colored in yellow, carbon atoms of the heavy chain in light blue and those of the light chain in pale green. Only those residues of the Fab’ that make contact to the peptide are shown in stick representation. Residues GlyP312 - AlaP316 form a five residue α-turn.
Figure 5.
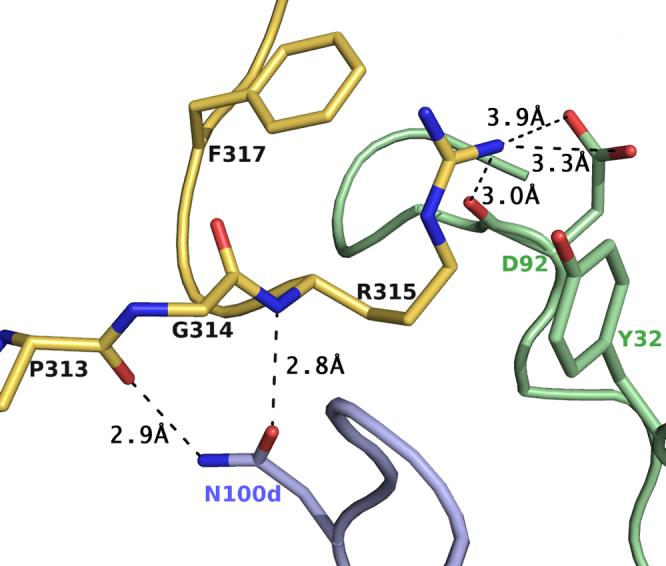
Detailed view of the exposed ArgP315 and its interactions with B4e8. Hydrogen bonds and salt bridges are shown by dashed lines. Color coding is as in Fig. 4. ArgP315 is sandwiched between PheP317 and TyrL32 and stabilized by a salt bridge and a hydrogen bond to the side chain and backbone of AspL92. The unusual α-turn conformation is stabilized by two hydrogen bonds between AsnH100d and the backbone of ArgP315 and ProP313, respectively.
The side chain of ArgP315 adopts an unusual rotamer that is sandwiched between TyrL32 (∼3.7 Å) and PheP317 (∼3.7 Å) (Fig. 5). The ArgP315 conformation is stabilized by a salt link and a hydrogen bond with AspL92. The only other major interaction of the Fab’ with a V3-peptide side chain involves IleP309, which is buried in a hydrophobic pocket formed by the CDR H2 loop and LeuL94, and complements the overall surface and charge complementarity between B4e8 and RP142 (Fig. 6). The shape correlation statistic (Sc) of 0.80 is similar to those observed for other Fab-V3-complexes, such as 2219 (PDB-ID: 2B0S, Sc=0.83), 447 52D (1Q1J, 0.77), 59.1 (1ACY, 0.80), 50.1 (1GGI, 0.79), 58.2 (1F58, 0.80) and 83.1 (1NAK, 0.78).
Figure 6.
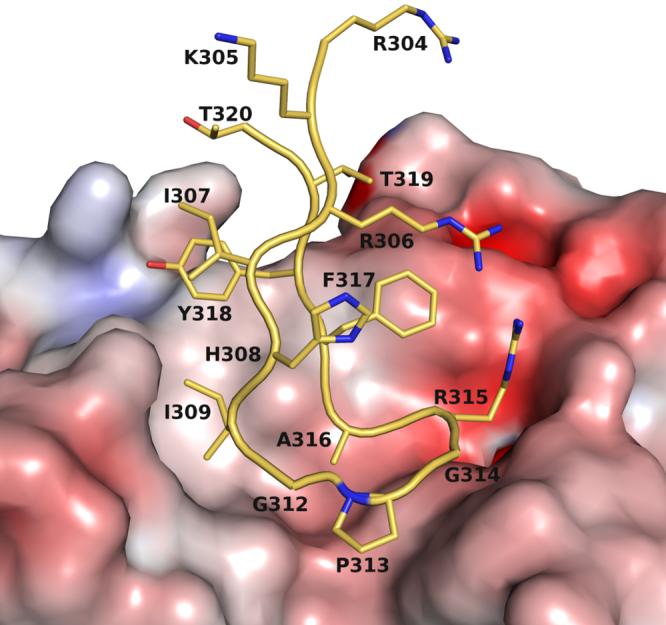
Molecular surface representation of the antigen-binding pocket colored by electrostatic potential, calculated in APBS63, and mapped onto the surface with the color code ranging from -10 kT/e (bright red) to +10 kT/e (dark blue). IleP309 is accommodated in a hydrophobic pocket, whereas ArgP315 forms a salt bridge with AspL92.
The side chain of HisP308 is involved in a crystal contact with CDR H2 via a bound Zn2+ ion, but does not participate in the B4e8-peptide interaction itself and the conformation of HisP308 is similar to those observed in other Fab-V3 complexes15,23,25-28.
Novel V3-conformation
V3 loop structures typically exhibit a β-hairpin conformation with a canonical β-turn for the central GPGR sequence at the tip15,23,25-28. The V3 peptide bound to B4e8 adopts this type of overall hairpin conformation, but forms an α-turn around its apex residues GlyP312 to AlaP316. α-Turns are defined as five residue turns, with a distance between Cα (i) and Cα (i+4) of less than 7 Å and a non-helical conformation40,41. Hence, the B4e8-peptide structure presented here depicts a novel V3 conformation, which does not exhibit the canonical β-turn for the central GPGR sequence at the tip. The extended N-terminal region of the peptide (RP304KRIHIP309) is structurally very similar to other Fab-bound V3-peptides or the gp120 structure, as reflected by the similarity of their dihedral angles. The subsequent loop region (GP312PGRAP316) is twisted, when compared to the 447-52D, 59.1 or the gp120 structures. A similar twist was observed in the 2219 structure, which resulted from a difference in the ψ angle of IleP309. Indeed, in the B4e8 structure, this angle is equivalently perturbed, resulting in conformational similarity of the V3-peptides bound to B4e8 and 2219, but only up to ProP313, as evident by their similar dihedral angles (Table 3, Fig.7).
Table 3.
Main chain torsion angles (°) bound to Fabs B4e8, 2219 and the V3-region in the gp120 core
| Peptide residue | Angle | B4e8 | 2219MN | gp120 | Average deviationa |
|---|---|---|---|---|---|
| RP304 | ϕ | -109 | -108 | 1 | |
| ψ | 51 | 24 | 148 | 49 | |
| KP305 | ϕ | -81 | -75 | -124 | 20 |
| ψ | 151 | -32 | -12 | 83 | |
| RP306 | ϕ | -94 | -61 | -147 | 31 |
| ψ | 120 | 131 | 146 | 9 | |
| IP307 | ϕ | -97 | -110 | -118 | 8 |
| ψ | 121 | 123 | 160 | 17 | |
| HP308 | ϕ | -91 | -94 | -129 | 16 |
| ψ | 126 | 106 | 121 | 8 | |
| IP309 | ϕ | -87 | -93 | -84 | 3 |
| ψ | -43 | -27 | 140 | 82 | |
| GP312 | ϕ | -149 | -157 | -158 | 4 |
| ψ | -169 | -178 | 131 | 25 | |
| PP313 | ϕ | -70 | -55 | -59 | 6 |
| ψ | 149 | 122 | 99 | 17 | |
| GP314 | ϕ | 77 | 70 | 100 | 12 |
| ψ | -136 | 8 | 8 | 96 | |
| RP315 | ϕ | -73 | -123 | -172 | 33 |
| ψ | 5 | 129 | 149 | 60 | |
| AP316 | ϕ | -82 | -147 | -131 | 25 |
| ψ | 66 | 150 | 167 | 41 | |
| FP317 | ϕ | -147 | -117 | -133 | 10 |
| ψ | 161 | 165 | 153 | 4 | |
| YP318 | ϕ | -133 | 53 | 60 | 76 |
| ψ | 151 | 41 | 65 | 44 | |
| TP319 | ϕ | -97 | -80 | -150 | 27 |
| ψ | 156 | 4 | -2 | 69 | |
| TP320 | ϕ | -141 | -62 | -101 | 26 |
| ψ | 47 | 0 |
The average deviation from the mean of the three torsion angles (from B4e8, 2219 and gp120)
Figure 7.

Comparison of V3-peptides RP142 and MN bound to B4e8 (atomic coloring with carbon atoms in yellow) and 2219 (carbon atoms in light blue), respectively. Identical residues in both peptides are labeled only for B4e8 (black). Identical hydrogen bonds are marked by dashed lines and shown only for B4e8. Due to the conversion from a type II β-turn in 2219 to an α-turn in B4e8, one hydrogen bond in the apex of RP142 is lost in the complex with B4e8. The β-hairpin conformation is maintained by a one-residue shift accompanied by a 180° rotation of AlaP316 and all subsequent residues. Residues P304 to P306 and P319 to 320 are omitted for clarity.
At the tip of the loop, residues GlyP312 to AlaP316 form an α-turn with a hydrogen bond between GlyP312 N (i) and AlaP316 O (i+4). A search of the PDB revealed that at least 45 other structures have α-turns with similar dihedral angles. In all V3 structures reported previously, the structural motif around residues GlyP312 to ArgP315 displays a type II or, in the case of 83.1, a type I β-turn. Thus, the structure of the B4e8-peptide complex depicts a novel V3 conformation that highlights its conformational flexibility. The change of the four-residue β-turn (GP312PGRP315) to a five-residue α-turn conformation spanning GlyP312 to AlaP316 is a result of the structural insertion of ArgP315 into the apex of the loop. The much wider turn at the loop tip renders the ArgP315 side chain unusually accessible and close to the CDR L1 and L3 loops, thus permitting numerous interactions with the Fab’ (Fig. 5).
As a result of the insertion of ArgP315 into the turn region of the β-hairpin conformation, the hydrogen bond pattern of the peptide is shifted by one residue compared to other V3-structures. This register shift is most obvious in a comparison of the peptides bound to B4e8 and human mAb 2219, where the position of the side chains of residues P316 to P318 in B4e8 is almost identical to residues P315 to P317 in 2219 (Fig. 7). In the context of gp120, careful inspection of the V3-containing gp120 structure15 suggests that the shift of the hydrogen bond pattern can be accommodated by the flexible stem region without major rearrangements of the conserved V3-base.
The one residue shift is a result of a 180° rotation of the ψ-angle of the central GlyP314 compared to the 2219 or the V3-containing gp120 core structure (Table 3). As a consequence, all subsequent residues are correspondingly rotated, which allows the insertion of ArgP315 into the turn region and the aforementioned displacement in hydrogen bonding pattern. A shift of the β-strands with respect to each other has been observed before in the NMR structure of a V3-peptide derived from the HIV strain IIIB in complex with mAb 447-52D42. Furthermore the B4e8 structure an additional interstrand hydrogen bond is found between LysP305 and ThrP320, thus extending each β-strand by two residues, compared to the 2219 complex structure27.
DISCUSSION
Human mAb B4e8 neutralizes a subset of viruses from HIV-1 subtypes B, C and D31. The structure of the B4e8-Fab’ in complex with the peptide RP142, which comprises the V3-loop sequence of the MN isolate HIV-1 envelope gp120, was determined to provide structural insight into the cross-reactivity of B4e8. Our results illustrate how this antibody can tolerate sequence variability at certain positions within V3. A previous study31 showed that the interaction of mAb B4e8 with the V3 loop is critically affected by mutating IleP309, ArgP315 or PheP317. IleP309 is buried in a hydrophobic pocket formed by CDR H2 and LeuL94. As a consequence, position 309 in V3 requires small, hydrophobic residues, such as Ile, Leu and Val, for high affinity antibody binding. Interestingly, the most commonly found residues at this position in V3 loops are Ile, Leu, Phe and Met43. Computer modeling suggests that the side chains of Phe and Met would be too large to be accommodated by the Fab’ hydrophobic pocket, in good agreement with the inability of B4e8 to neutralize subtype B viruses with a Met at position 30931.
The majority of interactions between B4e8 and V3 involve ArgP315. An Ala or Gln substitution at this position severely reduces the affinity of B4e8 for V331. Computer modeling of the Arg to Gln mutation suggests that Gln could maintain the hydrogen bond with AspL92, although most of the van der Waals contacts with TyrL32 and the salt bridge with AspL92 would not be formed. Interestingly, B4e8 was able to neutralize one subtype C and one subtype D virus, albeit at high concentrations, both of which have Gln at this position31. We speculate that the ability of F425-Be48 to neutralize the two non-B viruses in the previous study could be due to an optimal presentation of the V3 loop on these viruses to the antibody, thus, compensating for the fewer number of contacts with Gln315. The reason for the inability of B4e8 to neutralize the other viruses is not fully clear. It is certainly possible that the V3 region on these other viruses is not accessible to B4e8. Further studies will be required to adequately address this issue.
The third residue critical for B4e8 binding is PheP317. Its side chain participates only weakly in the interaction between Fab’ and peptide (Table 2). However, it is in close contact (∼3.7 Å) with the guanidino side chain of ArgP315 and presumably stabilizes the unusual rotamer conformation of ArgP315 by diminishing its structural flexibility (Fig. 6). Therefore, PheP317 participates in positioning ArgP315 for optimal recognition by B4e8.
The V3 conformation in the complex-structure with B4e8 has not been observed previously and illustrates the conformational flexibility inherent in certain regions of V3. Despite significant structural homology among all eight V3 crystal structures determined so far, it has become apparent that small changes can lead to variable accessibility for the same sets of residues, as well as differential accessibility for different regions of the V3 loop. This scenario can limit and vary the exposure of conserved residues for recognition by potential neutralizing antibodies.
Nevertheless, aside from B4e8, two other moderately broadly neutralizing antibodies, namely 447-52D and 2219, have been elicited against the V3 loop and have been structurally characterized. These three structures reveal three quite distinct modes of recognition for a plethora of viral isolates, through main-chain interactions between the CDR H3 loop and the N-sterminal part of V3 (447-52D), by binding to a conserved face of V3 that allows variation in the tip (2219), or by recognition of a unique V3 conformation (B4e8).
In summary, these results add substantially towards a complete understanding of the V3 loop as a potential target for neutralization of HIV-1. In the future, additional neutralizing and non-neutralizing V3 antibody structures will provide further insights into how such structural information can be utilized with regard to rational design of V3-based antigens that may elicit cross-reactive immune responses.
MATERIALS AND METHODS
Fab’ production and purification
Human IgG B4e8 (IgG2, κ) was prepared as previously reported30. Briefly, mononuclear splenocytes of an HIV-1 infected patient were transformed with Epstein Barr virus for 48 h and fused with HMMA human myeloma cells44. Hybridomas were selected on their specific reactivity with HIV-infected cells and their ability to capture a relatively high degree of virions in a virus capture assay30. Fab’ fragments were prepared by cleavage of B4e8 IgG with 1 % pepsin (w/w) for 1 h at 37°C and subsequent reduction with 1.5 mM dithiothreitol for 4 h at 37°C. The Fab’ fragment was then purified by size exclusion chromatography in 10 mM Hepes, pH 7.5 and 150 mM NaCl on a Superdex 200 16/60 column.
Crystallization and data collection
The peptide (YNKRKRIHIGPGRAFYTTKNIIGC) used for co-crystallization was previously termed RP14245. The B4e8-peptide complex was prepared by mixing the Fab’ and the peptide in a 1:5 molar ratio in 10 mM Hepes, pH 7.5 and 150 mM NaCl and incubating overnight at 4°C. Crystals were grown by sitting drop vapor diffusion. Drops containing 0.3 μl B4e8-peptide complex (9 mg/ml) and 0.3 μl well solution were equilibrated against 0.5 ml well solution of 16 % polyethylene glycol (PEG) 8000, 100 mM MES, pH 6.0, and 150 mM zinc acetate. Crystals appeared after two days and reached their final size of 80 × 20 × 5 μm3 after one week. Data to 2.80 Å resolution were collected at the Stanford Synchrotron Radiation Laboratory (SSRL) beamline 11-1, with an overall Rsym of 10.2 %, due to low overall I/σ, and a completeness of 99.0 %. Data were processed with the HKL2000 package46 (see Table 1 for complete data collection and refinement statistics).
Table 1.
Data collection and refinement statistics
| Data Collection | |
| Wavelength (Å) | 0.979 |
| Resolution (Å) | 2.80 (2.87 - 2.80)a |
| Space group, cell | P2 |
| dimension a, b, c (Å), β (°) | 82.9, 39.9, 98.0, 108.5 |
| Unique reflections | 15,169 (962) |
| Redundancy | 3.7 (3.2) |
| Completeness (%) | 99.0 (94.6) |
| Rsym (%)b | 10.2 (47.6) |
| Avg I/σ | 12.6 (2.3) |
| Refinement statistics for all reflections >0.0 σF | |
| Resolution | 50.0 - 2.80 |
| No. of reflections (working) | 14,414 |
| No. of reflections (test) | 755 |
| Rcryst (%)c | 21.9 (33.0) |
| Rfree (%)d | 26.4 (40.7) |
| No. of Fab atoms | 3,229 |
| No. of peptide atoms | 125 |
| No. of water molecules | 15 |
| No. of ions (Zn2+ / Cl-) | 4 / 4 |
| Overall B value (Å2): | |
| Variable domain | 49.3 |
| Constant domain | 49.4 |
| Peptide | 49.4 |
| Water | 35.9 |
| Ions | 53.6 |
| Sugar | 80.0 |
| Wilson B value (Å2) | 60.8 |
| Ramachandran plote (%) | |
| Favored | 96.7 |
| Allowed | 3.1 |
| Disallowedf | 0.2 |
| Root mean square deviations | |
| Bond length (Å) | 0.005 |
| Angle (°) | 0.95 |
Numbers in parenthesis are for the highest resolution shell.
Rsym = ∑hkl | I - <I> | / ∑hkl | I |
Rcryst = ∑hkl | Fobs - Fcalc | / ∑hkl | Fobs |
Rfree is the same as Rcryst except for 5% of the data excluded from the refinement.
Evaluated with MolProbity 39
ProH149, which is part of a conserved loop region and in clearly interpretable density.
The crystals were indexed in monoclinic space group P2 with unit cell dimensions a=82.9Å, b=39.9Å and c=98.0Å with β-=108.5° and one B4e8-peptide complex in the asymmetric unit. The Matthews’ coefficient47 was 3.0 Å3/Da corresponding to a solvent content of ∼59%.
Structure determination, refinement and analysis
The structure was determined by molecular replacement using MERLOT48 and Phaser49. The former program was used to search a library of 243 Fab molecules to obtain the best fitting variable domain. Phaser was used for the consecutive rotation and translation of the models. The Fab’ constant and variable domains were treated as separate search models using equivalent fragments from PDB entries 1N0X50 and 1JHL51, respectively. After initial rigid body and restrained refinement, Rcryst dropped to 35.4 % with an Rfree of 42.7 %. Several cycles of model building and restrained refinement, with maintenance of the same test set of 5 % of reflections throughout, were carried out using Coot52 and Refmac553, respectively. No interpretable density was found for residues H128 to H136 and residues H196 to H199; therefore, these residues were not built into the model. Final refinement statistics are outlined in Table 1. The quality of the structure was analyzed using the programs MolProbity54 and WHATIF55. Buried molecular surface areas were calculated with the program MS56 using a probe radius of 1.7 Å and standard van der Waals radii57. Shape correlation statistics (Sc) were calculated with CCP4 Sc, version 2.058. Hydrogen bonds were evaluated with the HBPLUS59 and van der Waals contacts were analyzed with Contacsym60,61. The elbow angle was determined to be 134° using RBOW62.
Protein Data Bank accession code
The atomic coordinates and structure factors have been deposited with the RCSB Protein Data Bank and are available under accession code 2QSC.
Supplementary Material
ACKNOWLEDGEMENT
We thank D. Shore and P. Verdino for helpful discussions and comments on the manuscript, H. Doherty for IgG purification, R. Aguilar-Sino and S. Ferguson for help with antibody fragmentation and SSRL Beamline 11-1 for synchrotron beamtime.
We gratefully acknowledge support by NIH grants GM46192 (I.A.W. and R.L.S.), AI33292 (D.R.B.), AI63986 (L.A.C.), the International AIDS Vaccine Initiative (IAVI) Neutralizing Antibody Consortium (I.A.W., D.R.B.), a Skaggs predoctoral fellowship (C.H.B.) and a German Academic Exchange Service (DAAD) fellowship (C.H.B.).
This is manuscript 19052-MB from The Scripps Research Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Wyatt R, Sodroski J. The HIV-1 envelope glycoproteins: fusogens, antigens, and immunogens. Science. 1998;280:1884–1888. doi: 10.1126/science.280.5371.1884. [DOI] [PubMed] [Google Scholar]
- 2.Cardoso RM, Zwick MB, Stanfield RL, Kunert R, Binley JM, Katinger H, Burton DR, Wilson IA. Broadly neutralizing anti-HIV antibody 4E10 recognizes a helical conformation of a highly conserved fusion-associated motif in gp41. Immunity. 2005;22:163–173. doi: 10.1016/j.immuni.2004.12.011. [DOI] [PubMed] [Google Scholar]
- 3.Zwick MB, Jensen R, Church S, Wang M, Stiegler G, Kunert R, Katinger H, Burton DR. Anti-human immunodeficiency virus type 1 (HIV-1) antibodies 2F5 and 4E10 require surprisingly few crucial residues in the membrane-proximal external region of glycoprotein gp41 to neutralize HIV-1. J. Virol. 2005;79:1252–1261. doi: 10.1128/JVI.79.2.1252-1261.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ofek G, Tang M, Sambor A, Katinger H, Mascola JR, Wyatt R, Kwong PD. Structure and mechanistic analysis of the anti-human immunodeficiency virus type 1 antibody 2F5 in complex with its gp41 epitope. J. Virol. 2004;78:10724–10737. doi: 10.1128/JVI.78.19.10724-10737.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Saphire EO, Parren PW, Pantophlet R, Zwick MB, Morris GM, Rudd PM, Dwek RA, Stanfield RL, Burton DR, Wilson IA. Crystal structure of a neutralizing human IgG against HIV-1: a template for vaccine design. Science. 2001;293:1155–1159. doi: 10.1126/science.1061692. [DOI] [PubMed] [Google Scholar]
- 6.Zhou T, Xu L, Dey B, Hessell AJ, Van Ryk D, Xiang SH, Yang X, Zhang MY, Zwick MB, Arthos J, Burton DR, Dimitrov DS, Sodroski J, Wyatt R, Nabel GJ, Kwong PD. Structural definition of a conserved neutralization epitope on HIV-1 gp120. Nature. 2007;445:732–737. doi: 10.1038/nature05580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Calarese DA, Scanlan CN, Zwick MB, Deechongkit S, Mimura Y, Kunert R, Zhu P, Wormald MR, Stanfield RL, Roux KH, Kelly JW, Rudd PM, Dwek RA, Katinger H, Burton DR, Wilson IA. Antibody domain exchange is an immunological solution to carbohydrate cluster recognition. Science. 2003;300:2065–2071. doi: 10.1126/science.1083182. [DOI] [PubMed] [Google Scholar]
- 8.Tugarinov V, Zvi A, Levy R, Hayek Y, Matsushita S, Anglister J. NMR structure of an anti-gp120 antibody complex with a V3 peptide reveals a surface important for co-receptor binding. Structure. 2000;8:385–395. doi: 10.1016/s0969-2126(00)00119-2. [DOI] [PubMed] [Google Scholar]
- 9.Binley JM, Wrin T, Korber B, Zwick MB, Wang M, Chappey C, Stiegler G, Kunert R, Zolla-Pazner S, Katinger H, Petropoulos CJ, Burton DR. Comprehensive cross-clade neutralization analysis of a panel of anti-human immunodeficiency virus type 1 monoclonal antibodies. J. Virol. 2004;78:13232–13252. doi: 10.1128/JVI.78.23.13232-13252.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gorny MK, Williams C, Volsky B, Revesz K, Cohen S, Polonis VR, Honnen WJ, Kayman SC, Krachmarov C, Pinter A, Zolla-Pazner S. Human monoclonal antibodies specific for conformation-sensitive epitopes of V3 neutralize human immunodeficiency virus type 1 primary isolates from various clades. J. Virol. 2002;76:9035–9045. doi: 10.1128/JVI.76.18.9035-9045.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gorny MK, Williams C, Volsky B, Revesz K, Wang XH, Burda S, Kimura T, Konings FA, Nadas A, Anyangwe CA, Nyambi P, Krachmarov C, Pinter A, Zolla-Pazner S. Cross-clade neutralizing activity of human anti-V3 monoclonal antibodies derived from the cells of individuals infected with non-B clades of human immunodeficiency virus type 1. J. Virol. 2006;80:6865–6872. doi: 10.1128/JVI.02202-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Basmaciogullari S, Babcock GJ, Van Ryk D, Wojtowicz W, Sodroski J. Identification of conserved and variable structures in the human immunodeficiency virus gp120 glycoprotein of importance for CXCR4 binding. J. Virol. 2002;76:10791–10800. doi: 10.1128/JVI.76.21.10791-10800.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hwang SS, Boyle TJ, Lyerly HK, Cullen BR. Identification of the envelope V3 loop as the primary determinant of cell tropism in HIV-1. Science. 1991;253:71–74. doi: 10.1126/science.1905842. [DOI] [PubMed] [Google Scholar]
- 14.Rizzuto CD, Wyatt R, Hernandez-Ramos N, Sun Y, Kwong PD, Hendrickson WA, Sodroski J. A conserved HIV gp120 glycoprotein structure involved in chemokine receptor binding. Science. 1998;280:1949–1953. doi: 10.1126/science.280.5371.1949. [DOI] [PubMed] [Google Scholar]
- 15.Huang CC, Tang M, Zhang MY, Majeed S, Montabana E, Stanfield RL, Dimitrov DS, Korber B, Sodroski J, Wilson IA, Wyatt R, Kwong PD. Structure of a V3-containing HIV-1 gp120 core. Science. 2005;310:1025–1028. doi: 10.1126/science.1118398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dragic T. An overview of the determinants of CCR5 and CXCR4 co-receptor function. J. Gen. Virol. 2001;82:1807–1814. doi: 10.1099/0022-1317-82-8-1807. [DOI] [PubMed] [Google Scholar]
- 17.Chen B, Vogan EM, Gong H, Skehel JJ, Wiley DC, Harrison SC. Structure of an unliganded simian immunodeficiency virus gp120 core. Nature. 2005;433:834–841. doi: 10.1038/nature03327. [DOI] [PubMed] [Google Scholar]
- 18.Chen B, Vogan EM, Gong H, Skehel JJ, Wiley DC, Harrison SC. Determining the structure of an unliganded and fully glycosylated SIV gp120 envelope glycoprotein. Structure. 2005;13:197–211. doi: 10.1016/j.str.2004.12.004. [DOI] [PubMed] [Google Scholar]
- 19.Kwong PD, Wyatt R, Majeed S, Robinson J, Sweet RW, Sodroski J, Hendrickson WA. Structures of HIV-1 gp120 envelope glycoproteins from laboratory-adapted and primary isolates. Structure. 2000;8:1329–1339. doi: 10.1016/s0969-2126(00)00547-5. [DOI] [PubMed] [Google Scholar]
- 20.Kwong PD, Wyatt R, Robinson J, Sweet RW, Sodroski J, Hendrickson WA. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature. 1998;393:648–659. doi: 10.1038/31405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ratner L, Fisher A, Jagodzinski LL, Mitsuya H, Liou RS, Gallo RC, Wong-Staal F. Complete nucleotide sequences of functional clones of the AIDS virus. AIDS Res. Hum. Retroviruses. 1987;3:57–69. doi: 10.1089/aid.1987.3.57. [DOI] [PubMed] [Google Scholar]
- 22.Rini JM, Stanfield RL, Stura EA, Salinas PA, Profy AT, Wilson IA. Crystal structure of a human immunodeficiency virus type 1 neutralizing antibody, 50.1, in complex with its V3 loop peptide antigen. Proc. Natl. Acad. Sci. U S A. 1993;90:6325–6329. doi: 10.1073/pnas.90.13.6325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stanfield R, Cabezas E, Satterthwait A, Stura E, Profy A, Wilson I. Dual conformations for the HIV-1 gp120 V3 loop in complexes with different neutralizing Fabs. Structure. 1999;7:131–142. doi: 10.1016/s0969-2126(99)80020-3. [DOI] [PubMed] [Google Scholar]
- 24.Ghiara JB, Ferguson DC, Satterthwait AC, Dyson HJ, Wilson IA. Structure-based design of a constrained peptide mimic of the HIV-1 V3 loop neutralization site. J. Mol. Biol. 1997;266:31–39. doi: 10.1006/jmbi.1996.0768. [DOI] [PubMed] [Google Scholar]
- 25.Ghiara JB, Stura EA, Stanfield RL, Profy AT, Wilson IA. Crystal structure of the principal neutralization site of HIV-1. Science. 1994;264:82–85. doi: 10.1126/science.7511253. [DOI] [PubMed] [Google Scholar]
- 26.Stanfield RL, Ghiara JB, Ollmann Saphire E, Profy AT, Wilson IA. Recurring conformation of the human immunodeficiency virus type 1 gp120 V3 loop. Virology. 2003;315:159–173. doi: 10.1016/s0042-6822(03)00525-7. [DOI] [PubMed] [Google Scholar]
- 27.Stanfield RL, Gorny MK, Zolla-Pazner S, Wilson IA. Crystal structures of human immunodeficiency virus type 1 (HIV-1) neutralizing antibody 2219 in complex with three different V3 peptides reveal a new binding mode for HIV-1 cross-reactivity. J. Virol. 2006;80:6093–6105. doi: 10.1128/JVI.00205-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stanfield RL, Gorny MK, Williams C, Zolla-Pazner S, Wilson IA. Structural rationale for the broad neutralization of HIV-1 by human monoclonal antibody 447-52D. Structure. 2004;12:193–204. doi: 10.1016/j.str.2004.01.003. [DOI] [PubMed] [Google Scholar]
- 29.Galanakis PA, Spyroulias GA, Rizos A, Samolis P, Krambovitis E. Conformational properties of HIV-1 gp120/V3 immunogenic domains. Curr. Med. Chem. 2005;12:1551–1568. doi: 10.2174/0929867054038982. [DOI] [PubMed] [Google Scholar]
- 30.Cavacini L, Duval M, Song L, Sangster R, Xiang SH, Sodroski J, Posner M. Conformational changes in env oligomer induced by an antibody dependent on the V3 loop base. AIDS. 2003;17:685–689. doi: 10.1097/00002030-200303280-00006. [DOI] [PubMed] [Google Scholar]
- 31.Pantophlet R, Aguilar-Sino RO, Wrin T, Cavacini LA, Burton DR. Analysis of the neutralization breadth of the anti-V3 antibody F425-B4e8 and re-assessment of its epitope fine specificity by scanning mutagenesis. Virology. 2007;364:441–453. doi: 10.1016/j.virol.2007.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kabat EA, Wu TT, Perry HM, Gottesman KS, Foeller C. Sequences of Proteins of Immunological Interest. 5th edit. National Institutes of Health; Bethesda, MD: 1991. [Google Scholar]
- 33.Al-Lazikani B, Lesk AM, Chothia C. Standard conformations for the canonical structures of immunoglobulins. J. Mol. Biol. 1997;273:927–948. doi: 10.1006/jmbi.1997.1354. [DOI] [PubMed] [Google Scholar]
- 34.Martin AC, Thornton JM. Structural families in loops of homologous proteins: automatic classification, modelling and application to antibodies. J. Mol. Biol. 1996;263:800–815. doi: 10.1006/jmbi.1996.0617. [DOI] [PubMed] [Google Scholar]
- 35.Vargas-Madrazo E, Paz-Garcia E. Modifications to canonical structure sequence patterns: Analysis for L1 and L3. Proteins. 2002;47:250–254. doi: 10.1002/prot.10187. [DOI] [PubMed] [Google Scholar]
- 36.Shirai H, Kidera A, Nakamura H. Structural classification of CDR-H3 in antibodies. FEBS Lett. 1996;399:1–8. doi: 10.1016/s0014-5793(96)01252-5. [DOI] [PubMed] [Google Scholar]
- 37.Huang L, Biolsi S, Bales KR, Kuchibhotla U. Impact of variable domain glycosylation on antibody clearance: an LC/MS characterization. Anal. Biochem. 2006;349:197–207. doi: 10.1016/j.ab.2005.11.012. [DOI] [PubMed] [Google Scholar]
- 38.Qian J, Liu T, Yang L, Daus A, Crowley R, Zhou Q. Structural characterization of N-linked oligosaccharides on monoclonal antibody cetuximab by the combination of orthogonal matrix-assisted laser desorption/ionization hybrid quadrupole-quadrupole time-of-flight tandem mass spectrometry and sequential enzymatic digestion. Anal. Biochem. 2007;364:8–18. doi: 10.1016/j.ab.2007.01.023. [DOI] [PubMed] [Google Scholar]
- 39.Stanfield RL, Wilson IA. Protein-peptide interactions. Curr. Opin. Struct. Biol. 1995;5:103–113. doi: 10.1016/0959-440x(95)80015-s. [DOI] [PubMed] [Google Scholar]
- 40.Dasgupta B, Pal L, Basu G, Chakrabarti P. Expanded turn conformations: Characterization and sequence-structure correspondence in α-turns with implications in helix folding. Proteins. 2004;44:7250–7258. doi: 10.1002/prot.20064. [DOI] [PubMed] [Google Scholar]
- 41.Chou KC. Prediction of tight turns and their types in proteins. Anal. Biochem. 2000;286:1–16. doi: 10.1006/abio.2000.4757. [DOI] [PubMed] [Google Scholar]
- 42.Rosen O, Chill J, Sharon M, Kessler N, Mester B, Zolla-Pazner S, Anglister J. Induced fit in HIV-neutralizing antibody complexes: evidence for alternative conformations of the gp120 V3 loop and the molecular basis for broad neutralization. Biochemistry. 2005;44:7250–7258. doi: 10.1021/bi047387t. [DOI] [PubMed] [Google Scholar]
- 43.Kuiken CL, Foley B, Hahn B, Marx PA, McCutchan F, Mellors JW, Mullins JI, Wolinsky S, Korber BE, Theoretical Biology and Biophysics Group . Human retroviruses and AIDS 1999: a compilation and analysis of nucleic acid and amino acid sequences. Los Alamos National Laboratory; Los Alamos, N. Mex.: 1999. [Google Scholar]
- 44.Posner MR, Barrach HJ, Elboim HS, Nivens K, Santos DJ, Chichester CO, Lally EV. The generation of hybridomas secreting human monoclonal antibodies reactive with type II collagen. Hybridoma. 1989;8:187–197. doi: 10.1089/hyb.1989.8.187. [DOI] [PubMed] [Google Scholar]
- 45.Javaherian K, Langlois AJ, LaRosa GJ, Profy AT, Bolognesi DP, Herlihy WC, Putney SD, Matthews TJ. Broadly neutralizing antibodies elicited by the hypervariable neutralizing determinant of HIV-1. Science. 1990;250:1590–1593. doi: 10.1126/science.1703322. [DOI] [PubMed] [Google Scholar]
- 46.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276A:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 47.Matthews BW. Determination of protein molecular weight, hydration, and packing from crystal density. Methods Enzymol. 1985;114:176–187. doi: 10.1016/0076-6879(85)14018-8. [DOI] [PubMed] [Google Scholar]
- 48.Fitzgerald PMD. MERLOT, an integrated package of computer programs for the determination of crystal structures by molecular replacement. J. Appl. Crystallogr. 1988;21:273–278. [Google Scholar]
- 49.McCoy AJ, Grosse-Kunstleve RW, Storoni LC, Read RJ. Likelihood-enhanced fast translation functions. Acta Crystallogr. Biol. Crystallogr. 2005;D61:458–464. doi: 10.1107/S0907444905001617. [DOI] [PubMed] [Google Scholar]
- 50.Saphire EO, Montero M, Menendez A, van Houten NE, Irving MB, Pantophlet R, Zwick MB, Parren PW, Burton DR, Scott JK, Wilson IA. Structure of a high-affinity “mimotope” peptide bound to HIV-1-neutralizing antibody b12 explains its inability to elicit gp120 cross-reactive antibodies. J. Mol. Biol. 2007;369:696–709. doi: 10.1016/j.jmb.2007.01.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chitarra V, Alzari PM, Bentley GA, Bhat TN, Eisele JL, Houdusse A, Lescar J, Souchon H, Poljak RJ. Three-dimensional structure of a heteroclitic antigen-antibody cross-reaction complex. Proc. Natl. Acad. Sci. USA. 1993;90:7711–7715. doi: 10.1073/pnas.90.16.7711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. Biol. Crystallogr. 2004;D60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 53.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. Biol. Crystallogr. 1997;D53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 54.Lovell SC, Davis IW, Arendall WB, 3rd, de Bakker PI, Word JM, Prisant MG, Richardson JS, Richardson DC. Structure validation by Calpha geometry: phi,psi and Cbeta deviation. Proteins. 2003;50:437–450. doi: 10.1002/prot.10286. [DOI] [PubMed] [Google Scholar]
- 55.Vriend G. WHAT IF: a molecular modeling and drug design program. J. Mol. Graph. 1990;8:52–56. doi: 10.1016/0263-7855(90)80070-v. [DOI] [PubMed] [Google Scholar]
- 56.Connolly ML. The molecular surface package. J. Mol. Graph. 1993;11:139–141. doi: 10.1016/0263-7855(93)87010-3. [DOI] [PubMed] [Google Scholar]
- 57.Gelin BR, Karplus M. Side-chain torsional potentials: effect of dipeptide, protein, and solvent environment. Biochemistry. 1979;18:1256–1268. doi: 10.1021/bi00574a022. [DOI] [PubMed] [Google Scholar]
- 58.Lawrence MC, Colman PM. Shape complementarity at protein/protein interfaces. J. Mol. Biol. 1993;234:946–950. doi: 10.1006/jmbi.1993.1648. [DOI] [PubMed] [Google Scholar]
- 59.McDonald IK, Thornton JM. Satisfying hydrogen bonding potential in proteins. J. Mol. Biol. 1994;238:777–793. doi: 10.1006/jmbi.1994.1334. [DOI] [PubMed] [Google Scholar]
- 60.Sheriff S, Hendrickson WA, Smith JL. Structure of myohemerythrin in the azidomet state at 1.7/1.3 Ǻ resolution. J. Mol. Biol. 1987;197:273–296. doi: 10.1016/0022-2836(87)90124-0. [DOI] [PubMed] [Google Scholar]
- 61.Sheriff S, Silverton EW, Padlan EA, Cohen GH, Smith-Gill SJ, Finzel BC, Davies DR. Three-dimensional structure of an antibody-antigen complex. Proc. Natl. Acad. Sci. USA. 1987;84:8075–8079. doi: 10.1073/pnas.84.22.8075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Stanfield RL, Zemla A, Wilson IA, Rupp B. Antibody elbow angles are influenced by their light chain class. J. Mol. Biol. 2006;357:1566–1574. doi: 10.1016/j.jmb.2006.01.023. [DOI] [PubMed] [Google Scholar]
- 63.Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. USA. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.