

Abstract
The renal medulla is sensitive to hypoxia, and a depression of medullary circulation, e.g. in response to angiotensin II (Ang II), could endanger the function of this zone. Earlier data on Ang II effects on medullary vasculature were contradictory. The effects of Ang II on total renal blood flow (RBF), and cortical and medullary blood flow (CBF and MBF: by laser-Doppler flux) were studied in anaesthetised rats. Ang II infusion (30 ng kg−1 min−1i.v.) decreased RBF 27 ± 2 % (mean ± s.e.m.), whereas MBF increased 12 ± 2 % (both P < 0.001). Non-selective blockade of Ang II receptors with saralasin (3 μg kg−1 min−1i.v.) increased RBF 12 ± 2 % and decreased MBF 8 ± 2 % (P < 0.001). Blockade of AT1 receptors with losartan (10 mg kg−1) increased CBF 10 ± 2 % (P < 0.002) and did not change MBF. Losartan given during Ang II infusion significantly increased RBF (53 ± 7 %) and decreased MBF (27 ± 7 %). Blockade of AT2 receptors with PD 123319 (50 μg kg−1 min−1i.v.) did not change CBF or MBF. Intramedullary infusion of PD 123319 (10 μg min−1) superimposed on intravenous Ang II infusion did not change RBF, but slightly decreased MBF (4 ± 2 %, P < 0.05). We conclude that in anaesthetised surgically prepared rats, exogenous or endogenous Ang II may not depress medullary circulation. In contrast to the usual vasoconstriction in the cortex, vasodilatation was observed, possibly related to secondary activation of vasodilator paracrine agents rather than to a direct action via AT2 receptors.
An appropriate perfusion of the renal medulla with blood is necessary for the functional integrity of this zone and for the normal excretory and regulatory function of the whole kidney. This is of major physiological and pathophysiological importance, as the medulla is widely viewed as having a crucial role in maintaining body fluid homeostasis and in the control of arterial pressure (Cowley, 1997).
The intrarenal vasculature can respond to neural and a variety of humoral stimuli with vasodilatation or vasoconstriction, resulting in increased or decreased perfusion of renal tissue, respectively. Such responses may have more serious functional consequences within the medulla than in the cortex. For instance, an increase in medullary blood flow (MBF) could result in a wash-out of solutes from the medullary interstitium, less efficient action of the countercurrent exchanger, and impaired ability of the kidney to concentrate urine (Thurau et al. 1962; Cupples & Sonnenberg, 1987). More recent studies have shown that this particular risk was exaggerated and that unrealistically large increases in MBF would be needed to significantly dissipate the medullary interstitial hypertonicity (Cupples & Sonnenberg, 1987; Sadowski et al. 1997).
A decrease in blood and oxygen supply to the renal medulla could present a more serious danger as, even at normal perfusion, oxygen tension in the medullary tissue is low and the medulla appears to function at the edge of anoxia (Heyman et al. 1997). Thus, a depression of medullary circulation, e.g. following a release of renin and increased generation of angiotensin II (Ang II), could seriously endanger the medullary tissue. On the other hand, evidence has accumulated over the years to indicate that perfusion of the renal medulla is controlled separately, independent of the control of blood flow through the cortex, which represents the greater part of the renal mass.
The present study was undertaken to examine whether changing activity of Ang II would affect the renal cortical and medullary circulation in a parallel fashion, and thus if high hormone activity would indeed compromise perfusion and, potentially, the function of the medulla. This subject has not yet been sufficiently elucidated, largely due to the doubtful value of early methods of measurement of the MBF or papillary blood flow (PBF). The progress in this area has accelerated following the introduction of laser-Doppler (LD) methodology, and newer studies have suggested that, unlike the cortex, the medulla may not respond with vasoconstriction to exogenous Ang II (Nobes et al. 1991; Ortíz et al. 1998). To simplify interpretation, in the present work, effects of high and low Ang II level on MBF (LD flux) were examined while renal perfusion pressure (RPP) was artificially maintained constant. The study focused on the role of two Ang II receptor types, AT1 and AT2. We found that the medullary vasculature did not constrict with increasing or dilate with decreasing hormone concentration or action, as was simultaneously observed in the cortex. The MBF changed in the direction opposite to changes in cortical blood flow (CBF), a response that was probably related to the secondary activation of the synthesis of paracrine agents, such as prostaglandins, kinins and nitric oxide (NO).
METHODS
Male Wistar rats weighing 280–330 g, maintained on a dry pellet diet and given free access to water, were anaesthetised with intraperitoneal thiopentabarbital (Thiopental, Biochemie GmbH, Vienna, Austria), 100 mg (kg body wt)−1. The experimental procedures were approved by the Ethical Committee of the Medical Research Centre, Polish Academy of Sciences. Throughout the surgical preparation and experimental procedures, body temperature was maintained at about 37 °C by means of a heated pad. In order to compensate for fluid losses, during surgical preparation 3 % bovine albumin in Ringer solution was infused at 0.22 ml kg−1 min−1.
Surgical preparations
A cannula was placed in the trachea to ensure free airways. Femoral and jugular veins were cannulated for infusion of fluids and drugs. For measurement of mean arterial blood pressure, a Teflon catheter, 0.4 mm in outer diameter, was introduced into the right femoral artery. The left kidney was exposed by a subcostal flank incision and placed in a plastic holder similar to that used for micropuncture experiments; the inside of the holder was padded in such a way that the kidney's dorsal curvature (and not the side surface) was facing upwards. The ureter was cannulated to ensure drainage of the left renal pelvis. In order to keep RPP stable, a screw-controlled snare was placed on the aorta above the left renal artery. The snare was tightened slightly to lower blood pressure distal to the constriction site by 10 mmHg. Later during experiments, the snare could be loosened or tightened as needed, so that the mean blood pressure below the snare, equivalent to RPP, was maintained constant. Thereafter, a flow probe, 1 mm in diameter, was placed on the renal artery and connected with a Transonic flow meter (type T106, Transonic Systems Inc., Ithaca, NY, USA) for measurement of total renal blood flow (RBF). Subsequently, a needle laser-Doppler (LD) probe (type PF 402, Perimed, Jarfalla, Sweden), for estimation of medullary blood flow (MBF), was inserted into the kidney from its dorsal surface along the cortico-papillary axis. In one series of experiments, parallel to the LD probe, a stainless steel cannula (25 G) was inserted for drug infusion directly into the renal medulla. In some experimental series, total RBF was not measured. Instead, a second LD probe (type PF 407/415) was placed on the kidney surface for estimation of superficial cortical blood flow (CBF; LD flux); this measurement would not detect changes in perfusion of juxtamedullary glomeruli. To ensure a stable contact of this probe with the kidney surface, its tip was weighted and the probe was suspended on its fibre-optic lead, fastened to a horizontal rod positioned 20 cm above the kidney. The surface probe was applied exclusively in experiments in which RPP was maintained constant.
In 20 preliminary experiments, the Transonic probe was placed on the renal artery and an LD probe was placed on the kidney surface for simultaneous measurement of total RBF and CBF. The percentage changes in both parameters, recorded after infusion of Ang II or saralasin, were highly correlated (r = 0.959, P < 0.001). This indicated that measurement of total RBF provided a reliable estimate of the changes in perfusion of the superficial cortex.
The LD probes were connected to a LD flow meter (Periflux 4001, Perimed). The system measures the LD flux (a product of the number of blood cells moving and of their mean velocity) within an area less than 1 mm3 beneath the tip of the probes. Results are expressed in arbitrary perfusion units (PU). Thus, only relative changes are measured, but a calibration procedure enabled comparison of the results between animals. Each probe was calibrated using a motility standard, a colloidal suspension of latex particles (supplied by the manufacturer). The Brownian motion of the suspension provides the standard value of 250 PU. The biological zero flow was determined at the end of experiments after clamping the renal artery. In agreement with accepted usage, throughout this paper the cortical and medullary LD fluxes are termed ‘flows’ (CBF and MBF, respectively).
After experiments, the rats were killed with an overdose of the anaesthetic. The position of the LD probe in the inner medulla (close to the border with the outer medulla) and of the parallel cannula for intramedullary infusion were verified after the experiments in kidney cross-sections.
Experimental procedures and protocols
At the end of surgical preparations, bovine serum albumin infusion was replaced by isotonic saline solution at 0.22 ml kg−1 min−1. After placement of blood flow probes for measurement of MBF and RBF or CBF, about 1 h was allowed for stabilisation, and RPP was adjusted to a value that was maintained constant throughout the experiment (with the exception of one experimental series, see below). We have shown that in this experimental preparation and at constant RPP, CBF and MBF are stable over at least 2 h (Dobrowolski et al. 1998; Kompanowska-Jezierska et al. 2001).
The baseline values were first recorded for 30 min during isotonic saline infusion. Thereafter, records were obtained during infusion of drugs and, whenever possible, after cessation of drug infusion.
Effects of exogenous Ang II on RBF and MBF
Infusion of Ang II (Hypertensin, Ciba-Geigy, Switzerland) at 30 ng kg−1 min−1i.v. was performed in two groups of rats. In eight rats (Group 1), RPP was allowed to change freely during the experiment. After obtaining control records, Ang II was infused for about 30 min, until stabilisation of MBF and RBF. Thereafter, Ang II was replaced by saline vehicle for about 20 min to record recovery of the haemodynamic parameters measured. In a large group of animals (Group 2, n = 39), Ang II was infused in the same dose as above while RPP was maintained constant. The usual baseline and Ang II recordings of RBF and MBF were performed. In these animals, Ang II infusion was later used as a background for administration of other drugs (see below). Therefore, post-angiotensin recovery records could not be obtained.
Effects of angiotensin converting enzyme (ACE) inhibition on CBF and MBF
The angiotensin converting enzyme (ACE) inhibitor captopril (E. R. Squibb & Sons Ltd, Hounslow, UK) was administered i.v. as a bolus of 1 mg kg−1 in 0.5 ml saline given over 5 min. Captopril was also added to the maintenance saline infusion to deliver 1 mg kg−1 h−1 over 30–40 min until stabilisation of CBF and MBF (n = 17).
Effects of non-selective blockade of Ang II receptors by saralasin on RBF and MBF
After baseline recordings, 1-Sar, 8-Ala Ang II (saralasin), a non-selective peptide inhibitor of Ang II receptors (Roehm Pharma GmbH, Darmstadt, Germany), was infused at 3 μg kg−1 min−1i.v. for 40 min while RPP was maintained constant (n = 13). In another group, the saralasin infusion was superimposed on Ang II infusion in the usual dose (n = 17).
Effects of selective blockade of Ang II receptors by losartan or PD 123319 on MBF and RBF or CBF
(1) After baseline recordings, losartan, a selective, non-peptide AT1 angiotensin receptor antagonist (Merck & Co., Rahway, NJ, USA), was injected i.v. as a bolus of 10 mg kg−1 in 0.5 ml saline (n = 10). In another group, losartan injection was made during Ang II infusion in the usual dose (n = 10). RBF or CBF and MBF were recorded for about 20 min. (2) PD 123319, a selective, non-peptide AT2 angiotensin receptor antagonist (Research Biochemicals International, Natick, MA, USA), was infused i.v. at 50 μg kg−1 min−1 for 20 min (n = 8). In another group, the drug was infused directly into the medulla at 5–10 μg min−1 (infusion rate: 17 μl min−1) for 20 min, superimposed on i.v. infusion of Ang II in the usual dose (n = 12).
Statistics
The significance of changes within one group over time was first evaluated by repeat measurement analysis of variance (ANOVA), followed by Student's t test for dependent variables. Differences in mean values between groups were evaluated by Student's t test for independent variables. Absolute values were used for statistical calculations, whereas percentage changes are shown in graphs. s.e.m. was used throughout as a measure of data dispersion; however, when the values were too small to be visible on graphs, the s.d. is shown and the appropriate information is given in the legend.
RESULTS
The data on the effect of exogenous Ang II on RBF and MBF are given in Fig. 1. Figure 1A shows the influence of Ang II in experiments in which RPP was allowed to increase significantly, from a control value of 113 ± 3 mmHg to 123 ± 2 mmHg under Ang II. After cessation of infusion, RPP decreased to 116 ± 2 mmHg. Ang II induced a significant 31 % decrease in RBF (from a control value of 7.7 ± 0.4 ml min−1), contrasted by a significant 17 % increase in MBF from a control value of 186 ± 10 PU. The recovery after withdrawal of Ang II was almost complete; however, RBF remained 4 % below control. Figure 1B shows the data pooled from all experimental groups in which Ang II was infused while RPP was maintained constant at 102 ± 1 mmHg. After Ang II, RBF decreased significantly (27 %), from a control value of 8.6 ± 0.3 ml min−1, whereas MBF increased significantly (12 %), from a control value of 171 ± 8 PU. These changes were similar to those noted without control of RPP. Ang II infusion was continued in these experiments to provide elevated background activity before administration of other drugs and post-angiotensin recovery could not be recorded.
Figure 1. Effect of i.v. Ang II infusion (30 ng kg−1 min−1) on total RBF and renal MBF (LD flux).
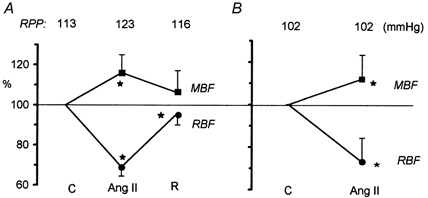
The RPP was allowed to increase (A, n = 8) or was maintained constant at 102 ± 1 mmHg (B, n = 39 for RBF and n = 31 for MBF). Data are presented as means ± s.d. as a percentage of the initial control value. C, control; R, post-Ang II recovery. *P < 0.05 or less versus control.
Effects of inhibition of Ang II generation with captopril on CBF and MBF in a large group of rats (n = 17) are shown in Fig. 2. CBF increased significantly about 10 %, from a control value of 539 ± 17 PU, whereas MBF remained stable at about 120 PU.
Figure 2. Effect of i.v. captopril administration on renal CBF and MBF (LD fluxes).
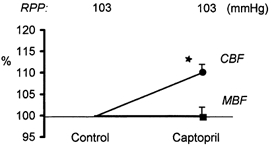
Captopril was given i.v. as a bolus, 1 mg kg−1, followed by an infusion of 1 mg kg−1 h−1i.v., at constant RPP (n = 17). Data are presented as means ± s.e.m., as a percentage of the initial control value. *P < 0.0001 versus control.
Sample records of RBF and MBF responses to saralasin, a non-selective inhibitor of Ang II receptors, observed without or with background infusion of Ang II, are shown in Fig. 3. The RBF first showed a clear decrease, which was more pronounced without background Ang II infusion. This was followed by an increase, which was slower under Ang II, but the final level achieved was higher than that seen without hormone infusion. In the absence of exogenous Ang II, RBF stabilised at an elevated level beginning from about the sixth minute of saralasin infusion. MBF decreased after saralasin, similarly in the presence or absence of exogenous Ang II; however, the recovery that followed was only partial, and MBF remained low throughout saralasin infusion.
Figure 3. Sample records of total RBF (A) and MBF (B) responses to i.v. saralasin (3 μg kg−1 min−1) in two experiments.
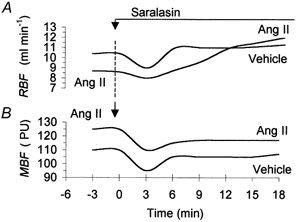
In one experiment, Ang II was continuously infused at 30 ng kg−1 min−1i.v. In the other, Ang II vehicle was given. PU, LD perfusion units.
All the data on the effects of saralasin infusion are collected in Fig. 4. In the absence of exogenous Ang II (Fig. 4A), RBF decreased about 15 %, from a control value of 8.0 ± 0.9 ml min−1, a change similar to that seen for MBF. Subsequently, RBF increased to a value significantly higher than control (8.7 ± 1.0 ml min−1), whereas MBF recovered only slightly and remained significantly lower (8 %) than control, at 117 ± 8 PU, throughout saralasin infusion. When saralasin was given on a background of Ang II infusion (Fig. 4B), the initial 8 % decrease in RBF, from a control value of 6.2 ± 0.5 ml min−1, was significant but significantly smaller than in the absence of exogenous Ang II. In the second phase, RBF increased to 8.3 ± 0.5 ml min−1, significantly above control, a change greater than that seen without Ang II infusion. MBF decreased in the first phase by about 17 %, from a control value of 180 ± 7 PU, but then tended to recover, and at the end of saralasin infusion was not significantly different from control.
Figure 4. Biphasic effect of i.v. saralasin (S, 3 μg kg−1 min−1) on total RBF and MBF.
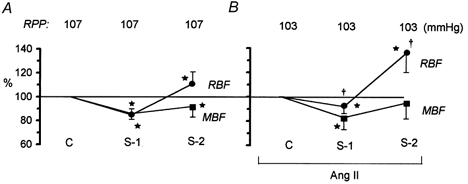
A, data recorded with Ang II vehicle (n = 13). B, data recorded with background Ang II infusion, 30 ng kg−1 min−1i.v. (n = 17). The RPP was maintained constant. S-1, S-2, early and final effects of saralasin, respectively. Means ± s.d. for values expressed as a percentage of the pre-saralasin control value (C). *P < 0.05 or less versus pre-saralasin control. †P < 0.002 or less versus the change in the corresponding value measured without background Ang II infusion (A).
Selective inhibition of angiotensin AT1 receptors with losartan moderately but significantly increased CBF, by 10 % from a control value of 440 ± 23 PU, without affecting MBF (Fig. 5A). Losartan was also administered during infusion of Ang II; in this series, total RBF was measured instead of CBF (Fig. 5B). After losartan, RBF increased 53 %, from a control value of 6.4 ± 0.6 ml min−1, significantly more than did CBF in the absence of exogenous Ang II. In this series, MBF decreased significantly (21 %), from a control value of 163 ± 13 PU.
Figure 5. Effects of i.v. losartan on renal haemodynamics.
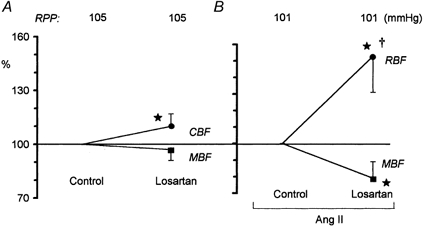
A, effects of losartan, 10 mg kg−1i.v. (n = 10), on renal CBF and MBF. B, total RBF and MBF after losartan given in the same dose during Ang II infusion, 30 ng kg−1 min−1i.v. (n = 10). Data are presented as means ± s.d., as a percentage of the pre-losartan control value. *P < 0.05 or less versus pre-losartan control. †P < 0.001 versus the CBF value measured after losartan alone (as shown in A).
PD 123319, a relatively selective inhibitor of angiotensin AT2 receptors, did not change RBF or CBF, either with i.v. PD 123319 infusion or with the intramedullary infusion superimposed on i.v. Ang II infusion (data not shown). After i.v. PD 123319, MBF tended to decrease (not significant; Fig. 6). After intramedullary infusion of the drug, a slight (4 %) decrease in MBF, from 187 ± 17 to 179 ± 16 PU, proved significant (P = 0.03; Fig. 6).
Figure 6. The renal MBF responses to PD 123319 (PD).
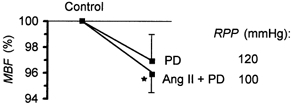
PD 123319 was given i.v., 50 μg kg−1 min−1 (PD; n = 8), or infused directly into the medullary interstitium, 5–10 μg min−1, during background infusion of Ang II (30 ng kg−1 min−1i.v.) (Ang II + PD; n = 12). The RPP was maintained constant in both groups. Data are presented as means ± s.e.m. as a percentage of the pre-PD control value. *P < 0.05 versus pre-PD control.
DISCUSSION
The present study extensively documents increased perfusion of the renal medulla in response to a moderate pressor dose of Ang II, in contrast to the well established vasoconstrictor action of the hormone within the cortex. Both when RPP was allowed to increase, and when it was maintained constant, the total RBF, representing largely perfusion of the cortex, decreased after Ang II, whereas the MBF showed a significant increase.
Previous data on the response of the intrarenal circulation to angiotensin were not uniform. In agreement with our results, all studies reported a dose-dependent decrease in total RBF or CBF. When perfusion of the medulla was measured separately, a number of workers reported no change in MBF or PBF (Huang et al. 1991; Mattson et al. 1991; Nobes et al. 1991; Parekh & Zou 1996). A medullary vasodilatation in response to Ang II was also seen. Nobes et al. (1991), who positioned an LD probe over the exposed papilla of young rats, noted an increase in PBF, albeit only with the highest dose used (300 ng kg−1 min−1); no change was seen at 100 ng kg−1 min−1. Using similar methodology (an external LD probe over the exposed papilla) in adult rats, Ortiz et al. (1998) were able to demonstrate a dose-dependent increase in PBF both at 100 and 300 ng kg−1 min−1. It is apparent that in studies involving exposure of the papilla, variability of the results may relate to the use of rats of different size (age) or strain. As in the two studies mentioned above (Nobes et al. 1991; Ortiz et al. 1998), we measured regional renal perfusion by the LD technique. However, we used an internal probe placed in the inner (white) medulla, close to the border with the outer medullary zone. This approach does not require exposure of the papilla, a manoeuvre that can alter papillary circulation and its responsiveness to any stimuli (Pallone et al. 1990). Perhaps due to a relatively high sensitivity of our experimental preparation, we were able to demonstrate a definite increase in MBF with an Ang II dose as low as 30 ng kg−1 min−1.
The absence of a depression of medullary circulation after Ang II, as documented in the above quoted and our own whole kidney studies, does not accord with evidence from work using a variety of other experimental preparations, such as split hydronephrotic kidney, isolated perfused juxtaglomerular nephron, isolated renal arterial vessels of different calibre, as well as from measurements of blood cell velocity in visualised vasa recta of an exposed papilla; virtually all of these studies showed constriction of medullary vessels after Ang II (reviewed in Pallone et al. 1990; Navar et al. 1996; Arendshorst et al. 1999). This indicates that the medullary vasculature has the potential to respond to Ang II in the same way as is seen in the cortex; however, vasoconstriction does not manifest itself in many whole animal studies, with the kidney functioning in the natural milieu of the organism.
The majority of data indicate that inhibition of ACE results in dilatation of both the cortical and medullary vasculature (reviewed in Pallone et al. 1990). This is at variance with our present finding of no change of MBF after captopril, as opposed to a clear increase in CBF. However, since ACE is also a kininase, the concentration of vasodilator kinins would increase after captopril administration. Indeed, there is long-standing evidence for interaction of Ang II and kinins in the control of PBF (Roman et al. 1988). Since in our experience, exogenous Ang II dilated medullary vasculature, elimination of the hormone after captopril could be expected to cause vasoconstriction. Possibly, such an action was, indeed, induced but the effect was offset by an accumulation of the vasodilator bradykinin, and the final result was no change in MBF. In a recent study in the dog, a blockade of bradykinin B2 receptors abolished the effect of ACE inhibition on MBF (Omoro et al. 2000).
In order to further explore the role of endogenous Ang II in the control of regional renal circulation, we examined effects of non-selective blockade of both AT1 and AT2 angiotensin receptors with saralasin. It is remarkable that an expected increase of RBF after saralasin was preceded by a significant decrease. This probably reflected the known agonistic properties of saralasin and other similar peptides (Hollenberg, 1979; Harris et al. 1986; Walker et al. 1986); the agonistic action was reported to be more pronounced when endogenous Ang II levels were low and to subside within about 15 min. These observations agree with our data showing that the initial decrease in RBF was less pronounced when saralasin was given during Ang II infusion and was, indeed, transient (Fig. 4). It is remarkable that the biphasic, first agonistic and later antagonistic, action of saralasin did not become apparent in the medulla, an observation that confirms the view of different responsiveness of this zone to Ang II. The ultimate effect of non-selective blockade of Ang II receptors with saralasin – vasodilatation within the cortex and a tendency towards, or overt, vasoconstriction within the medulla – fits the overall pattern of dissociated responses of the cortical versus the medullary vasculature, which emerged from our studies with exogenous Ang II and with captopril. Collectively, the data provide coherent evidence that the net vasoconstrictor action of both exogenous and endogenous hormone is confined to the cortical vasculature.
The mechanisms underlying the unusual influence of Ang II on medullary vasculature are not clear. One possibility is the hormone's action via AT2 receptors. Whereas earlier literature emphasised their role in growth processes in the fetus, there is now increasing evidence that AT2 receptors can mediate systemic vasodilatation and lower arterial blood pressure (de Gasparo & Siragy, 1999). However, a direct angiotensin-dependent vasodilatation in the medulla might become apparent only if the action mediated by AT2 receptors prevailed over vasoconstriction dependent on stimulation of AT1 receptors. This is unlikely, as the density of AT2 receptors in the kidney seems to be much lower than that of AT1. Thus, AT2 receptor-dependent vasodilatation could probably be demonstrated only when AT1 receptors have been blocked (Csikós et al. 1998; Matsubara, 1998; Arendshorst et al. 1999).
Ang II-induced vasodilatation, as seen in the medulla, could also have been indirect, dependent on stimulation by this hormone of the biosynthesis of vasodilator agents, such as prostaglandins, kinins or NO. It has been established that the rate of synthesis and tissue concentration of prostaglandins are much higher in the medulla compared with the cortex, and Ang II stimulates prostaglandin synthesis via AT1 receptors (Siragy & Carey, 1996). A potential vasodilator role of kinins is suggested by the observation that an increase in PBF in response to Ang II was prevented by inhibition of kallikrein (Nobes et al. 1991). Still another possible mediator of vasodilatation could be NO. First, exogenous Ang II was reported to increase NO concentration in renal tissue more effectively in the medulla (Zou et al. 1997). Furthermore, inhibition of NO synthesis prevented an increase in perfusion of the medulla after Ang II (Zou et al. 1997; Ortíz et al. 1998). It was proposed that stimulation of NO synthesis by Ang II is mediated by AT2 receptors (Siragy & Carey, 1997); however, there is also evidence that AT1 and other ill-defined Ang II or angiotensin (1–7) receptors may also be involved (Ferrario et al. 1998; Thorup et al. 1999).
In order to further investigate the mechanism of medullary vasodilatation after angiotensin, we examined renal regional circulatory responses to losartan, a selective antagonist of AT1 receptors, and to PD 123319, a relatively selective AT2 antagonist. As expected, losartan increased perfusion of the cortex and, as observed with saralasin, the increase was greater in the presence of high Ang II concentration induced by i.v. infusion (Fig. 4 and Fig. 5).
In this study, both saralasin and losartan caused a decrease in medullary perfusion, an effect compatible with the increase seen in the present studies after exogenous Ang II. Obviously, the decrease in MBF after losartan cannot be explained by abolition of a direct action of Ang II on AT1 receptors in the medullary vasculature, and indirect mechanisms discussed above must be invoked. The blockade of these receptors would also eliminate stimulation by Ang II of the biosynthesis of prostaglandin E2 (PGE2) and remove its tonic vasodilator influence on medullary vasculature, leading ultimately to vasoconstriction. Indeed, losartan was found to decrease renal interstitial fluid PGE2 concentration (Siragy et al. 1996). The effect would be more pronounced at high baseline Ang II concentration, as was, indeed, observed when losartan was administered during Ang II infusion (Fig. 5). There is ample evidence for a tonic vasodilator influence of prostaglandins on the medullary circulation (Knox & Granger, 1992); in our acute experiments with anaesthetised rats, indomethacin always reduced MBF (Sadowski et al. 1997; Kompanowska-Jezierska et al. 1999). On the other hand, Nobes et al. (1991) showed that, at least in young rats with exposed papilla, Ang II-induced increase in PBF could not be prevented by indomethacin. Since the stimulation of kinins by Ang II seems to be mediated by non-AT1, possibly by AT2, receptors (Siragy et al. 1996; Siragy, 2000), the decrease of MBF after losartan cannot be explained by the elimination of a tonic vasodilator action of kinins.
It is not clear why losartan decreased MBF distinctly more than did saralasin; obviously, a confounding factor was that the latter displayed a transition in time from AT1 agonistic to antagonistic properties as described above.
Effects of blockade of angiotensin AT2 receptors on regional renal circulation detected in this study were non-existent to modest. After i.v. infusion of PD 123319, a selective AT2 antagonist, neither RBF nor MBF changed. In order to provide a fairly high concentration of the drug in the medulla, without provoking possible extrarenal effects resulting from a high concentration in systemic blood, PD 123319 was infused directly into the medulla. Moreover, to enhance any potential effect of receptor blockade, the baseline plasma concentration of Ang II was first raised by its i.v. infusion. Under such conditions, a slight decrease in MBF (but not in RBF) was seen and proved significant. These results suggest that in acute experiments with anaesthetised surgically prepared rats, the vasodilator activity of AT2 receptors, whether direct or mediated by NO and/or kinins, is minor. Nevertheless, this is the first demonstration of an effect of AT2 antagonism on regional RBF in normal rats. A tendency (insignificant) to a decrease in RBF after an i.v. dose of PD 123319 sixfold higher than we used was reported in the anaesthetised dog (Keiser et al. 1992). It should be noted that in this study, a dose comparable to ours appeared biologically active; for instance, it significantly reduced urine osmolality. This result is not compatible with our observation of a decrease in MBF: such a change could possibly enhance urine concentration due to a more effective action of the countercurrent exchanger and an increase in medullary hypertonicity.
In summary, our results document opposite vascular effects of exogenous and endogenous Ang II on the circulation in the renal cortex and medulla. The focus of the study was on the role of AT1 and AT2 receptors; it was shown that differential regional responses cannot simply be explained by constriction or relaxation of the vascular smooth muscle as a direct consequence of the different activation status of two angiotensin receptor types. The present data suggest very strongly that activation of the renin-angiotensin system, as observed under conditions of arterial hypotension, hypovolaemia or sodium deficit or following a variety of stressful stimuli, would probably not compromise perfusion of the renal medulla with blood. This is of major importance, as even under normal conditions, the medullary tissue has only a small reserve of oxygen and, indeed, appears to function on the verge of hypoxia (Heyman et al. 1997).
Acknowledgments
Losartan was kindly supplied by Dr Ronald D. Smith of Merck Research Laboratories, Rahway, NJ, USA.
REFERENCES
- Arendshorst WJ, Brännström K, Ruan X. Action of angiotensin II on the renal microvasculature. Journal of the American Society of Nephrology. 1999;10:S149–161. [PubMed] [Google Scholar]
- Cowley AW. Role of the renal medulla in volume and arterial blood pressure regulation. American Journal of Physiology. 1997;273:R1–15. doi: 10.1152/ajpregu.1997.273.1.R1. [DOI] [PubMed] [Google Scholar]
- Csikós T, Chung O, Unger Th. Receptors and their classification: Focus on angiotensin II and the AT2 receptor. Journal of Human Hypertension. 1998;12:311–318. doi: 10.1038/sj.jhh.1000639. [DOI] [PubMed] [Google Scholar]
- Cupples WA, Sonnenberg H. Renal medullary plasma flow rate and reabsorption of salt and water from inner medullary collecting duct. Canadian Journal of Physiology and Pharmacology. 1987;65:2415–2421. doi: 10.1139/y87-383. [DOI] [PubMed] [Google Scholar]
- de Gasparo M, Siragy HM. The AT2 receptor: fact, fancy and fantasy. Regulatory Peptides. 1999;81:11–24. doi: 10.1016/s0167-0115(99)00023-3. [DOI] [PubMed] [Google Scholar]
- Dobrowolski L, Badzynska B, Walkowska A, Sadowski J. Osmotic hypertonicity of the renal medulla during changes in renal perfusion pressure in the rat. Journal of Physiology. 1998;508:929–935. doi: 10.1111/j.1469-7793.1998.929bp.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrario CM, Chappel MC, Dean RH, Iyer SN. Novel angiotensin peptides regulate blood pressure, endothelial function, and natriuresis. Journal of the American Society of Nephrology. 1998;9:1716–1722. doi: 10.1681/ASN.V991716. [DOI] [PubMed] [Google Scholar]
- Harris PJ, Mitchell KD, Munro JO. Sar 1-leu 8-angiotensin II reverses the effect of captopril on renal function in rats. Clinical and Experimental Pharmacology and Physiology. 1986;13:77–82. doi: 10.1111/j.1440-1681.1986.tb00318.x. [DOI] [PubMed] [Google Scholar]
- Heyman SN, Rosen S, Brezis M. The renal medulla: Life at the edge of anoxia. Blood Purification. 1997;15:232–242. doi: 10.1159/000170341. [DOI] [PubMed] [Google Scholar]
- Hollenberg NK. Pharmacologic interruption of the renin-angiotensin system. Annual Review of Pharmacology and Toxicology. 1979;19:559–582. doi: 10.1146/annurev.pa.19.040179.003015. [DOI] [PubMed] [Google Scholar]
- Huang C, Davis G, Johns EJ. A study of the action of angiotensin II on perfusion through the cortex and papilla of the rat kidney. Experimental Physiology. 1991;76:787–798. doi: 10.1113/expphysiol.1991.sp003544. [DOI] [PubMed] [Google Scholar]
- Keiser JA, Bjoerk FA, Hodges JC, Taylor DG., Jr Renal hemodynamic and excretory responses to PD 123319 and losartan, nonpeptide AT1 and AT2 subtype-specific angiotensin II ligands. Journal of Pharmacology and Experimental Therapeutics. 1992;262:1154–1160. [PubMed] [Google Scholar]
- Knox FG, Granger JP. Control of sodium excretion: An integrative approach. In: Windhager EE, editor. Renal Physiology. New York and Oxford: Oxford University Press; 1992. pp. 944–945. [Google Scholar]
- Kompanowska-Jezierska E, Walkowska A, Johns EJ, Sadowski J. Early effects of renal denervation in the anaesthetised rat: Natriuresis and increased cortical blood flow. Journal of Physiology. 2001;531:527–534. doi: 10.1111/j.1469-7793.2001.0527i.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kompanowska-Jezierska E, Walkowska A, Sadowski J. Exaggerated volume expansion natriuresis in rats preloaded with hypertonic saline: A paradoxical enhancement by inhibition of prostaglandin synthesis. Acta Physiologica Scandinavica. 1999;167:189–194. doi: 10.1046/j.1365-201x.1999.00604.x. [DOI] [PubMed] [Google Scholar]
- Matsubara H. Pathophysiological role of angiotensin II type 2 receptor in cardiovascular and renal diseases. Circulation Research. 1998;83:1182–1191. doi: 10.1161/01.res.83.12.1182. [DOI] [PubMed] [Google Scholar]
- Mattson DL, Raff H, Roman RJ. Influence of angiotensin II on pressure natriuresis and renal hemodynamics in volume-expanded rats. American Journal of Physiology. 1991;260:R1200–1209. doi: 10.1152/ajpregu.1991.260.6.R1200. [DOI] [PubMed] [Google Scholar]
- Navar LG, Inscho EW, Majid DSA, Imig JD, Harrison-Bernard LM, Mitchell KD. Paracrine regulation of the renal microcirculation. Physiological Reviews. 1996;76:425–536. doi: 10.1152/physrev.1996.76.2.425. [DOI] [PubMed] [Google Scholar]
- Nobes MS, Harris PJ, Yamada H, Mendelsohn FAO. Effects of angiotensin on renal cortical and papillary blood flows measured by laser-Doppler flowmetry. American Journal of Physiology. 1991;261:F998–1006. doi: 10.1152/ajprenal.1991.261.6.F998. [DOI] [PubMed] [Google Scholar]
- Omoro SA, Majid DS, El Dahr SS, Navar LG. Roles of Ang II and bradykinin in the renal regional blood flow responses to ACE inhibition in sodium-depleted dogs. American Journal of Physiology – Renal Physiology. 2000;279:F289–293. doi: 10.1152/ajprenal.2000.279.2.F289. [DOI] [PubMed] [Google Scholar]
- Ortíz MC, Fortepiani LA, Ruiz-Marcos FM, Atucha NM, García-Estań J. Role of AT1 receptors in the renal papillary effects of acute and chronic nitric oxide inhibition. American Journal of Physiology. 1998;274:R760–766. doi: 10.1152/ajpregu.1998.274.3.R760. [DOI] [PubMed] [Google Scholar]
- Pallone TL, Robertson CR, Jamison RL. Renal medullary microcirculation. Physiological Reviews. 1990;70:907–912. doi: 10.1152/physrev.1990.70.3.885. [DOI] [PubMed] [Google Scholar]
- Parekh N, Zou AP. Role of prostaglandins in renal medullary circulation: Responses to different vasoconstrictors. American Journal of Physiology. 1996;271:F653–658. doi: 10.1152/ajprenal.1996.271.3.F653. [DOI] [PubMed] [Google Scholar]
- Roman RJ, Kaldunski ML, Scicli AG, Carretero OA. Influence of kinins and angiotensin II on the regulation of papillary blood flow. American Journal of Physiology. 1988;255:F690–698. doi: 10.1152/ajprenal.1988.255.4.F690. [DOI] [PubMed] [Google Scholar]
- Sadowski J, Kompanowska-Jezierska E, Dobrowolski L, Walkowska A, Badzynska B. Simultaneous recording of tissue ion content and blood flow in rat renal medulla: Evidence on interdependence. American Journal of Physiology. 1997;273:F658–662. doi: 10.1152/ajprenal.1997.273.4.F658. [DOI] [PubMed] [Google Scholar]
- Siragy HM. AT1 and AT2 receptors in the kidney; role in disease and treatment. American Journal of Kidney Diseases. 2000;6:S4–S9. doi: 10.1053/ajkd.2000.9684. [DOI] [PubMed] [Google Scholar]
- Siragy HM, Carey RM. The subtype-2 (AT2) angiotensin receptor regulates renal cyclic guanosine 3,5-monophosphate and AT1 receptor-mediated prostaglandin E2 production in conscious rats. Journal of Clinical Investigation. 1996;97:1978–1982. doi: 10.1172/JCI118630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siragy HM, Carey RM. The subtype 2 (AT2) angiotensin receptor mediates renal production of nitric oxide in conscious rats. Journal of Clinical Investigation. 1997;100:264–269. doi: 10.1172/JCI119531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siragy HM, Jaffa AA, Margolius HS, Carey RM. Renin-angiotensin system modulates renal bradykinin production. American Journal of Physiology. 1996;271:R1090–1095. doi: 10.1152/ajpregu.1996.271.4.R1090. [DOI] [PubMed] [Google Scholar]
- Thorup C, Kornfeld M, Goligorsky MS, Moore LC. AT1 receptor inhibition blunts angiotensin II-stimulated nitric oxide release in renal arteries. Journal of the American Society of Nephrology. 1999;10:S220–224. [PubMed] [Google Scholar]
- Thurau K, Deetjen P, Günzler H. Die Diurese bei arteriellen Drucksteigerungen. Bedeutung der Haemodynamik des Nierenmarks für die Harnkonzentrierung. Pflügers Archiv für die Gesamte Physiologie des Menschen und Tiere. 1962;274:567–580. [PubMed] [Google Scholar]
- Walker LA, Gellai M, Valtin H. Renal response to pentobarbital anesthesia in rats: Effect of interrupting the renin-angiotensin system. Journal of Pharmacology and Experimental Therapeutics. 1986;236:721–728. [PubMed] [Google Scholar]
- Zou A, Wu F, Cowley AW., Jr Protective effect of angiotensin II-induced increase in nitric oxide in the renal medullary circulation. Hypertension. 1997;31:271–276. doi: 10.1161/01.hyp.31.1.271. [DOI] [PubMed] [Google Scholar]