

Abstract
Type IV pili are a major virulence factor of the obligate human pathogen Neisseria gonorrhoeae (Gc). Pili facilitate bacterial adherence to epithelial cells, but their participation in later steps of epithelial infection, particularly intracellular replication and exit, is poorly understood. Using polarized T84 cells as a model for mature mucosal epithelia, pilus dynamics in piliated, Opa-expressing Gc were examined over time. T84 infection was characterized by a several-hour delay in the growth of cell-associated bacteria and by non-directional exit of Gc, the first time these phenomena have been reported. During infection, non-piliated progeny arose stochastically from piliated progenitors. Piliated and non-piliated Gc replicated and exited from T84 cell monolayers equally well, demonstrating that piliation did not influence Gc survival during epithelial infection. The frequency with which pilin variants arose from a defined piliated progenitor during T84 cell infection was found to be sufficiently high to account for the extensive pilin variation reported during human infection. However, the repertoire of variants appearing in association with T84 cells was similar to what was seen in the absence of cells, demonstrating that polarized epithelial cells can support Gc replication without selecting for a subset of pilin variants or piliation states.
Keywords: gonorrhea, recombination, pilus, epithelial cells
Introduction
Neisseria gonorrhoeae (the gonococcus; Gc) is an obligate human pathogen and the sole causative agent of the sexually transmitted disease gonorrhea. Gonococcal infection typically presents as an acute urethritis in men and an often asymptomatic cervicitis in women, but the anorectum, pharynx, or conjunctiva can also be involved (Edwards and Apicella, 2004). Establishment of a successful infection requires colonization of these mucosal epithelial surfaces, which has been modeled in human fallopian tube explants and recapitulated in primary cells of the urethra, cervix, and endometrium and in transformed epithelial cell lines (reviewed in Merz and So, 2000). Initial attachment of Gc to the apical or luminal surface of epithelial cells utilizes the neisserial type IV pili (McGee et al., 1981). Binding partners for pili include CD46, I domain-containing integrins, and additional membrane proteins that have not yet been identified (Kallstrom et al., 1997; Edwards and Apicella, 2005; Kirchner and Meyer, 2005). Microcolonies of piliated Gc and the retractile forces they produce initiate localized actin polymerization and signal transduction cascades in epithelial cells (Giardina et al., 1998; Kallstrom et al., 1998; Merz et al., 1999; Binnicker et al., 2003; Howie et al., 2005). These pilus-mediated interactions are critical to the establishment of symptomatic urethral infection in male volunteers (Cohen and Cannon, 1999). Intimate attachment and subsequent bacterial internalization are mediated by the interaction of Gc opacity-associated (Opa) proteins with members of the carcinoembryonic antigen cell adhesion molecule (CEACAM) family of receptors or heparan sulfate proteoglycans (reviewed in Dehio et al., 1998). Gc strains carry at least 11 opa genes, each of which is phase-variable (Dehio et al., 1998). Opa+ Gc are recovered from male volunteers infected urethrally with Opa- bacteria and are frequently isolated from patients with symptomatic urethral and cervical infection (James and Swanson, 1978; Draper et al., 1980; Schwalbe et al., 1985; Swanson et al., 1988; Jerse et al., 1994).
In most epithelial cell types, internalized Gc reside within vacuoles with phagolysosomal characteristics (Evans, 1977; Ayala et al., 1998; Wang et al., 1998; Booth et al., 2003; Timmerman et al., 2005), although bacteria are found in the cytoplasm of Hec-1-B cervical cells and exfoliated urethral epithelial cells (Apicella et al., 1996; Griffiss et al., 1999). It is assumed that Gc replicate within epithelial cells, although this has not been directly demonstrated. Gc exit into the basal medium of fallopian tube explants and polarized epithelial cells after 16 to 24 h of continuous apical colonization, which occurs without disruption of the epithelial barrier and without compromising epithelial cell viability (McGee et al., 1981; Merz et al., 1996; Wang et al., 1998). Transcytosis (apical-to-basal movement) of Gc is aided by expression of either pili or CEACAM-binding Opa proteins (Merz et al., 1996; Wang et al., 1998). By gaining access to deeper tissues, basally exited Gc can enable the spread of infection within a host, which can result in the rare sequelae of arthritis and endocarditis (McGee et al., 1981).
In addition to host cell adherence, type IV pili confer on Gc the properties of twitching motility, natural competence for DNA transformation, and uptake of antibiotics and heme-containing compounds (Biswas et al., 1977; Wolfgang et al., 1998; Chen et al., 2004; Zhao et al., 2005). The major subunit of the type IV pilus is the ∼18 kD pilin protein, encoded by the pilE gene (Meyer et al., 1984). Pilin monomers assemble along with minor pilus components in the periplasm and are extruded through the outer membrane secretin PilQ (Drake and Koomey, 1995). One of these minor components, PilC, was originally characterized as an epithelial adhesin, and it has recently been shown to aid in pilus presentation on the cell surface by countering pilus retractile forces (Rudel et al., 1995; Scheuerpflug et al., 1999; Morand et al., 2004; Kirchner and Meyer, 2005).
The pilin protein undergoes high-frequency changes in amino acid sequence due to pilin antigenic variation (Av), a gene conversion system in which unexpressed or silent pilin coding information found in storage loci (pilS) on the Gc chromosome is unidirectionally recombined into pilE (reviewed in(Kline et al., 2003). Each pilS copy carries unique sequences interspersed with short (1−34 bp) stretches of sequence shared with each other and with pilE (Haas and Meyer, 1986; Segal et al., 1986; Haas et al., 1992; Howell-Adams and Seifert, 2000; Criss et al., 2005). pilE recombination events give rise to antigenically distinct pilin monomers, producing piliated Gc whose colonies have a “P+” morphology (Segal et al., 1986; Swanson et al., 1987a). Pilin Av can also generate proteins that are poorly assembled into pili, leading to non-piliated Gc that exhibit a “P−” colony morphology (Segal et al., 1985; Bergstrom et al., 1986; Swanson et al., 1986; Koomey et al., 1987). Pilin Av is considered a specialized form of homologous recombination and requires the function of RecA and a gonococcal RecF-like pathway (Kline et al., 2003). Pilin Av also depends upon a cis-acting sequence in an intergenic region upstream of pilE (Sechman et al., 2005). We established a molecular assay for pilin Av in the Gc strain FA1090 that measures the number of recombination events occurring in a large number of clones arising from a defined pilE progenitor. In rapidly growing cultures of the pilin variant 1−81-S2, the rate of pilin Av was 4 × 10−3 recombination events per colony-forming unit (CFU) per generation, the highest reported for any pathogenic gene conversion system (Criss et al., 2005).
Although both piliated and non-piliated Gc arise from a piliated progenitor in vitro, only piliated bacteria are recovered from infected individuals (Kellogg et al., 1963; Swanson et al., 1987b; Seifert et al., 1994; Hamrick et al., 2001). Furthermore, male subjects experimentally inoculated with a specific Gc pilin variant produce bacteria that encode non-parental pilin proteins after 24 h of infection (Swanson et al., 1987b; Seifert et al., 1994; Hamrick et al., 2001). These observations may either suggest that the frequency of pilin Av is stimulated in vivo, perhaps through host-mediated iron limitation (Serkin and Seifert, 2000), or that there is a selection for pilin variants conferring a P+ morphology during human infection. These questions were addressed in the semi-polarized Hec-1-B cervical cell line, where a high frequency of pilin Av was measured in basally exited Gc, but instead of a selection for piliated bacteria, the majority of clones exhibited P− colony morphology (Ilver et al., 1998). This phenotype has not been recapitulated in other cell lines (Merz et al., 1996; Wang et al., 1998).
To further characterize the function of pili during epithelial infection, we have used polarized monolayers of T84 cells to directly address the survival of piliated vs. non-piliated bacteria in association with epithelial cells, the importance of pili to bacterial transit of epithelial cells, and the frequency of pilin Av during epithelial infection. Polarized monolayers of T84 epithelial cells are an excellent model for mature epithelial tissues and have been extensively used to study epithelial interactions with mucosal pathogens, including Neisseria (Madara et al., 1987; Merz et al., 1996; Wang et al., 1998). Here, cells were infected with a P+, Opa+ variant of Gc strain FA1090, allowing pilus dynamics to be assessed in the presence of other bacterial surface structures expressed during human infection. Our findings reveal that T84 cells support the growth of Gc in a pilus-independent manner, yielding bacteria that have undergone pilin Av at a frequency similar to what has been measured in the absence of host cells.
Results
Infection of polarized T84 epithelial cells by Gc
We measured the invasion, cell-associated growth, and exit of Gc from polarized monolayers of T84 epithelial cells, which are characterized by their high transepithelial electrical resistance and discrete apical and basal surfaces (Madara et al., 1987). Infected T84 cells were subjected to a modified gentamicin-protection assay, where internalized bacteria survive after exposure to the antibiotic and extracellular bacteria are killed (Elsinghorst, 1994). After gentamicin treatment, antibiotic-free medium was added back to T84 cells, and at different time points colony-forming units (CFU) were enumerated from cell lysates and from the apical and basal media. This allowed a defined population of cell-associated, gentamicin-protected Gc to be followed over time.
Using this assay, we examined the invasion profile of a P+, Opa+ derivative of Gc strain FA1090 encoding the 1−81-S2 pilin variant (Seifert et al., 1994) (Fig. 1A). 45% of the Gc inoculum was associated with the apical surface of T84 cell monolayers after 4 h, equivalent to 8 CFU for each of the 106 cells in the monolayer. Treatment with gentamicin for 2 h resulted in a > 1000-fold reduction in the number of viable CFU recovered from the T84 cells. These results are consistent with a majority of the bacteria remaining extracellular and with the low frequency of internalization of Gc, as previously reported in organ culture models and epithelial cell lines (Shaw and Falkow, 1988; Merz et al., 1996; Griffiss et al., 1999). For the first 4 to 6 h following gentamicin treatment, the number of Gc recovered from T84 cell lysates held constant at 103 CFU per monolayer. After this lag phase the number of cell-associated CFU began to increase, reaching a maximum of 108 CFU per monolayer at 25 h post-infection.
Figure 1. Profile of Gc infection of polarized T84 cells.

Panel A: Cell-associated Gc. Polarized monolayers of 106 T84 cells were apically infected in triplicate with P+ FA1090 Gc for 4 h, then treated with gentamicin for 2 h (dotted line). At the indicated times, T84 lysates were serially diluted and plated for colony-forming units (CFU). The average number of CFU ± standard error of the mean (SEM) for three replicate monolayers per time point is presented. Panel B: Exited Gc. At the indicated intervals, the apical (AP; black bars) and basal (BL; white bars) medium was removed from infected T84 cells and CFU enumerated from each. *, P < 0.01 between apical and basal CFU in the specified interval (Student's t-test). Panel C: Gc are detected within T84 cells. T84 cells were fixed at 12 h post-infection and intracellular and extracellular Gc were discriminated from one another by differential antibody accessibility and examined by confocal laser scanning miscroscopy. Green, intracellular Gc; turquoise (blue + green), extracellular Gc; epithelial F-actin, red. In the top image, arrows point to two intracellular bacteria, one of which is adjacent to an extracellular bacterium (arrowhead). Asterisk denotes a potentially intracellular bacterium at the edge of a microcolony. In the bottom image, an X-Z section was taken through the slice indicated in the top image by the dotted line and shows an internalized, subapical bacterium in the middle of the field, flanked by two apically adherent, extracellular bacteria. Scale bars, 5 μm. Panels D-E: Gc reside within vacuoles in T84 cells. T84 cells were fixed at 12 h post-infection and processed for thin-section transmission electron microscopy. Arrows indicate intracellular bacteria; N, nucleus; MVB, multivesicular body. Scale bars, 1 μm.
To further examine the gentamicin-protected population of Gc, infected T84 cells were fixed at various times and processed for thin-section electron microscopy and for confocal laser scanning microscopy, the latter using a procedure that distinguishes internalized from extracellular bacteria based upon their accessibility to a Gc-specific antibody before and after host cell permeabilization (see Experimental Procedures for details). In these images extracellular bacteria appear turquoise (blue + green), while intracellular bacteria appear green only. In support of the gentamicin-protection data in Fig. 1A, intracellular bacteria were rarely detected in T84 cells infected for 4 h (data not shown). In contrast, intracellular Gc were readily observed at 12 h post-infection. By confocal laser scanning microscopy, internalized bacteria appeared as fluorescent particles that localized near the apical surface (arrows, Fig. 1C), sometimes in cells with extracellular, adherent bacteria (arrowhead, Fig. 1C). In thin-section electron micrographs of T84 cells processed at this time point, bacteria-sized structures were observed within vacuoles near the cell nucleus and near late endosomal/lysosomal multivesicular bodies (Fig. 1E, F); uninfected T84 cells did not contain these vacuoles (data not shown). Cytoplasmically localized Gc were not detected at this or any other time point of T84 cell infection. Extracellular microcolonies containing several Gc were also observed at 12 h infection. Although occasional bacteria at the edge of a microcolony appeared intracellular from their fluorescence pattern (asterisk, Fig. 1C), their close association with the other bacteria may have prevented recognition by the extracellular antibody. Similar observations were made in confocal sections processed at 25 h post-infection, except that the extracellular aggregates were larger (data not shown). These results indicate that a subpopulation of Gc are internalized by T84 cells into subapical vacuoles. We conclude that the T84 cell-associated Gc consists of both intracellular and intimately associated, surface-adherent bacterial populations.
Although Gc can exit into the basal medium of fallopian tube explants and polarized epithelial monolayers, apical release has not been reported (Merz et al., 1996; Ilver et al., 1998; Wang et al., 1998). Our modified gentamicin-protection assay was used to examine the timing and directionality of Gc release from infected T84 cells. Bacterial growth in the tissue culture medium was limited by changing the apical and basal medium on each monolayer every 2−3 h. Therefore, Gc exit was reported as the number of CFU recovered in the medium for each of these intervals. Gc were first released from T84 cells between 12−14 h post-infection, and the number of exited CFU increased thereafter, with a maximum rate of exit from T84 cells, between 27 and 30 h infection, of 5 × 106 CFU per h (Fig. 1B). We found that Gc exited into the apical as well as the basal medium at all intervals examined. At times through 27 h of infection, 10-fold more CFU were cultured from the basal medium than the apical medium, after which equal numbers of CFU were recovered. T84 monolayers retained high transepithelial electrical resistance and barrier function for the duration of the experiment, indicating that intercellular junctions remained intact and that the apical and basal medium had not mixed (data not shown). Bacteria were not observed between the lateral membranes of adjacent T84 cells, implying that movement of Gc across T84 cells was transcellular and not paracellular. In summary, infection of polarized T84 cells by P+, Opa+ Gc is characterized by avid adherence to the apical plasma membrane, a low frequency of bacterial internalization, delayed replication of cell-associated bacteria, and non-directional release into the surrounding medium.
Bacterial replication and exit from T84 cells does not select for piliation states
The majority of bacteria isolated from individuals with acute gonorrhea exhibit a P+ colony morphology, suggesting a selection for pilus expression in vivo (Kellogg et al., 1963; Swanson et al., 1987b; Seifert et al., 1994; Hamrick et al., 2001). If growth in association with epithelial cells were to impose this selection, few non-piliated Gc should arise from a piliated progenitor during T84 cell infection. Instead, we observed that the percentage of P− CFU in the cell-associated and exited populations increased with time (Fig. 2A and data not shown). The percentage of P− CFU varied among replicate monolayers collected at each time point and became more pronounced as the infection progressed (Fig. 2B). As one example, predominantly P+ CFU were recovered from the lysates of monolayers A and C at 30 h post-infection, but in monolayer B nearly 50% of the cell-associated CFU had P− morphology. In contrast, the apically exited Gc in monolayer C were mostly of P− morphology but predominantly P+ in monolayers A and B. Importantly, the percentage of P− CFU did not affect the total number of CFU recovered in each of these populations (Fig. 2B). These results argue against a selection for pilus-dependent colony morphologies during T84 cell infection and instead indicate that P− Gc arise stochastically in association with epithelial cells.
Figure 2. P− Gc variants arise from a P+ progenitor during T84 cell infection.
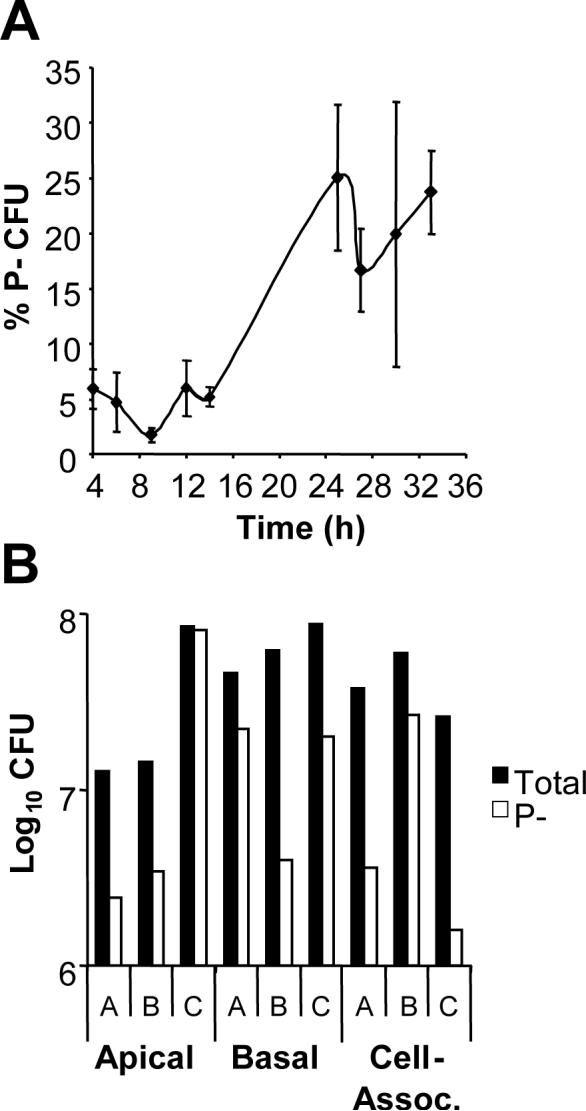
Panel A: Percentage of cell-associated P− CFU. The pilus-dependent colony morphology of CFU arising over time in T84 cells infected with P+ FA1090 Gc was examined using a stereomicroscope. The average percentage of P− CFU ± SEM was determined for three replicate monolayers per time point. Panel B: P− Gc arise stochastically during epithelial infection. Three monolayers of T84 cells, denoted A-C, were infected with P+ FA1090. The P+ (black bars) and P− (white bars) CFU present in the cell-associated population of each monolayer at 30 h and entering the apical and basal medium from each monolayer between 27 and 30 h were enumerated as in Figs. 1A and B.
The majority of P− Gc grown in vitro are the result of changes in pilE sequence, but phase variation of pilC, L-pilin production (the formation of an overly long pilin protein that does not assemble into pili) and deletion of the pilE locus also contribute (Hagblom et al., 1985; Segal et al., 1985; Swanson et al., 1986; Jonsson et al., 1991; Manning et al., 1991; Long et al., 1998; Criss et al., 2005). We found that pilin Av was the predominant mechanism for generating P−CFU at early times (< 24 h) of T84 cell infection, which was observed in both cell-associated and exited populations (Table 1 and data not shown). The P− pilin variants encoded either truncated pilin proteins or full-length but assembly-deficient pilins, as found for P− CFU arising from the 1−81-S2 pilin variant after passage on solid medium (Criss et al., 2005). After 24 h, a high percentage of cell-associated P− CFU encoded pilin antigenic variants, but PilC phase variation became an important contributor to the P− population. PilC phase variants lacked PilC expression by Western blot and retained the 1−81-S2 pilE sequence (data not shown). None of the cell-associated P− CFU that were examined had undergone both pilin Av and PilC phase variation. P− CFU with a deletion of the pilE locus were rarely recovered, and the percentage of deletions did not increase with time; no CFU were recovered that encoded L-pilins. Because similar results were observed in vitro for pilin variant 1−81-S2 (Criss et al., 2005), we conclude that the mechanisms by which P− Gc arise during epithelial infection parallel those occurring during growth on solid medium.
Table 1.
| Time (h) |
Pilin Av |
PilCoff |
pilE deletion |
Total |
|---|---|---|---|---|
| 6 | 75% (6) | 25% (2) | 0% (0) | 8 |
| 7.5 | 50 (4) | 37.5 (3) | 12.5 (1) | 8 |
| 9 | 67 (4) | 33 (2) | 0 (0) | 6 |
| 10.5 | 70 (7) | 30 (3) | 0 (0) | 10 |
| 12 | 71 (5) | 29 (2) | 0 (0) | 7 |
| 14 | 100 (6) | 0 (0) | 0 (0) | 6 |
| 25 | 0 (0) | 100 (7) | 0 (0) | 7 |
| 27 | 40 (2) | 60 (3) | 0 (0) | 5 |
| 30 | 25 (2) | 75 (6) | 0 (0) | 8 |
| 33 | 73 (8) | 27 (3) | 0 (0) | 11 |
Gc CFU exhibiting a P− colony morphology were collected from lysates of infected T84 cells at the indicated times. The percentage and number of CFU exhibiting changes in pilE sequence (Pilin Av), phase variation of PilC (PilCoff), and deletion of the pilE locus (pilE deletion) were determined.
To directly address a requirement for piliation during epithelial infection, P+ 1−81-S2 Gc were compared to an isogenic P− pilin variant in infection of T84 cells. Although P− Gc adhered ten-fold less well than P+ Gc to the apical surface of T84 cells, each yielded approximately equal numbers of gentamicin-resistant CFU, and no significant difference in cell-associated CFU was detected thereafter (Fig. 3A). Both P+ and P− Gc were released into the apical and basal medium (Fig. 3B). Slightly higher numbers of CFU were recovered from the apical medium of T84 cells infected with the P− variant, a difference that was statistically significant at the 29−32 h interval but not in subsequent experiments with these clones (data not shown). Infection with a P− PilC phase variant yielded similar results (data not shown).
Figure 3. Piliation is dispensable for Gc replication and exit from T84 cells.
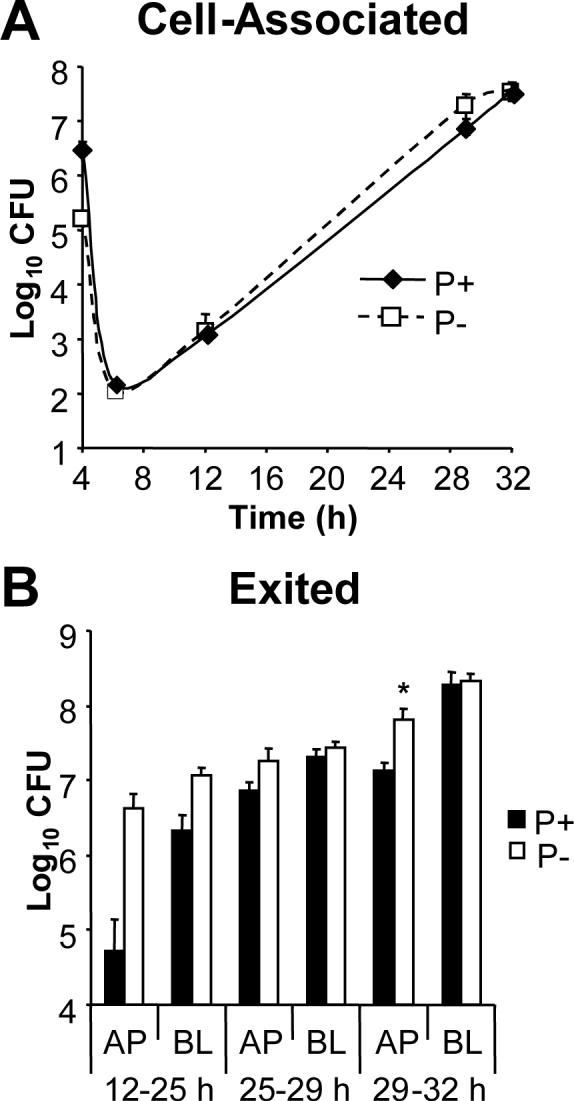
T84 cells were apically infected with P+ FA1090 Gc (solid lines, black bars) or an isogenic P− pilin variant (P−; dotted lines, white bars). Panel A: Cell-associated CFU. The CFU associated with the T84 cells (Panel A) were enumerated as in Fig. 1A. Panel B: Exited CFU. CFU exiting into the apical (AP) and basal (BL) medium (Panel B) were enumerated as in Fig. 1B. *, P < 0.05 by Student's t-test.
Since the P− variant produced colonies of both P− and P+ colony morphologies during T84 cell infection, we definitively tested whether pili were necessary for Gc to replicate and exit from T84 cells using a clone that was constitutively P− due to a non-reverting deletion at pilE (P−n; Swanson et al., 1985). The presence of pili enhanced bacterial adherence to the apical surface of T84 cells, with a 50-fold difference in CFU noted between the P+ and P−n strains after 4 h of attachment, but similar numbers of P+ and P−n CFU were recovered from T84 cells after gentamicin treatment and through 14 h of infection (Fig. 4A). At 22, 26, and 30 h post-infection, approximately 10-fold more P−n Gc than P+ Gc were associated with T84 cells (Fig. 4A). However, this difference was only statistically significant at the 30 h time point, and differences between the P+ and P−n strains were smaller in subsequent experiments (data not shown). As seen with the varying P− strain, P−n Gc exited into the apical medium of T84 cells more readily than the P+ parent and in a statistically significant manner; although 10-fold more P−n than P+ Gc were recovered in the basal medium of T84 cells at both intervals examined, this difference was not significant (Fig. 4B). We conclude that Gc do not need to retain a piliated state in order to multiply and exit from T84 cells and that the absence of pili may in fact facilitate these processes.
Figure 4. Gc that are nonvarying in P+ or P− phenotype can replicate and exit from T84 cells similar to the P+ parental strain.

Panels A-B: Comparison of varying P+ to nonvarying P− FA1090 Gc. T84 cells were apically infected with the P+ FA1090 parental strain (solid lines; black bars) or an isogenic P−n mutant with a deletion in the pilE locus (dotted lines; white bars). The number of cell-associated (Panel A) and exited (Panel B) CFU were enumerated over time as in Figs. 1A and B, respectively. Panels C-D: Comparison of varying P+ to nonvarying P+ FA1090. T84 cells were apically infected with the P+ FA1090 parent (solid lines; black bars) or an isogenic P+nv mutant that does not undergo pilin Av due to the presence of a transposon insertion upstream of pilE (dotted lines; white bars). The number of cell-associated (Panel C) and exited (Panel D) CFU were enumerated over time as in Figs. 1A and B, respectively. *, P < 0.05 between strains at the indicated time point by Student's t-test.
These results led us to speculate that switching from a P+ to a P− colony morphology during T84 cell infection could aid bacterial replication and exit, an observation that had been reported with a different strain of Gc in Hec-1-B cells (Ilver et al., 1998). We therefore compared T84 cell infection of the varying P+ parent strain to a non-varying P+ mutant, in which a cis-acting transposon inserted in the pilE locus blocks pilin Av without disrupting other pilus-dependent processes (P+nv; Sechman et al., 2005). The two strains attached to and were internalized equally well by T84 cells (Fig. 1C). Interestingly, P+nv Gc replicated in association with T84 cells slightly better than the varying parent, with statistically significant differences noted at 10.5, 14, and 24 h of infection, but at 28 and 32 h of infection there was no difference in cell-associated CFU between the two clones (Fig. 4C), nor was there a difference in their ability to exit from cells (Fig. 4D). P− Gc that had undergone PilC phase variation were isolated from T84 cells infected with the P+nv strain, but they arose stochastically and the majority of the isolates retained a P+ morphology (data not shown). All clones retained their Opa+ phenotype during infection (data not shown). These data show that Gc do not need to vary in piliation state in order to replicate and exit from T84 cells. We conclude that attributes of Gc other than type IV pili facilitate bacterial growth and exit from T84 cells, implying that epithelial cells tested under standard in vitro culture conditions do not provide the selection for P+ Gc observed during human infection.
Extensive pilin Av in Gc in contact with epithelial cells
Gc isolated from infected individuals can encode different pilin proteins from the parental strain, and it has been suggested that a high level of pilin Av occurs during human infection (Swanson et al., 1987b; Seifert et al., 1994; Hamrick et al., 2001). Pilin variation has been suggested to aid in evasion of the host humoral immune response and in infection of different host tissues (Swanson et al., 1987b; Kline et al., 2003). We asked whether interaction with epithelial cells influences pilin Av, either by altering the frequency or by modifying the repertoire of variants produced, by sequencing the pilE genes of Gc growing in association with T84 cells (Criss et al., 2005). The frequency of pilin Av measured at 6 h post-infection was 0.12 recombination events per CFU, and the proportion of Gc encoding recombinant pilE increased at all time points thereafter (Fig. 5A). The change in the frequency of pilin Av for a given time interval did not always correlate with the increase in CFU in that interval. For instance, when the number of cell-associated CFU began to rise between 9 and 14 h post-infection, the frequency of pilin Av remained relatively constant; conversely, the number of cell-associated CFU reached a plateau at 25 h post-infection, but the frequency of pilin Av doubled between 25 and 33 h. At 25 h the frequency of pilin Av was similar to what was measured in pilin variants recovered from experimentally infected male volunteers (Seifert et al., 1994). Gc that had exited from T84 cells also exhibited a high frequency of pilin Av, averaging 0.8−0.9 recombination events per CFU at each interval, and no significant difference was measured in the frequency of pilin Av between the apically and basally exited Gc (Fig. 5B). We estimated the rate of pilin Av for cell-associated Gc between 14 and 25 h post-infection, where the bacteria underwent 6 generations of net growth and the change in frequency of pilin Av was 0.28 events per CFU, as 4.5 × 10−2 recombination events per CFU per generation. This estimate assumes that Gc in different compartments in T84 cells grow at similar rates. Notably, when the inoculum used to infect T84 cells was simultaneously grown in liquid medium in the absence of epithelial cells, the rate of pilin Av was similar (4.1 × 10−2 events per CFU per generation). These results show that Gc undergo extensive pilin Av during epithelial infection and suggest that the rate is not significantly different from Gc grown in the absence of epithelial cells.
Figure 5. Extensive pilin antigenic variation occurs in Gc associated with T84 cells.
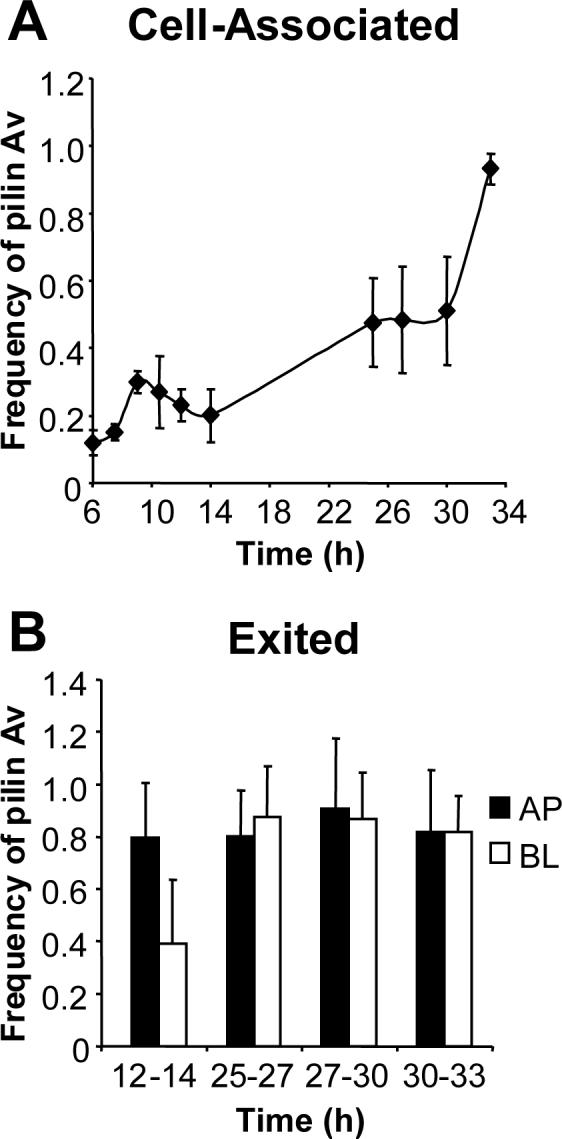
Panel A: Cell-associated CFU. T84 cells were infected with P+ FA1090, and CFU of varying pilus-dependent morphologies were collected from cell lysates. Panel B: Exited CFU. CFU were collected from the apical (AP; black bars) and basal (BL; white bars) medium of infected T84 cells at the indicated intervals. The pilE genes of the collected CFU were amplified, sequenced, and examined for changes from the parental 1−81-S2 sequence. The frequency of pilin Av at each time point or interval is defined as the number of recombination events detected divided by the number of pilE genes sequenced. The frequency was measured separately for the P+ and PCFU in each population, and the overall frequency of pilin Av was calculated by the formula: Frequency = (Frequency P+)(% P+ CFU) + (Frequency P−)(% P− CFU). Data are presented as the average frequency of pilin Av ± SEM measured in triplicate T84 cell monolayers. From the data in Table 1, the actual frequency of pilin Av at each time point is approximately 55% of the reported values.
Since FA1090 1−81-S2 Gc constitutively undergo pilin Av, some of the recombination events that were detected could have occurred during bacterial growth after isolation from T84 cells, and the contribution of colonial growth to pilin Av has remained an open question in previous studies. We therefore compared the frequency of pilin Av in plate-grown colonies to that directly measured from infected T84 cells. Lysates of cells infected for 24 h were plated on solid medium, and CFU were collected and analyzed for pilE variants as above. Chromosomal DNA was extracted from the remainder of the lysates, PCR was performed using pilE-specific primers, and the resulting products were directly cloned into E. coli. No PCR product was obtained from chromosomal DNA extracted from uninfected T84 cells (data not shown). Greater than 35 E. coli clones and Gc isolates were sequenced from each of 3 replicate infected T84 monolayers. While the frequency of pilin Av in Gc isolates at this time was 0.59, the frequency of pilin Av in the E. coli pilE clones was 0.33, a statistically significant difference (Table 2). We conclude that variation during growth on solid media contributes to the measured frequency of pilin Av, but a majority of the recorded recombination events occurred during T84 cell infection.
Table 2.
| |
Recombination events in Gc grown in vitro after T84 cell infection |
Recombination events in pilE genes amplified from T84 cell lysates |
||||
|---|---|---|---|---|---|---|
| Monolayer |
# pilE Clones |
# Recomb. Events |
Frequency of pilin Av |
# pilE Clones |
# Recomb. Events |
Frequency of pilin Av |
| A | 37 | 19 | 0.51 | 37 | 12 | 0.32 |
| B | 45 | 27 | 0.60 | 36 | 10 | 0.27 |
| C |
47 |
31 |
0.66 |
37 |
15 |
0.41 |
| Average | 43 | 26 | 0.59 | 37 | 12 | 0.33 |
T84 cells were infected in triplicate with 1−81-S2 as described in Fig. 1. 24 h post-infection, CFU were enumerated from cell lysates (left columns). Chromosomal DNA was used as the template in pilE-specific PCR reactions and cloned into E. coli (right columns). In each population, the number of pilE sequences analyzed, the number of pilE recombination events, and the resulting frequency of pilin Av per monolayer are presented. The difference in the frequency of pilin Av measured in the two populations was statistically significant (P < 0.01 by Student's t-test).
To address whether growth in association with epithelial cells led to a selection for particular pilin sequences, we compared the repertoire of pilS copies in pilin variants arising after growth in association with T84 cells and in liquid medium. Similar pilS copies contributed to the pilE sequences carried in these two populations at 9, 12, and 14 h of growth, demonstrating that there was no epithelial selection for pilin sequences (data not shown). We also examined the non-parental pilE sequences carried by Gc associated with T84 cells at later times of infection. The pilin variants appearing after 24 h were an expanded version of the population seen up through 14 h of infection. These variants changed in each of the replicate monolayers collected per time point and also changed over time (data not shown). Had there been a selection for particular pilin variants during epithelial infection, a select subset of non-parental pilE sequences would have been detected at each time, a prediction not supported by these data. We conclude that the frequency of pilin Av and the repertoire of pilin variants that are produced are not altered in Gc that have been internalized by T84 cells and that pilin Av during epithelial infection would be capable of generating the spectrum of variants seen during human disease.
Discussion
While type IV pili have been identified as a key virulence factor of Gc from their role in bacterial attachment to mucosal epithelial surfaces, their importance to events occurring after internalization, particularly replication and exit, is less clear. For instance, it was plausible that Gc localized within vacuoles lose their pili in order to transit across epithelial cells. Here we have shown that P− CFU arise stochastically from a P+, Opa+ progenitor when associated with polarized T84 cells and that non-varying P+ and P− Gc replicate and exit from T84 cells equally well. We conclude that pili do not confer an advantage or disadvantage to Gc in epithelial survival in this model system.
Because Gc is an obligate human pathogen, studies defining the importance of host and bacterial factors to infection have been limited to certain models. Experimental urethral infection of male volunteers best mimics natural gonococcal infection but only permits examination of the bacteria released during infection, not those remaining associated with the epithelium (Cohen and Cannon, 1999). Infection studies have therefore been carried out ex vivo with explants from physiologically relevant epithelial tissues such as fallopian tube and with primary epithelial cells from the male urethra and female cervix and endometrium (Edwards and Apicella, 2004). In conjunction with previous studies, we have shown that the T84 colorectal cell line reproduces many of the aspects of Gc adherence, invasion, and exit first identified in these other systems. Importantly, this study has revealed previously unappreciated characteristics of Gc-epithelial cell interactions, including that replication of cell-associated bacteria occurs after several hours and that exit occurs non-directionally. Since our electron micrographs did not reveal multiple bacteria within a single vacuole, it is possible that Gc primarily multiplied after vacuolar exocytosis, when tightly apposed to the plasma membrane. We anticipate further examination of these aspects of infection in other epithelial cell lines and organ culture.
In contrast to our observation that piliation did not affect the numbers of Gc associated with or released from polarized T84 cells, P− Gc with a deletion in pilE were reported to be unable to transit semi-polarized Hec-1-B cervical cells (Ilver et al., 1998). One explanation for this discrepancy is differences in Opa expression in the strains used in the two studies: the Hec-1-B study was performed with an Opa- variant of strain MS11, whereas our experiments used Opa+ Gc, the phenotype of the majority of clinical isolates. Previous reports have shown that P−, Opa- Gc do not adhere to or invade Hec-1-B or other epithelial cells effectively (Griffiss et al., 1999). Expression of either pili or Opa proteins is also known to aid Gc in transiting T84 cells (Merz et al., 1996; Wang et al., 1998). Additionally, Opa expression may direct Gc into a different intracellular compartment, which could affect piliated vs. non-piliated bacteria differently. A second possibility is that T84 and Hec-1-B cells exert different selection pressures on internalized Gc. Whereas we and others have observed bacteria inside vacuoles in T84 cells (Merz et al., 1996; Wang et al., 1998), Gc in Hec-1-B cells are mainly localized to the cytoplasm (Shaw and Falkow, 1988; Ilver et al., 1998). It is possible that the epithelial cytoplasm differentially affects the growth or survival of piliated and non-piliated Gc, but the cause of this selection is minimized or absent in the Gc-containing vacuole. In the Hec-1-B study, this could explain both why the pilE deletion mutant did not exit and why the majority of the P+ inoculum had switched to a P− morphology after transit. The possibility that cytoplasmic vs. vacuolar localization influences Gc survival remains to be examined.
Because we observed that P+ and P− Gc exited equally well from T84 cells, epithelial cells may not provide the selective pressure that produces P+ CFU in mucosal secretions and in urine during human infection (Kellogg et al., 1963; Swanson et al., 1987b; Cohen and Cannon, 1999). We can propose at least five alternative mechanisms that could drive the selection for P+ Gc. First, piliated bacteria may be more readily recovered from the urogenital tract. This may stem from the increased adherence of piliated Gc to the exfoliated epithelial cells, polymorphonuclear leukocytes (PMN), and macrophages found in gonorrheal exudates (Rest and Shafer, 1989; Apicella et al., 1996; Knepper et al., 1997). This may also be due to autoagglutination of P+ Gc, allowing bacteria to be more easily cultured as piliated aggregates than as individual, non-piliated particles. Second, a selection for P+ Gc may occur during the inflammatory response of gonorrheal disease. Piliated Gc may be enriched in secretions and urine during symptomatic disease because they are more resistant to the bactericidal mechanisms of leukocytes (Rest et al., 1982). Third, P+ Gc may survive better in nutrient-limiting conditions in primary epithelial cells or the urogenital lumen (but not T84 cells), since the pilus can mediate acquisition of heme-containing compounds and potentially other nutrients (Chen et al., 2004). Fourth, epithelial cells in the urogenital tract may exert different selection pressures on Gc than do T84 cells, since differences in infection have been noted between primary epithelial cells and their transformed derivatives (Edwards and Apicella, 2004). For example, the P+ phenotype could be selected for by bactericidal components of primary epithelial cells such as defensins or Nox-family oxidases, which may not be expressed by polarized T84 cells. Finally, an environmental factor may be present during urogenital infection that directs the production of only piliated progeny, although such a factor has not been identified in any in vitro studies of pilin Av. All these possibilities warrant further investigation.
The importance of pilin Av to gonococcal infection is underscored by the finding that a vaccine raised against a particular pilin variant is not protective in individuals infected with Gc expressing other pilin variants (Boslego et al., 1991). Human studies have further shown that extensive pilin Av occurs during acute gonococcal disease, with non-parental pilin variants recovered from experimentally infected male volunteers 24 h after inoculation (Swanson et al., 1987b; Seifert et al., 1994; Hamrick et al., 2001). Combined with the observation that pilin Av is stimulated under iron-limiting conditions as could be found in the urogenital tract, it was suggested that the frequency of pilin Av is stimulated during human infection (Serkin and Seifert, 2000). The data presented here demonstrate for the first time that liquid-grown and T84 cell-associated Gc exhibit a similar frequency of pilin Av, implying that Gc can acquire sufficient host cell iron during epithelial infection (Bonnah et al., 2000). Importantly, the Gc recovered from T84 cells after 24 h infection, like those isolated from male volunteers, encoded nonparental pilE sequences that had undergone 1−2 recombination events (Hamrick et al., 2001). We conclude that the inherently high frequency of pilE recombination can generate all the pilin variants arising during natural human infection.
Taken together, the results from this study conceptualize the urogenital epithelium as an environment that is permissive for Gc replication and provides protection from the innate immune response, thereby facilitating the generation of progeny expressing diverse pilin sequences and piliation states. The ability to undergo pilus variation has been suggested to be important for colonization of secondary anatomical sites by Gc, but tissue-specific adherence of particular variants, independent of their piliation state, has never been clearly demonstrated (Lambden et al., 1980; Rudel et al., 1992; Jonsson et al., 1994). We would instead support the model that pilin Av generates an ever-changing pool of variants that ensures continual reinfection of a core group who remain immunologically naïve to the newly arising variants with which they are presented (Tramont et al., 1979; Zak et al., 1984). Our work supports this model in demonstrating that epithelial infection does not limit the frequency or spectrum of pilin variants that would be available for transmission to other hosts. Pilin Av therefore allows Gc to retain an effective foothold in this core group of individuals and aids in the persistence of this sexually transmitted pathogen in the human population.
Experimental Procedures
Bacterial strains
FA1090 1−81-S2 is a P+ Gc strain isolated from an experimentally infected male volunteer that contains a defined pilE sequence (Seifert et al., 1994). An opaque derivative isolated after in vitro passage was found to express OpaA, OpaB/D, and OpaF by immunoblotting with a panel of Opa-specific monoclonal antibodies (a gift from J. Cannon, Univ. of North Carolina). Two P− variants, 2c4-A and PilC-, were isolated after non-selective passage of 1−81-S2. In 2c4-A, 175 nucleotides of pilS2 copy 4 sequence replaced nucleotides 226−399 of 1−81-S2, shifting the pilE reading frame to produce a stop codon after amino acid 95 (Criss et al., 2005). This variant did not react with an antibody against the pilin N-terminus by Western blot. PilC- retained 1−81-S2 pilE sequence and did not express PilC by Western blot. The P−n mutant was generated by transformation of 1−81-S2 with chromosomal DNA from a strain with a deletion in pilE (Chen et al., 2004) using standard techniques (Stohl and Seifert, 2001). The P+nv mutant was generated by transformation of 1−81-S2 with chromosomal DNA from a strain with a mini-Tn5 transposon (Epicentre) inserted upstream of the pilE open reading frame; this mutant is abrogated for pilin Av without affecting DNA repair or the natural competence of Gc (Sechman et al., 2005). All mutants retained the parental Opa expression profile, and all piliated strains encoded the 1−81-S2 pilE sequence.
Gc were grown on GCB agar (GC Medium Base (Difco) with an additional 1.25 g L−1 select agar (Invitrogen) containing Kellogg's supplements (Kellogg et al., 1963)) at 37 °C and 5% CO2 for 16 h. For invasion assays, ½ of a single colony was passaged on GCB agar, and the remainder was used as template for pilE-specific PCR and sequencing to ensure that the colony retained the correct pilE sequence (see below). The number of CFU arising on GCB agar was quantified by resuspending the bacteria in liquid medium and comparing the resulting OD550 to a predetermined growth curve.
T84 epithelial cells
The T84 colorectal carcinoma cell line was obtained from J. Casanova (Univ. of Virginia, Charlottesville). T84 cells were cultured in DMEM:Ham's F12 medium with 6% heat-inactivated fetal bovine serum and antibiotic-antimycotic (all from Invitrogen) as described (McCormick et al., 1993). To produce polarized monolayers of T84 cells, 12 mm diameter Transwell filter supports with 3.0 μm pores (Costar) were coated with 10 μg per cm2 rat tail collagen (Upstate) dissolved in 0.1M NaHCO3, pH 8.3, and T84 cells were seeded at confluency (1 × 106 cells) on each support. Monolayers were used 8−12 days after plating. The integrity of the monolayers was assessed prior to each experiment by monitoring the transepithelial electrical resistance with an EVOM voltohmmeter (World Precision Instruments, used with permission of K. Satchell, Northwestern Univ.), which exceeded 2000 Ω × cm2.
Modified T84 gentamicin-protection assay
T84 cells were incubated in cell-culture medium lacking antibiotics for > 16 h, then equilibrated in “T84 cell medium” (antibiotic-free medium supplemented with 5 μg ml−1 holo-human transferrin (Serologicals Corp.) for 30 min prior to infection. Gc were washed twice and resuspended at 2.5 × 108 CFU ml−1 in T84 cell medium, and 0.2 ml of this culture was added to the apical surface of monolayers containing 106 T84 cells to yield a multiplicity of infection of 50. After 4 h at 37 °C, monolayers were washed twice and lysed in 2% saponin (Sigma) in PBS for 15 min, and the lysate was serially diluted and plated on GCB agar. The remaining monolayers were incubated for 2 h at 37 °C in T84 cell medium containing 200 μg ml−1 gentamicin (Invitrogen). Cells were then washed three times and replaced in T84 cell medium lacking antibiotics (1 ml in each of the apical and basal compartments). Immediately after gentamicin treatment and at selected times post-infection, the apical and basal medium were collected, the monolayers were lysed, and all were serially diluted and plated. The medium was changed on all monolayers starting at 14 h post-infection and at each time point thereafter. The number of CFU ml−1 arising on GCB agar and the percentage of CFU with P+ morphology were determined for each compartment (apical, basal, cell-associated) at each time point. Transepithelial electrical resistances remained above 1500 Ω × cm2 for the duration of the experiment. Infections were carried out in triplicate for each time point, and assays were performed at least three times per variant. Because day-to-day variability in T84 monolayers and in bacterial growth was observed, comparisons between variants were only made within a given experiment.
Thin-section transmission electron microscopy
T84 monolayers infected with Gc were washed thoroughly in ice-cold 0.2 M phosphate buffer, pH 7.3, then fixed in 2.5% glutaraldehyde in phosphate buffer. Samples were treated with 2% osmium tetroxide, dehydrated in increasing grades of ethanol, and embedded in Epon resin (Electron Microscopy Sciences). 100 nm thin sections were mounted on Formvar carbon-coated grids (Electron Microscopy Sciences), negatively stained with uranyl acetate and lead citrate, and examined with a JEOL 1220 transmission electron microscope at an accelerating voltage of 60 kV. Images were acquired with a Kodak 1.6 digital camera using the Advantage software (AMT) and processed with Adobe Photoshop 7.0.
Confocal laser scanning microscopy
At 4, 12, and 25 h post-infection, T84 cells were fixed in 4% paraformaldehyde in 0.2 M phosphate buffer, pH 7.3, then blocked in phosphate-buffered saline containing 10% normal goat serum (Invitrogen) without permeabilization. Extracellular Gc were detected using a polyclonal anti-Gc antiserum (Biodesign) followed by Alexa Fluor 647-coupled goat anti-rabbit IgG (Molecular Probes). Cells were then re-fixed in paraformaldehyde, followed by permeabilization with 0.1% Triton X-100 in PBS for 5 min at room temperature. The cells were incubated with the polyclonal anti-Gc antiserum followed by fluorescein isothiocyanate-coupled goat anti-rabbit IgG (Jackson Immunoresearch) in blocking buffer containing 0.2% saponin. The actin cytoskeleton was recognized with Alexa Fluor 568-coupled phalloidin (Molecular Probes). Cells were examined with a LSM 510 confocal microscope (Zeiss) and digital images were processed using the manufacturer's software.
Western blotting
Lysates of Gc were prepared as described by Long et al. (Long et al., 1998) except for detecting heat-modifiable changes in Opa protein mobility, where half of the lysate was instead heated at 37 °C for 60 min as described (Black et al., 1984). Samples were separated by SDS-polyacrylamide gel electrophoresis (10% polyacrylamide for analysis of PilC, 15% for Opa and pilin) and transferred to polyvinylidene difluoride (Millipore). Blots were blocked for 16 h at 4 °C in Tris-buffered saline (for PilC) or PBS (for Opa and pilin) containing 0.05% Tween-20 and 5% nonfat dry milk. PilC and pilin were detected with polyclonal antisera as recently described (Criss et al., 2005). Individual Opa proteins of Gc strain FA1090 were detected using a panel of monoclonal antibodies (gift from J. Cannon), followed by goat anti-mouse IgG coupled to horseradish peroxidase (Chemicon).
pilE sequencing
DNA sequencing was performed essentially as described (Criss et al., 2005). Briefly, 16−32 CFU isolated from each of three replicate infected monolayers per time point were individually frozen at −80 °C. Both P+ and P− CFU were collected, and the colony morphology of each isolate was noted. The pilE gene of each CFU was amplified by Taq-based PCR using the PILRBS-SP3A primer pair (Seifert et al., 1994) and sequenced. pilE sequences were aligned with the parental pilE sequence as well as all pilS copies in the FA1090 genome, and all sequence changes observed corresponded to one or more pilS copies. The frequency of pilin Av is defined as the number of recombination events occurring in a population of pilE sequences divided by the total number of pilE sequences analyzed. The frequency of pilin Av was determined separately for the P+ and P− populations, and an overall frequency was calculated by multiplying the frequency from each population by the percentage of CFU with that colony morphology. Data are presented as the average of the frequencies measured from replicate monolayers ± SEM.
Growth of Gc in vitro
0.2 ml of the Gc suspension used for T84 cell infection (at 2.5 × 108 CFU ml−1) was diluted in 0.8 ml of fresh medium and placed in a 37 °C, 5% CO2 incubator. Gc were vigorously mixed by pipetting and diluted 1:10 into fresh medium every 3 h through 12 h. An aliquot of each culture was removed after 3, 6, 9, 12, and 14 h and total CFU were enumerated as above.
Cloning of pilE from T84 cell lysates
T84 cells infected in triplicate for 24 h were lysed in saponin and CFU enumerated as above. The remainder of the lysates (∼0.2 ml) were mixed with 0.3 ml of TES (50 mM Tris-HCl, pH 8.0, 10 mM EDTA, 50 mM NaCl), to which was added 50 μl of 10% SDS and 3 μl of 10 mg ml−1 RNase A. Chromosomal DNA was phenol:chloroform extracted, precipitated with isopropanol, and washed in 70% ethanol. pilE was amplified from the chromosomal DNA templates with Pfu polymerase using forward primer CONSTF2 (Seifert et al., 1994) with reverse primer SP3A, both of which were 5’-phosphorylated. The 5’ end of pilE is not amplified with this primer pair, thereby avoiding toxicity associated with overexpression of full-length pilin in E. coli (H.S. Seifert, unpublished observations). No PCR product was generated when chromosomal DNA isolated from uninfected cells served as template. The PCR products were directly ligated into pSmart-LC-Amp and transformed into E. cloni electrocompetent cells (both from Lucigen) according to the manufacturer's instructions. At least 35 ampicillin-resistant clones containing an insert of the correct molecular weight were sequenced per lysate, using CONSTF2 as primer.
Statistical analysis
Matched time points were compared between two strains in a given experiment by two-tailed Student's t-test. At least 3 monolayers per time point per strain were compared. Student's t-test was also used to compare the frequency of pilin Av measured in Gc clones grown from T84 cells to pilE sequences directly amplified from infected T84 cell lysates. Data were considered significant if the P value was ≤ 0.05.
Acknowledgments
We thank Janne Cannon for the monoclonal anti-Opa antibodies, Teng-Leong Chew for advice on confocal microscopy, James Casanova for the T84 cells, Lennell Reynolds Jr. for assistance with electron microscopy, Karla Fullner-Satchell for the use of the EVOM voltohmmeter, and Elizabeth Stohl for critical reading of the manuscript. This research was supported by Public Health Service Grants R37 AI033493 and RO1 AI044239 to H.S.S. and Ruth L. Kirchstein National Research Service Award F32 AI056681 to A.K.C.
References
- Apicella MA, Ketterer M, Lee FK, Zhou D, Rice PA, Blake MS. The pathogenesis of gonococcal urethritis in men: confocal and immunoelectron microscopic analysis of urethral exudates from men infected with Neisseria gonorrhoeae. J Infect Dis. 1996;173:636–646. doi: 10.1093/infdis/173.3.636. [DOI] [PubMed] [Google Scholar]
- Ayala P, Lin L, Hopper S, Fukuda M, So M. Infection of epithelial cells by pathogenic neisseriae reduces the levels of multiple lysosomal constituents. Infect Immun. 1998;66:5001–5007. doi: 10.1128/iai.66.10.5001-5007.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergstrom S, Robbins K, Koomey JM, Swanson J. Piliation control mechanisms in Neisseria gonorrhoeae. Proc Natl Acad Sci USA. 1986;83:3890–3894. doi: 10.1073/pnas.83.11.3890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binnicker MJ, Williams RD, Apicella MA. Infection of human urethral epithelium with Neisseria gonorrhoeae elicits an upregulation of host anti-apoptotic factors and protects cells from staurosporine-induced apoptosis. Cell Microbiol. 2003;5:549–560. doi: 10.1046/j.1462-5822.2003.00300.x. [DOI] [PubMed] [Google Scholar]
- Biswas GD, Sox T, Blackman E, Sparling PF. Factors affecting genetic transformation of Neisseria gonorrhoeae. J Bacteriol. 1977;129:983–992. doi: 10.1128/jb.129.2.983-992.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black WJ, Schwalbe RS, Nachamkin I, Cannon JG. Characterization of Neisseria gonorrhoeae protein II phase variation by use of monoclonal antibodies. Infect Immun. 1984;45:453–457. doi: 10.1128/iai.45.2.453-457.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnah RA, Lee SW, Vasquez BL, Enns CA, So M. Alteration of epithelial cell transferrin-iron homeostasis by Neisseria meningiditis and Neisseria gonorrhoeae. Cell Microbiol. 2000;2:207–218. doi: 10.1046/j.1462-5822.2000.00042.x. [DOI] [PubMed] [Google Scholar]
- Booth JW, Telio D, Liao EH, McCaw SE, Matsuo T, Grinstein S, Gray-Owen SD. Phosphatidylinositol 3-kinases in carcinoembryonic antigen-related cellular adhesion molecule-mediated internalization of Neisseria gonorrhoeae. J Biol Chem. 2003;278:14037–14045. doi: 10.1074/jbc.M211879200. [DOI] [PubMed] [Google Scholar]
- Boslego JW, Tramont EC, Chung RC, McChesney DG, Ciak J, Sadoff JC, et al. Efficacy trial of a parenteral gonococcal pilus vaccine in men. Vaccine. 1991;9:154–162. doi: 10.1016/0264-410x(91)90147-x. [DOI] [PubMed] [Google Scholar]
- Chen CJ, Tobiason DM, Thomas CE, Shafer WM, Seifert HS, Sparling PF. A mutant form of the Neisseria gonorrhoeae pilus secretin protein PilQ allows increased entry of heme and antimicrobial compounds. J Bacteriol. 2004;186:730–739. doi: 10.1128/JB.186.3.730-739.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen MS, Cannon JG. Human experimentation with Neisseria gonorrhoeae: progress and goals. J Infect Dis. 1999;179(Suppl 2):S375–379. doi: 10.1086/513847. [DOI] [PubMed] [Google Scholar]
- Criss AK, Kline KA, Seifert HS. The frequency and rate of pilin antigenic variation in Neisseria gonorrhoeae. Mol Microbiol. 2005;58:510–519. doi: 10.1111/j.1365-2958.2005.04838.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehio C, Gray-Owen SD, Meyer TF. The role of neisserial Opa proteins in interactions with host cells. Trends Microbiol. 1998;6:489–495. doi: 10.1016/s0966-842x(98)01365-1. [DOI] [PubMed] [Google Scholar]
- Drake SL, Koomey M. The product of the pilQ gene is essential for the biogenesis of type IV pili in Neisseria gonorrhoeae. Mol Microbiol. 1995;18:975–986. doi: 10.1111/j.1365-2958.1995.18050975.x. [DOI] [PubMed] [Google Scholar]
- Draper DL, James JF, Brooks GF, Sweet RL. Comparison of virulence markers of peritoneal and fallopian tube isolates with endocervical Neisseria gonorrhoeae isolates from women with acute salpingitis. Infecti Immun. 1980;27:882–888. doi: 10.1128/iai.27.3.882-888.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards JL, Apicella MA. The molecular mechanisms used by Neisseria gonorrhoeae to initiate infection differ between men and women. Clin Microbiol Rev. 2004;17:965–981. doi: 10.1128/CMR.17.4.965-981.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards JL, Apicella MA. I-domain-containing integrins serve as pilus receptors for Neisseria gonorrhoeae adherence to human epithelial cells. Cell Microbiol. 2005;7:1197–1211. doi: 10.1111/j.1462-5822.2005.00547.x. [DOI] [PubMed] [Google Scholar]
- Elsinghorst EA. Measurement of invasion by gentamicin resistance. Meth Enzymol. 1994;236:405–420. doi: 10.1016/0076-6879(94)36030-8. [DOI] [PubMed] [Google Scholar]
- Evans BA. Ultrastructural study of cervical gonorrhea. J Infect Dis. 1977;136:248–255. doi: 10.1093/infdis/136.2.248. [DOI] [PubMed] [Google Scholar]
- Giardina PC, Williams R, Lubaroff D, Apicella MA. Neisseria gonorrhoeae induces focal polymerization of actin in primary human urethral epithelium. Infect Immun. 1998;66:3416–3419. doi: 10.1128/iai.66.7.3416-3419.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiss JM, Lammel CJ, Wang J, Dekker NP, Brooks GF. Neisseria gonorrhoeae coordinately uses Pili and Opa to activate HEC-1- B cell microvilli, which causes engulfment of the gonococci. Infect Immun. 1999;67:3469–3480. doi: 10.1128/iai.67.7.3469-3480.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas R, Meyer TF. The repertoire of silent pilus genes in Neisseria gonorrhoeae: evidence for gene conversion. Cell. 1986;44:107–115. doi: 10.1016/0092-8674(86)90489-7. [DOI] [PubMed] [Google Scholar]
- Haas R, Veit S, Meyer TF. Silent pilin genes of Neisseria gonorrhoeae MS11 and the occurrence of related hypervariant sequences among other gonococcal isolates. Mol Microbiol. 1992;6:197–208. doi: 10.1111/j.1365-2958.1992.tb02001.x. [DOI] [PubMed] [Google Scholar]
- Hagblom P, Segal E, Billyard E, So M. Intragenic recombination leads to pilus antigenic variation in Neisseria gonorrhoeae. Nature. 1985;315:156–158. doi: 10.1038/315156a0. [DOI] [PubMed] [Google Scholar]
- Hamrick TS, Dempsey JA, Cohen MS, Cannon JG. Antigenic variation of gonococcal pilin expression in vivo: analysis of the strain FA1090 pilin repertoire and identification of the pilS gene copies recombining with pilE during experimental human infection. Microbiol. 2001;147:839–849. doi: 10.1099/00221287-147-4-839. [DOI] [PubMed] [Google Scholar]
- Howell-Adams B, Seifert HS. Molecular models accounting for the gene conversion reactions mediating gonococcal pilin antigenic variation. Mol Microbiol. 2000;37:1146–1159. doi: 10.1046/j.1365-2958.2000.02067.x. [DOI] [PubMed] [Google Scholar]
- Howie HL, Glogauer M, So M. The N. gonorrhoeae type IV pilus stimulates mechanosensitive pathways and cytoprotection through a pilT-dependent mechanism. PLoS Biol. 2005;3:e100. doi: 10.1371/journal.pbio.0030100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilver D, Kallstrom H, Normark S, Jonsson AB. Transcellular passage of Neisseria gonorrhoeae involves pilus phase variation. Infect Immun. 1998;66:469–473. doi: 10.1128/iai.66.2.469-473.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James JF, Swanson J. Studies on gonococcus infection. XIII. Occurrence of color/opacity colonial variants in clinical cultures. Infect Immun. 1978;19:332–340. doi: 10.1128/iai.19.1.332-340.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jerse AE, Cohen MS, Drown PM, Whicker LG, Isbey SF, Seifert HS, Cannon JG. Multiple gonococcal opacity proteins are expressed during experimental urethral infection in the male. J Exp Med. 1994;179:911–920. doi: 10.1084/jem.179.3.911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonsson A-B, Ilver D, Falk P, Pepose J, Normark S. Sequence changes in the pilus subunit lead to tropism variation of Neisseria gonorrhoeae to human tissue. Mol Microbiol. 1994;13:403–416. doi: 10.1111/j.1365-2958.1994.tb00435.x. [DOI] [PubMed] [Google Scholar]
- Jonsson AB, Nyberg G, Normark S. Phase variation of gonococcal pili by frameshift mutation in pilC, a novel gene for pilus assembly. EMBO J. 1991;10:477–488. doi: 10.1002/j.1460-2075.1991.tb07970.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kallstrom H, Liszewski MK, Atkinson JP, Jonsson AB. Membrane cofactor protein (MCP or CD46) is a cellular pilus receptor for pathogenic Neisseria. Mol Microbiol. 1997;25:639–647. doi: 10.1046/j.1365-2958.1997.4841857.x. [DOI] [PubMed] [Google Scholar]
- Kallstrom H, Islam MS, Berggren PO, Jonsson AB. Cell signaling by the type IV pili of pathogenic Neisseria. J Biol Chem. 1998;273:21777–21782. doi: 10.1074/jbc.273.34.21777. [DOI] [PubMed] [Google Scholar]
- Kellogg DS, Jr., Peacock WL, Deacon WE, Brown L, Pirkle CI. Neisseria gonorrhoeae. I. Virulence genetically linked to clonal variation. Journal of Bacteriology. 1963;85:1274–1279. doi: 10.1128/jb.85.6.1274-1279.1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirchner M, Meyer TF. The PilC adhesin of the Neisseria type IV pilus-binding specificities and new insights into the nature of the host cell receptor. Mol Microbiol. 2005;56:945–957. doi: 10.1111/j.1365-2958.2005.04600.x. [DOI] [PubMed] [Google Scholar]
- Kline KA, Sechman EV, Skaar EP, Seifert HS. Recombination, repair and replication in the pathogenic Neisseriae: the 3 R's of molecular genetics of two human-specific bacterial pathogens. Mol Microbiol. 2003;50:3–13. doi: 10.1046/j.1365-2958.2003.03679.x. [DOI] [PubMed] [Google Scholar]
- Knepper B, Heuer I, Meyer TF, van Putten JP. Differential response of human monocytes to Neisseria gonorrhoeae variants expressing pili and opacity proteins. Infect Immun. 1997;65:4122–4129. doi: 10.1128/iai.65.10.4122-4129.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koomey M, Gotschlich EC, Robbins K, Bergstrom S, Swanson J. Effects of recA mutations on pilus antigenic variation and phase transitions in Neisseria gonorrhoeae. Genetics. 1987;117:391–398. doi: 10.1093/genetics/117.3.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambden PR, Robertson JN, Watt PJ. Biological properties of two distinct pilus types produced by isogenic variants of Neisseria gonorrhoeae P9. J Bacteriol. 1980;141:393–396. doi: 10.1128/jb.141.1.393-396.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long CD, Madraswala RN, Seifert HS. Comparisons between colony phase variation of Neisseria gonorrhoeae FA1090 and pilus, pilin, and S-pilin expression. Infect Immun. 1998;66:1918–1927. doi: 10.1128/iai.66.5.1918-1927.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madara JL, Stafford J, Dharmsathaphorn K, Carlson S. Structural analysis of a human intestinal epithelial cell line. Gastroenterol. 1987;92:1133–1145. doi: 10.1016/s0016-5085(87)91069-9. [DOI] [PubMed] [Google Scholar]
- Manning PA, Kaufmann A, Roll U, Pohlner J, Meyer TF, Haas R. L-pilin variants of Neisseria gonorrhoeae MS11. Mol Microbiol. 1991;5:917–926. doi: 10.1111/j.1365-2958.1991.tb00766.x. [DOI] [PubMed] [Google Scholar]
- McCormick BA, Colgan SP, Delp-Archer C, Miller SI, Madara JL. Salmonella typhimurium attachment to human intestinal epithelial monolayers: transcellular signalling to subepithelial neutrophils. J Cell Biol. 1993;123:895–907. doi: 10.1083/jcb.123.4.895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGee ZA, Johnson AP, Taylor-Robinson D. Pathogenic mechanisms of Neisseria gonorrhoeae: observations on damage to human fallopian tubes in organ culture by gonococci of colony type 1 or type 4. J Infect Dis. 1981;143:413–422. doi: 10.1093/infdis/143.3.413. [DOI] [PubMed] [Google Scholar]
- Merz AJ, So M. Interactions of pathogenic neisseriae with epithelial cell membranes. Annu Rev Cell Dev Biol. 2000;16:423–457. doi: 10.1146/annurev.cellbio.16.1.423. [DOI] [PubMed] [Google Scholar]
- Merz AJ, Enns CA, So M. Type IV pili of pathogenic Neisseriae elicit cortical plaque formation in epithelial cells. Mol Microbiol. 1999;32:1316–1332. doi: 10.1046/j.1365-2958.1999.01459.x. [DOI] [PubMed] [Google Scholar]
- Merz AJ, Rifenbery DB, Arvidson CG, So M. Traversal of a polarized epithelium by pathogenic Neisseriae: facilitation by type IV pili and maintenance of epithelial barrier function. Mol Med. 1996;2:745–754. [PMC free article] [PubMed] [Google Scholar]
- Meyer TF, Billyard E, Haas R, Storzbach S, So M. Pilus genes of Neisseria gonorrhoeae: chromosomal organization and DNA sequence. Proc Natl Acad Sci USA. 1984;81:6110–6114. doi: 10.1073/pnas.81.19.6110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morand PC, Bille E, Morelle S, Eugene E, Beretti JL, Wolfgang M, et al. Type IV pilus retraction in pathogenic Neisseria is regulated by the PilC proteins. EMBO J. 2004;23:2009–2017. doi: 10.1038/sj.emboj.7600200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rest RF, Shafer WM. Interactions of Neisseria gonorrhoeae with human neutrophils. Clin Microbiol Rev. 1989;2(Suppl):S83–S91. doi: 10.1128/cmr.2.suppl.s83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rest RF, Fischer SH, Ingham ZZ, Jones JF. Interactions of Neisseria gonorrhoeae with human neutrophils: effects of serum and gonococcal opacity on phagocyte killing and chemiluminescence. Infect Immun. 1982;36:737–744. doi: 10.1128/iai.36.2.737-744.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudel T, Scheurerpflug I, Meyer TF. Neisseria PilC protein identified as type-4 pilus tip-located adhesin. Nature. 1995;373:357–359. doi: 10.1038/373357a0. [DOI] [PubMed] [Google Scholar]
- Rudel T, van Putten JPM, Gibbs CP, Haas R, Meyer TF. Interaction of two variable proteins (PilE and PilC) required for pilus-mediated adherence of Neisseria gonorrhoeae to human epithelial cells. Mol Microbiol. 1992;6:3439–3450. doi: 10.1111/j.1365-2958.1992.tb02211.x. [DOI] [PubMed] [Google Scholar]
- Scheuerpflug I, Rudel T, Ryll R, Pandit J, Meyer TF. Roles of PilC and PilE proteins in pilus-mediated adherence of Neisseria gonorrhoeae and Neisseria meningitidis to human erythrocytes and endothelial and epithelial cells. Infect Immun. 1999;67:834–843. doi: 10.1128/iai.67.2.834-843.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwalbe RS, Sparling PF, Cannon JG. Variation of Neisseria gonorrhoeae protein II among isolates from an outbreak caused by a single gonococcal strain. Infect Immun. 1985;49:250–252. doi: 10.1128/iai.49.1.250-252.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sechman EV, Rohrer MS, Seifert HS. A genetic screen identifies genes and sites involved in pilin antigenic variation in Neisseria gonorrhoeae. Mol Microbiol. 2005;57:468–463. doi: 10.1111/j.1365-2958.2005.04657.x. [DOI] [PubMed] [Google Scholar]
- Segal E, Hagblom P, Seifert HS, So M. Antigenic variation of gonococcal pilus involves assembly of separated silent gene segments. Proc Natl Acad Sci USA. 1986;83:2177–2181. doi: 10.1073/pnas.83.7.2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal E, Billyard E, So M, Storzbach S, Meyer TF. Role of chromosomal rearrangement in N. gonorrhoeae pilus phase variation. Cell. 1985;40:293–300. doi: 10.1016/0092-8674(85)90143-6. [DOI] [PubMed] [Google Scholar]
- Seifert HS, Wright CJ, Jerse AE, Cohen MS, Cannon JG. Multiple gonococcal pilin antigenic variants are produced during experimental human infections. J Clin Invest. 1994;93:2744–2749. doi: 10.1172/JCI117290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serkin CD, Seifert HS. Iron availability regulates DNA recombination in Neisseria gonorrhoeae. Mol Microbiol. 2000;37:1075–1086. doi: 10.1046/j.1365-2958.2000.02058.x. [DOI] [PubMed] [Google Scholar]
- Shaw JH, Falkow S. Model for invasion of human tissue culture cells by Neisseria gonorrhoeae. Infect Immun. 1988;56:1625–1632. doi: 10.1128/iai.56.6.1625-1632.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stohl EA, Seifert HS. The recX gene potentiates homologous recombination in Neisseria gonorrhoeae. Mol Microbiol. 2001;40:1301–1310. doi: 10.1046/j.1365-2958.2001.02463.x. [DOI] [PubMed] [Google Scholar]
- Swanson J, Robbins K, Barrera O, Koomey JM. Gene conversion variations generate structurally distinct pilin polypeptides in Neisseria gonorrhoeae. J Exp Med. 1987a;165:1016–1025. doi: 10.1084/jem.165.4.1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson J, Barrera O, Sola J, Boslego J. Expression of outer membrane protein II by gonococci in experimental gonorrhea. J Exp Med. 1988;168:2121–2129. doi: 10.1084/jem.168.6.2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson J, Bergstr:om S, Barrera O, Robbins K, Corwin D. Pilus- gonococcal variants. Evidence for multiple forms of piliation control. J Exp Med. 1985;162:729–744. doi: 10.1084/jem.162.2.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson J, Bergstrom S, Robbins K, Barrera O, Corwin D, Koomey JM. Gene conversion involving the pilin structural gene correlates with pilus+ in equilibrium with pilus-changes in Neisseria gonorrhoeae. Cell. 1986;47:267–276. doi: 10.1016/0092-8674(86)90449-6. [DOI] [PubMed] [Google Scholar]
- Swanson J, Robbins K, Barrera O, Corwin D, Boslego J, Ciak J, et al. Gonococcal pilin variants in experimental gonorrhea. J Exp Med. 1987b;165:1344–1357. doi: 10.1084/jem.165.5.1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmerman MM, Shao JQ, Apicella MA. Ultrastructural analysis of the pathogenesis of Neisseria gonorrhoeae endometrial infection. Cell Microbiol. 2005;7:627–636. doi: 10.1111/j.1462-5822.2005.00491.x. [DOI] [PubMed] [Google Scholar]
- Tramont EC, Hodge WC, Gilbreath MJ, Ciak J. Differences in attachment antigens of gonococci in reinfection. J Lab Clin Med. 1979;93:730–735. [PubMed] [Google Scholar]
- Wang J, Gray-Owen SD, Knorre A, Meyer TF, Dehio C. Opa binding to cellular CD66 receptors mediates the transcellular traversal of Neisseria gonorrhoeae across polarized T84 epithelial cell monolayers. Mol Microbiol. 1998;30:657–671. doi: 10.1046/j.1365-2958.1998.01102.x. [DOI] [PubMed] [Google Scholar]
- Wolfgang M, Lauer P, Park H-S, Brossay L, Hebert J, Koomey M. PilT mutations lead to simultaneous defects in competence for natural transformation and twitching motility in piliated Neisseria gonorrhoeae. Mol Microbiol. 1998;29:312–330. doi: 10.1046/j.1365-2958.1998.00935.x. [DOI] [PubMed] [Google Scholar]
- Zak K, Diaz J-L, Jackson D, Heckels JE. Antigenic variation during infection with Neisseria gonorrhoeae: Detection of antibodies to surface proteins in sera of patients with gonorrhea. J Infect Dis. 1984;149:166–174. doi: 10.1093/infdis/149.2.166. [DOI] [PubMed] [Google Scholar]
- Zhao S, Tobiason DM, Hu M, Seifert HS, Nicholas RA. The penC mutation conferring antibiotic resistance in Neisseria gonorrhoeae arises from a mutation in the PilQ secretin that interferes with multimer stability. Mol Microbiol. 2005;57:1238–1251. doi: 10.1111/j.1365-2958.2005.04752.x. [DOI] [PMC free article] [PubMed] [Google Scholar]