

Abstract
Cytokines are an integral part of the adaptive and innate immune responses. The signalling pathways triggered by receptor engagement translate exposure to cytokine into a coordinated biological response. To contain these responses, the initiation, duration and magnitude of the signal is controlled at multiple levels. SOCS (suppressor of cytokine signalling) proteins act in a negative feedback loop to inhibit signal transduction. Mice with a deletion of SOCS3 die at midgestion due to placental insufficiency. SOCS3-null placentae have increased numbers of mature trophoblast giant cells, disruption of the labyrinthine layer and a decrease in the spongiotrophoblast layer. Genetic crosses have revealed that the phenotype is due to dysregulation of signalling downstream of the leukaemia inhibitory factor (LIF) receptor alpha (LIFRα) and that the ligand responsible for this, LIF, is produced by embryonic tissues and acts in a paracrine fashion. These observations highlight the role of LIF as an extrinsic factor regulating trophoblast differentiation in vivo. The creation of mice with conditional deletion of SOCS3 in different tissues has also uncovered critical roles for SOCS3 in the regulation of IL-6, G-CSF and leptin signalling.
1. Introduction: the SOCS proteins
In recent years, the Suppressor of Cytokine signalling (SOCS) proteins, which were first identified as rapidly inducible negative regulators of cytokine signalling, have emerged as key modulators of the cellular response to diverse biological stimuli. The eight SOCS family proteins (SOCS1-7 and CIS) are characterised by structural features that include a conserved C-terminal SOCS box, a central SH2 domain and an N-terminal region of variable length and sequence (Hilton et al., 1998). The paradigm for specific inhibition of cytokine signalling by SOCS proteins is as follows. Cytokine binding induces receptor oligomerization, allowing transphosphorylation and activation of cytoplasmic kinase proteins called Janus Kinases (JAKs). JAKs phosphorylate tyrosine residues within the receptor cytoplasmic domain leading to the recruitment of Signal Transducer and Activators of Transcription (STAT) proteins. Phosphorylated STAT dimers translocate to the nucleus where they mediate cytokine-regulated gene transcription, including transcription of SOCS genes. SOCS proteins are recruited to specific phosphotyrosine residues in activated cytokine receptors or in JAKs and inhibit their activity. In addition, the SOCS box can interact with elongins B and C, proteins that form part of an E3 ubiquitin ligase complex that polyubiquitinates proteins, targeting them for proteasomal degradation (Kamura et al., 1998; Zhang et al., 1999). Thus, the SOCS proteins regulate the duration of the cytokine signalling response by both specific and non-specific regulatory mechanisms.
Transcription of SOCS3 is induced in numerous cell types by a diverse array of cytokines and other stimuli. In addition, overexpression of SOCS3 in cell lines has shown SOCS can inhibit many cytokine-induced responses (Alexander and Hilton, 2004). SOCS3 interacts with gp130, a common cytokine receptor for interleukin (IL)-6, LIF and other members of the IL-6 family of cytokines, and the interaction site has been mapped to phosphotyrosine (pTyr) 757 (Nicholson et al., 2000; Schmitz et al., 2000). SOCS3 has been reported also to interact with JAK2, the leptin receptor, erythropoietin receptor, insulin receptor, IL-12β receptor and granulocyte-colony stimulating factor receptor (Bjorbak et al., 2000; Emanuelli et al., 2000; Eyckerman et al., 2000; Hortner et al., 2002; Sasaki et al., 1999, 2000; Ueki et al., 2004a; Yamamoto et al., 2003). Studies to date have focussed mainly on the role of SOCS3 as an inhibitor of the JAK-STAT pathway; however, there is evidence that SOCS3 can regulate also the stability of signalling molecules including IRS proteins, the RAS inhibitor RasGAP and the adaptor proteins Nck and Crk-L.
To dissect the in vivo function and specificity of SOCS proteins, our laboratory has focussed on gene deletion studies in mice. The results have revealed remarkable cytokine specificity for different SOCS molecules (reviewed in Alexander and Hilton, 2004). Here, we review the results of studies of murine models with genetic deletion of SOCS3, focussing on the role of SOCS3 in placentation.
2. SOCS3-null embryos die of placental insufficiency
Genetic deletion of SOCS3 resulted in embryonic lethality due to placental insufficiency at around embryonic day (E)13 (Marine et al., 1999; Roberts et al., 2001; Takahashi et al., 2003). In the SOCS3 null placenta, chorio-allantoic fusion occurred normally, but the labyrinthine and spongiotrophoblast layers of the placenta were poorly formed, whilst trophoblast giant cells were increased in number and in size. Consistent with these histological abnormalities, cells expressing Pl-1 or Limk, genes expressed in trophoblast giant cells, were increased in number whilst cells expressing markers of spongiotrophoblast, such as Tpbp, were decreased. Expression of the helix-loop-helix transcription factor Hand1, which promotes differentiation of trophoblast stem cells, was increased whilst that of Mash1, which maintains trophoblast stem cells, was decreased. SOCS3 expression was high in cells at the edge of the chorionic plate, a region thought to give rise to syncytiotrophoblast cells, and was also detectable in labyrinthine, spongiotrophoblast and trophoblast giant cells and in fetal vasculature of the developing placenta (Roberts et al., 2001; Takahashi et al., 2003). The embryonic lethality associated with absence of SOCS3 could be rescued by provision of wild type extraembryonic tissues, using either complementation via aggregation with tetraploid embryos (which contribute to extra-embryonic tissues but not to embryo proper) or generation of chimeras composed of SOCS3-null embryos and wild-type trophoblast stem cells (Takahashi et al., 2003, 2006). These experiments established definitively that the embryonic lethality due to absence of SOCS3 was caused by placental insufficiency. Moreover, initial reports that SOCS3-null embryos had excess erythrocytosis were not supported by later analyses (Roberts et al., 2001; Takahashi et al., 2003).
The abnormalities observed in SOCS3-null placentae suggested a perturbation in trophoblast differentiation. This hypothesis was supported by experiments using the rat trophoblast cell line Rcho-1. The Rcho-1 cell line can be induced to differentiate to trophoblast giant cells in culture. SOCS3 expression was high in undifferentiated cells and decreased following induction of differentiation. In Rcho-1 subclones constitutively expressing SOCS3, differentiation, manifest by changes in gene expression, DNA endoreduplication, actin filament reorganisation and sensitivity to trypsin, was suppressed. Moreover, trophoblast stem cell lines lacking SOCS3 underwent precocious differentiation, even when maintained in FGF4 and conditioned media (Takahashi et al., 2003).
3. Genetic reduction of embryonic LIF signalling rescues placentation in SOCS3-null embryos
Using a genetic cross, Takahashi et al (2003) demonstrated that the defects in SOCS3-null placentae could be rescued by deletion of the LIFRα chain. LIFRα–null embryos were reported to die in the perinatal period with multi-system defects, including abnormal placentation. In contrast to SOCS3 placentae, the number of trophoblast giant cell numbers were decreased in LIFRα–null placentae (Ware et al., 1995). Mice heterozygous for deletion of SOCS3 and LIFRα were intercrossed and the offspring examined at E19, just prior to birth. Unlike SOCS3-null embryos, which died by E13, SOCS3-null/LIFRα–null embryos survived to E19 and histological analysis revealed a marked reduction in placental abnormalities (Takahashi et al., 2003).
Multiple IL-6-type cytokines including cardiotropin-1, neuropoietin, LIF, oncostatin M, ciliary neurotropic factor and cardiotropin-like cytokine-C can signal via the LIFRα chain which, upon ligand binding, complexes with gp130. A major signalling cascade activated by the LIFRα-gp130 receptor complex is the JAK-STAT pathway. To test if dysregulated LIF signals were responsible for the SOCS3-null placental phenotype, mice with heterozygosity for a null mutation of the LIF gene and for SOCS3 were intercrossed and E19 offspring were examined. E19 SOCS3-null/LIF-null embryos were present in expected numbers and histological examination of their placentae showed they were indistinguishable from the placentae of wild-type littermates. This established that, in vivo, absence of SOCS3 leads to dysregulated signalling in response to embryonic LIF production and that this alters trophoblast differentiation (Figure 1). The source of LIF production by embryonic tissues has not been established. Underscoring the exquisite sensitivity of trophoblast differentiation to LIF, it was observed that SOCS3-null embryos were also rescued if LIF signalling was reduced by heterozygosity for the null allele. SOCS3-null/LIF-heterozygous embryos appeared normal, but the placental histology was mildly abnormal, with increased trophoblast giant cells and reduced, abnormally distributed spongiotrophoblast cells (Robb et al., 2005).
Figure 1.
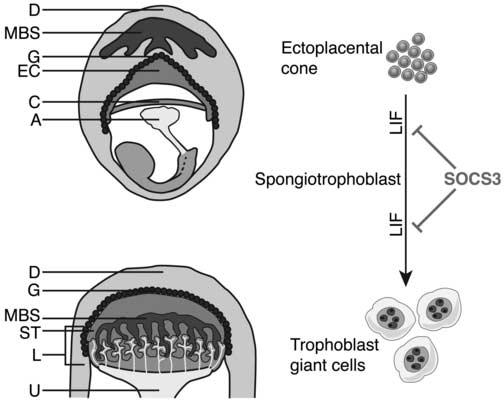
Precursor trophoblast cells within the ectoplacental cone (EC) mature to form the spongiotrophoblast layer of the placenta. Diagrammatic representation of an E8 embryo and extraembryonic tissues within the maternal decidua (top) and an E10 placenta (bottom). The spongiotrophoblast layer lies between the outer secondary trophoblast giant cells and the inner labyrinthine layer. (Some trophoblast giant cells may arise from a cell type within the ectoplacental cone that does not express spongiotrophoblast markers [not shown] (Simmons and Cross, 2005). LIF acts to promote differentiation of trophoblasts to giant cells and SOCS3 negatively regulates signalling downstream of LIF. A, allantois; C, chorionic plate; D, decidua; EC, ectoplacental cone; G, trophoblast giant cells; L, labyrinth; ST, spongiotrophoblast; U, umbilicus; MBS, maternal blood supply
4. SOCS3 expression in human placental tissues
Normal placental function is crucial for pregnancy success. In addition, substantial evidence supports the idea that a compromised in utero environment may not only influence fetal wellbeing but also affect human health in postnatal life (Barker et al., 1989). Comparative studies of mouse and human placentae demonstrate striking similarities in cellular mechanisms and tissue framework (Georgiades et al., 2002). Trophoblast generates surfaces for nutrient exchange and provides a barrier between maternal and fetal circulation. In addition, trophoblast cells interact with the uterus and produce hormones that affect maternal physiology and placental blood flow. Could alterations in trophoblast caused by changes in SOCS3 expression or function be responsible for some human placental pathologies? SOCS1, SOCS2 and SOCS3 mRNA and protein are detectable in preterm and term villous placenta and immunohistochemical analysis localised all three SOCS proteins to all decidual cells (Blumenstein et al., 2002, 2005). LIFRα expression was detected in villous and extravillous trophoblast throughout pregnancy and LIF expression was found in decidual leukocytes at the implantation site (Sharkey et al., 1999). Therefore, it would be valuable to examine alterations in SOCS3 expression or function in human placental disease.
5. Conditional deletion reveals additional key physiological regulatory roles for SOCS3
A conditional gene targeting strategy using the Cre recombinase and loxP system has enabled the generation of viable mice with tissue-specific deletion of SOCS3. Liver-specific or macrophage-specific deletion of SOCS3 resulted in prolonged activation of STAT3 in hepatocytes following IL-6 stimulation, indicating that SOCS3 negatively regulates IL-6 responses in vivo. In the absence of SOCS3, IL-6 also prolonged STAT1 phosphorylation which increased the upregulation of IFNγ-responsive genes, suggesting that, in addition to regulating the duration of signalling, SOCS3 may also regulate the specificity and balance of the biological response (Croker et al., 2003; Lang et al., 2003; Yasukawa et al., 2003).
Mice with a neuronal-specific deletion of SOCS3, or heterozygosity for a SOCS3-null allele, have clarified a role for SOCS3 in endocrine systems (Howard et al., 2004; Mori et al., 2004). Prolonged, leptin-stimulated activation of STAT3 was observed in neurons from mice with a brain-specific deletion of SOCS3 and these mice exhibited elevated leptin sensitivity and resistance to diet-induced obesity. SOCS3 has been shown to associate with the insulin receptor and SOCS1 and SOCS3 are involved in degradation of IRS-1 and IRS-2. In keeping with this, adipocytes established from SOCS3-deficient mice fibroblasts were significantly protected from TNF-α-induced insulin resistance, although a role of SOCS3 in systemic insulin resistance was not shown (reviewed in (Howard and Flier, 2006)).
Compared with wild-type mice, systemic administration of granulocyte(G)-CSF to mice with deletion of SOCS3 in haemopoietic and endothelial cells resulted in increased neutrophilia, splenomegaly and neutrophil infiltrates in multiple tissues, indicating that SOCS3 is also a key negative regulator of G-CSF signalling (Croker et al., 2004).
In murine autoimmune and inflammatory disease models, manipulation of local or whole animal levels of SOCS3 has been shown to affect disease severity. For example, increased expression of SOCS3 in the liver of wild type mice caused insulin resistance and metabolic syndrome, whilst inhibition of SOCS3 ameliorated disease in obese diabetic mice (Ueki et al., 2004b). Conversely, mice lacking SOCS3 in haematopoietic cells showed marked exacerbation of disease in an IL-1-dependent inflammatory arthritis model whilst intraarticular administration of SOCS3 protected against experimental arthritis (Shouda et al., 2001; Wong et al., 2006).
6. Molecular mechanism of SOCS3 regulation of LIF signalling
When the response to LIF signalling was measured in tissues of SOCS-null mice, prolonged STAT1/3 phosphorylation was consistently observed. STAT1/3-activation is one of a number of signalling pathways activated downstream of gp130. In SOCS3 null embryonic stem (ES) cells, LIF stimulation not only induced increased and prolonged STAT1/3 phosphorylation but also sustained induction of SHP-2 phosphorylation, leading to increased activation of the Ras-ERK1/2-MAPK signalling pathway (Forrai et al., 2006). SOCS3 and SHP-2 both bind to pTyr757 in gp130, therefore in SOCS3-null ES cells the absence of competition for binding to this activated residue resulted in increased SHP-2 activation (Figure 2). STAT1/3 and SHP-2-Ras-ERK signalling via gp130 are key regulators of cellular homeostasis. In SOCS3-null ES cells, alteration of this balance resulted in a spontaneous differentiation of the cells to endoderm, despite LIF addition, a phenotypic change that was prevented by addition of a Ras-MAPK inhibitor. Absence of SOCS3 in the trophoblast cells of the placenta may also perturb the balance of multiple signalling cascades downstream of the receptor. Preliminary evidence to support this hypothesis comes from our examination of the placentae of mice bearing a mutation of pTyr757 of gp130 (Tebbutt et al., 2002; L Robb, M Ernst, unpublished data). These placentae appear normal, suggesting that, as in SOCS3-null ES cells, increased Ras-MAPK signalling may affect the biological outcome of trophoblast differentiation in response to LIF.
Figure 2.
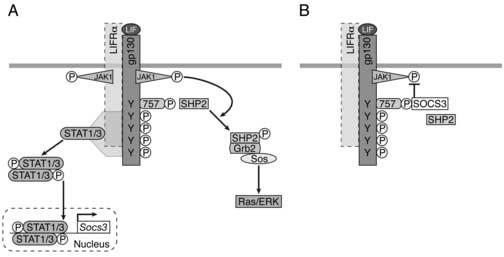
SOCS3 negatively regulates signalling via gp130. A) Binding of LIF to the multimeric receptor complex of LIFRα and gp130 results in transphosphorylation of constitutively associated JAK family kinases, leading to phosphorylation of receptor tyrosines and activation of two major signalling cascades: STAT1/3 and SHP2-Ras-ERK-MAPK. STAT1 and STAT3 bind to phosphorylated tyrosines on gp130 and LIFRα, become phosphorylated, form dimers and translocate to the nucleus where they bind DNA and activate target genes including SOCS3. Receptor-bound SHP-2 is tyrosine phosphorylated by JAKs, binds to Grb2, which is constitutively associated with the GDP/GTP Ras-exchange protein SOS, and GTP-bound Ras triggers ERK-MAPK activation. B) SOCS3 binds pTyr 757 on gp130, inhibiting JAK kinase activity and downregulating STAT1/3 signalling. In addition, SOCS3 may promote proteosomal degradation of ligand-occupied receptor complexes. SHP-2, which also binds to pTyr 757, is outcompeted by SOCS3 binding to this residue, resulting in attenuation of ERK-MAPK signalling. In the absence of SOCS3, there is prolonged activity of both pathways. For simplicity, signalling via LIFRα is not shown.
Conclusion
Gene deletion studies in mice have helped pinpoint key roles for SOCS3 in maintaining the balance between the beneficial and harmful effects of cytokine signalling. The key role for LIF signalling during murine placental development identifies this factor as an extrinsic regulator of trophoblast differentiation. Experiments in SOCS3-null ES cells show that absence of SOCS3 results in increased activation of both JAK/STAT and Ras-ERK1/2-MAPK signalling pathways in response to LIF and that this results in cellular differentiation. Further experiments will determine how dysregulated LIF signalling affects downstream signalling pathways and alters transcription of target genes in trophoblast cells.
Acknowledgements
Work in the authors' laboratory was funded by the National Health and Medical Research Council of Australia program grant 257500 and NIH grant CA22556.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alexander WS, Hilton DJ. The role of suppressors of cytokine signaling (SOCS) proteins in regulation of the immune response. Annu. Rev. Immunol. 2004;22:503–529. doi: 10.1146/annurev.immunol.22.091003.090312. [DOI] [PubMed] [Google Scholar]
- Barker DJ, Osmond C, Golding J, Kuh D, Wadsworth ME. Growth in utero, blood pressure in childhood and adult life, and mortality from cardiovascular disease. Brit. Med. J. 1989;298:564–567. doi: 10.1136/bmj.298.6673.564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjorbak C, Lavery HJ, Bates SH, Olson RK, Davis SM, Flier JS, Myers MG., Jr. SOCS3 mediates feedback inhibition of the leptin receptor via Tyr985. J. Biol. Chem. 2000;275:40649–40657. doi: 10.1074/jbc.M007577200. [DOI] [PubMed] [Google Scholar]
- Blumenstein M, Bowen-Shauver JM, Keelan JA, Mitchell MD. Identification of suppressors of cytokine signaling (SOCS) proteins in human gestational tissues: differential regulation is associated with the onset of labor. J. Clin. Endocrinol. Metab. 2002;87:1094–1097. doi: 10.1210/jcem.87.3.8463. [DOI] [PubMed] [Google Scholar]
- Blumenstein M, Keelan JA, Bowen-Shauver JM, Mitchell MD. Suppressors of cytokine signaling proteins in human preterm placental tissues. J Mol Endocrinol. 2005;35:165–175. doi: 10.1677/jme.1.01767. [DOI] [PubMed] [Google Scholar]
- Croker BA, Krebs DL, Zhang JG, Wormald S, Willson TA, Stanley EG, Robb L, Greenhalgh CJ, Forster I, Clausen BE, et al. SOCS3 negatively regulates IL-6 signaling in vivo. Nat. Immunol. 2003;4:540–545. doi: 10.1038/ni931. [DOI] [PubMed] [Google Scholar]
- Croker BA, Metcalf D, Robb L, Wei W, Mifsud S, DiRago L, Cluse LA, Sutherland KD, Hartley L, Williams E, et al. SOCS3 is a critical physiological negative regulator of G-CSF signaling and emergency granulopoiesis. Immunity. 2004;20:153–165. doi: 10.1016/s1074-7613(04)00022-6. [DOI] [PubMed] [Google Scholar]
- Emanuelli B, Peraldi P, Filloux C, Sawka-Verhelle D, Hilton D, Van Obberghen E. SOCS-3 is an insulin-induced negative regulator of insulin signaling. J. Biol. Chem. 2000;275:15985–15991. doi: 10.1074/jbc.275.21.15985. [DOI] [PubMed] [Google Scholar]
- Eyckerman S, Broekaert D, Verhee A, Vandekerckhove J, Tavernier J. Identification of the Y985 and Y1077 motifs as SOCS3 recruitment sites in the murine leptin receptor. FEBS Lett. 2000;486:33–37. doi: 10.1016/s0014-5793(00)02205-5. [DOI] [PubMed] [Google Scholar]
- Forrai A, Boyle K, Hart AH, Hartley L, Rakar S, Willson TA, Simpson KM, Roberts AW, Alexander WS, Voss AK, Robb L. Absence of suppressor of cytokine signalling 3 reduces self-renewal and promotes differentiation in murine embryonic stem cells. Stem Cells. 2006;24:604–614. doi: 10.1634/stemcells.2005-0323. [DOI] [PubMed] [Google Scholar]
- Georgiades P, Ferguson-Smith AC, Burton GJ. Comparative developmental anatomy of the murine and human definitive placentae. Placenta. 2002;23:3–19. doi: 10.1053/plac.2001.0738. [DOI] [PubMed] [Google Scholar]
- Hilton DJ, Richardson RT, Alexander WS, Viney EM, Willson TA, Sprigg NS, Starr R, Nicholson SE, Metcalf D, Nicola NA. Twenty proteins containing a C-terminal SOCS box form five structural classes. Proc. Natl. Acad. Sci. USA. 1998;95:114–119. doi: 10.1073/pnas.95.1.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hortner M, Nielsch U, Mayr LM, Johnston JA, Heinrich PC, Haan S. Suppressor of cytokine signaling-3 is recruited to the activated granulocyte-colony stimulating factor receptor and modulates its signal transduction. J. Immunol. 2002;169:1219–1227. doi: 10.4049/jimmunol.169.3.1219. [DOI] [PubMed] [Google Scholar]
- Howard JK, Cave BJ, Oksanen LJ, Tzameli I, Bjorbaek C, Flier JS. Enhanced leptin sensitivity and attenuation of diet-induced obesity in mice with haploinsufficiency of SOCS3. Nat. Med. 2004;10:734–738. doi: 10.1038/nm1072. [DOI] [PubMed] [Google Scholar]
- Howard JK, Flier JS. Attenuation of leptin and insulin signaling by SOCS proteins. Trends Endocrinol. Metab. 2006;17:365–371. doi: 10.1016/j.tem.2006.09.007. [DOI] [PubMed] [Google Scholar]
- Kamura T, Sato S, Haque D, Liu L, Kaelin WG, Jr., Conaway RC, Conaway JW. The elongin BC complex interacts with the conserved SOCS-box motif present in members of the SOCS, ras, WD-40 repeat, and ankyrin repeat families. Genes Dev. 1998;12:3872–3881. doi: 10.1101/gad.12.24.3872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang R, Pauleau AL, Parganas E, Takahashi Y, Mages J, Ihle JN, Rutschman R, Murray PJ. SOCS3 regulates the plasticity of gp130 signaling. Nat. Immunol. 2003;4:546–550. doi: 10.1038/ni932. [DOI] [PubMed] [Google Scholar]
- Marine JC, McKay C, Wang D, Topham DJ, Parganas E, Nakajima H, Pendeville H, Yasukawa H, Sasaki A, Yoshimura A, Ihle JN. SOCS3 is essential in the regulation of fetal liver erythropoiesis. Cell. 1999;98:617–627. doi: 10.1016/s0092-8674(00)80049-5. [DOI] [PubMed] [Google Scholar]
- Mori H, Hanada R, Hanada T, Aki D, Mashima R, Nishinakamura H, Torisu T, Chien KR, Yasukawa H, Yoshimura A. SOCS3 deficiency in the brain elevates leptin sensitivity and confers resistance to diet-induced obesity. Nat. Med. 2004;10:739–743. doi: 10.1038/nm1071. [DOI] [PubMed] [Google Scholar]
- Nicholson SE, De Souza D, Fabri LJ, Corbin J, Willson TA, Zhang JG, Silva A, Asimakis M, Farley A, Nash AD, et al. Suppressor of cytokine signaling-3 preferentially binds to the SHP-2-binding site on the shared cytokine receptor subunit gp130. Proc. Natl. Acad. Sci. USA. 2000;97:6493–6498. doi: 10.1073/pnas.100135197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robb L, Boyle K, Rakar S, Hartley L, Lochland J, Roberts AW, Alexander WS, Metcalf D. Genetic reduction of embryonic leukemia-inhibitory factor production rescues placentation in SOCS3-null embryos but does not prevent inflammatory disease. Proc. Natl. Acad. Sci. USA. 2005;102:16333–16338. doi: 10.1073/pnas.0508023102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts AW, Robb L, Rakar S, Hartley L, Cluse L, Nicola NA, Metcalf D, Hilton DJ, Alexander WS. Placental defects and embryonic lethality in mice lacking suppressor of cytokine signaling 3. Proc. Natl. Acad. Sci. USA. 2001;98:9324–9329. doi: 10.1073/pnas.161271798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki A, Yasukawa H, Shouda T, Kitamura T, Dikic I, Yoshimura A. CIS3/SOCS-3 suppresses erythropoietin (EPO) signaling by binding the EPO receptor and JAK2. J. Biol. Chem. 2000;275:29338–29347. doi: 10.1074/jbc.M003456200. [DOI] [PubMed] [Google Scholar]
- Sasaki A, Yasukawa H, Suzuki A, Kamizono S, Syoda T, Kinjyo I, Sasaki M, Johnston JA, Yoshimura A. Cytokine-inducible SH2 protein-3 (CIS3/SOCS3) inhibits Janus tyrosine kinase by binding through the N-terminal kinase inhibitory region as well as SH2 domain. Genes Cells. 1999;4:339–351. doi: 10.1046/j.1365-2443.1999.00263.x. [DOI] [PubMed] [Google Scholar]
- Schmitz J, Weissenbach M, Haan S, Heinrich PC, Schaper F. SOCS3 exerts its inhibitory function on interleukin-6 signal transduction through the SHP2 recruitment site of gp130. J. Biol. Chem. 2000;275:12848–12856. doi: 10.1074/jbc.275.17.12848. [DOI] [PubMed] [Google Scholar]
- Sharkey AM, King A, Clark DE, Burrows TD, Jokhi PP, Charnock-Jones DS, Loke YW, Smith SK. Localization of leukemia inhibitory factor and its receptor in human placenta throughout pregnancy. Biol. Reprod. 1999;60:355–364. doi: 10.1095/biolreprod60.2.355. [DOI] [PubMed] [Google Scholar]
- Shouda T, Yoshida T, Hanada T, Wakioka T, Oishi M, Miyoshi K, Komiya S, Kosai K, Hanakawa Y, Hashimoto K, et al. Induction of the cytokine signal regulator SOCS3/CIS3 as a therapeutic strategy for treating inflammatory arthritis. J. Clin. Invest. 2001;108:1781–1788. doi: 10.1172/JCI13568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons DG, Cross JC. Determinants of trophoblast lineage and cell subtype specification in the mouse placenta. Dev. Biol. 2005;284:12–24. doi: 10.1016/j.ydbio.2005.05.010. [DOI] [PubMed] [Google Scholar]
- Takahashi Y, Carpino N, Cross JC, Torres M, Parganas E, Ihle JN. SOCS3: an essential regulator of LIF receptor signaling in trophoblast giant cell differentiation. EMBO J. 2003;22:372–384. doi: 10.1093/emboj/cdg057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi Y, Dominici M, Swift J, Nagy C, Ihle JN. Trophoblast stem cells rescue placental defect in SOCS3-deficient mice. J. Biol. Chem. 2006;281:11444–11445. doi: 10.1074/jbc.C600015200. [DOI] [PubMed] [Google Scholar]
- Tebbutt NC, Giraud AS, Inglese M, Jenkins B, Waring P, Clay FJ, Malki S, Alderman BM, Grail D, Hollande F, et al. Reciprocal regulation of gastrointestinal homeostasis by SHP2 and STAT-mediated trefoil gene activation in gp130 mutant mice. Nat. Med. 2002;8:1089–1097. doi: 10.1038/nm763. [DOI] [PubMed] [Google Scholar]
- Ueki K, Kondo T, Kahn CR. Suppressor of cytokine signaling 1 (SOCS-1) and SOCS-3 cause insulin resistance through inhibition of tyrosine phosphorylation of insulin receptor substrate proteins by discrete mechanisms. Mol. Cell. Biol. 2004a;24:5434–5446. doi: 10.1128/MCB.24.12.5434-5446.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueki K, Kondo T, Tseng YH, Kahn CR. Central role of suppressors of cytokine signaling proteins in hepatic steatosis, insulin resistance, and the metabolic syndrome in the mouse. Proc. Natl. Acad. Sci. USA. 2004b;101:10422–10427. doi: 10.1073/pnas.0402511101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ware CB, Horowitz MC, Renshaw BR, Hunt JS, Liggitt D, Koblar SA, Gliniak BC, McKenna HJ, Papayannopoulou T, Thoma B, et al. Targeted disruption of the low-affinity leukemia inhibitory factor receptor gene causes placental, skeletal, neural and metabolic defects and results in perinatal death. Development. 1995;121:1283–1299. doi: 10.1242/dev.121.5.1283. [DOI] [PubMed] [Google Scholar]
- Wong PK, Egan PJ, Croker BA, O'Donnell K, Sims NA, Drake S, Kiu H, McManus EJ, Alexander WS, Roberts AW, Wicks IP. SOCS-3 negatively regulates innate and adaptive immune mechanisms in acute IL-1-dependent inflammatory arthritis. J. Clin. Invest. 2006;116:1571–1581. doi: 10.1172/JCI25660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto K, Yamaguchi M, Miyasaka N, Miura O. SOCS-3 inhibits IL-12-induced STAT4 activation by binding through its SH2 domain to the STAT4 docking site in the IL-12 receptor beta2 subunit. Biochem. Biophys. Res. Commun. 2003;310:1188–1193. doi: 10.1016/j.bbrc.2003.09.140. [DOI] [PubMed] [Google Scholar]
- Yasukawa H, Ohishi M, Mori H, Murakami M, Chinen T, Aki D, Hanada T, Takeda K, Akira S, Hoshijima M, et al. IL-6 induces an anti-inflammatory response in the absence of SOCS3 in macrophages. Nat. Immunol. 2003;4:551–556. doi: 10.1038/ni938. [DOI] [PubMed] [Google Scholar]
- Zhang JG, Farley A, Nicholson SE, Willson TA, Zugaro LM, Simpson RJ, Moritz RL, Cary D, Richardson R, Hausmann G, et al. The conserved SOCS box motif in suppressors of cytokine signaling binds to elongins B and C and may couple bound proteins to proteasomal degradation. Proc. Natl. Acad. Sci. USA. 1999;96:2071–2076. doi: 10.1073/pnas.96.5.2071. [DOI] [PMC free article] [PubMed] [Google Scholar]