

Abstract
The importance of specific protein kinase C (PKC) sites for modulation of the inhibitory coupling of 5-HT1A receptors to N-type Ca2+ channels was examined using patch-clamp techniques in F11 rat dorsal root ganglion × mouse neuroblastoma hybrid cells. The PKC activator phorbol 12-myristate 13-acetate (PMA, 10 nm) reduced by 28.6 ± 6.8% 5-HT-mediated, but not GTP-γ-S-induced, inhibition of Ca2+ current, whereas a higher concentration of PMA (500 nm) inhibited both the actions of 5-HT and GTP-γ-S. 5-HT1A receptor expression plasmids with or without mutation of a single PKC site in the second intracellular loop (i2, T149A) or of three PKC sites located in the third intracellular loop (i3, T229A-S253G-T343A) were stably transfected into F11 cells. The T149A 5 HT1A receptor inhibited forskolin-stimulated cyclic AMP levels but was largely uncoupled from Ca2+ channel modulation. In one (i2) clone a response rate to 5-HT of 31.6% was obtained. The T149A mutant displayed markedly reduced sensitivity to PMA (10 nm) compared to wild-type 5-HT1A receptors, with only a 13.4 ± 3% reduction in 5-HT-induced channel inhibition; when exposed to 500 nm PMA, reductions in the action of 5-HT were comparable to those of the wild-type receptor. By contrast, the i3 mutant displayed comparable sensitivity to the wild-type 5-HT1A receptor to either concentration of PMA. PMA at 10 nm exhibited a similar uncoupling effect on the response of the endogenous opiate receptor to the agonist d-alanine-5-leucine-enkephalin (DADLE) in wild-type and T149A mutant-expressing clones. The T149 site of the 5-HT1A receptor is crucial for receptor uncoupling by sub-maximal PKC activation while at maximal PKC activation, downstream sites uncouple G proteins from the N-type Ca2+ channel.
Protein kinase C (PKC) is a crucial site of integration for a variety of receptor-mediated signals within the brain. Phosphatidyl inositol turnover induced by activation of phospholipases C and D generates the dual second messengers calcium and diacylglycerol that together activate PKC (Nishizuka, 1995). These phospholipases are activated by a large variety of G protein-coupled receptors, as well as by tyrosine kinase receptors and adhesion molecules, leading to activation of PKC. In addition to mediating the intracellular actions of these receptors, PKC activation leads to negative regulation of both G protein-coupled and tyrosine kinase receptors.
We have examined the inhibitory actions of PKC in a biologically relevant system, namely PKC-mediated uncoupling of 5-HT1A receptors (Swartz, 1993; Lembo & Albert, 1995; Chen & Penington, 1996), which regulate calcium channels via Gi/Go proteins (Ikeda & Dunlap, 1999). Multiple potential sites of PKC action have been identified along the 5-HT1A signalling pathway that have been demonstrated to be phosphorylated by PKC including the 5-HT1A receptor (Raymond, 1991), the G protein (Strassheim & Malbon, 1994; Kozasa & Gilman, 1996; Fan et al. 1998; Macek et al. 1998) and certain calcium channel subunits (Ahlijanian et al. 1991; Zamponi et al. 1997). In Ltk− fibroblasts we found that mutation of three PKC sites in the third intracellular (i3) loop of the 5-HT1A receptor was required for maximal (> 70%) attenuation of phorbol 12-myristate 13-acetate (PMA)-induced uncoupling of 5-HT1A receptor-induced calcium mobilization (Lembo & Albert, 1995). Resistance of the mutant receptor to PKC-induced uncoupling could be overcome by higher concentrations of PMA (1 μm), suggesting a role for other PKC sites at high levels of PKC activation.
The general approach to address the role of the receptor in the effect of PMA was to determine whether removing the sites of PKC action on the receptor molecule by site-directed mutagenesis would alter the effect of PKC on coupling to Ca2+ channels. We assessed the PMA sensitivity of 5-HT-induced calcium channel modulation by 5-HT1A receptors mutated at three PKC sites in the i3 domain or a single PKC site in the i2 domain of the receptor. These sites represent all of the consensus intracellular PKC sites of the 5-HT1A receptor and their removal influences the PKC sensitivity of receptor coupling to other effectors (Lembo & Albert, 1995; Lembo et al. 1997). It is not easy to do this in a serotonergic neuron, as it is difficult to suppress the synthesis and expression of wild-type receptors during transfection of a mutated version of the PKC target receptor. Our approach required growth of the cells in tissue culture. The current study circumvented these difficulties by the use of an electrically excitable neuronal cell line (F11, a dorsal root ganglion neuron crossed with a neuroblastoma cell, Platika et al. 1985) that does not express the 5-HT1A receptor (Banerjee et al. 1993), but in which it can be stably expressed by transfection.
METHODS
Tissue culture
Cells (a gift from Dr P. Banerjee, City University of New York, USA; Banerjee et al. 1993) were grown as monolayers in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum at 37°C in a humidified atmosphere of 95% O2 −5% CO2. The medium was changed every day. F11 cells were differentiated by removal of serum from the medium. After 2 days the cells stopped dividing and began extending processes, at which time they were gently re-plated onto glass coverslips. This had the effect of removing the long processes and it facilitated voltage clamp. The current recording was performed one day after re-plating.
Before differentiation the stably transfected F11 cells were treated with 10 μg ml−1 puromycin for 24 h to select for the transfected clones. Adding dibutylryl cyclic AMP (cAMP) or, indeed, using any method to speed the differentiation of clones with either form of the mutated 5-HT1A DNA resulted in explosive cell division followed by extensive cell death. This unusual response was not seen in cells transfected with the wild-type 5-HT1A receptor DNA. For this reason differentiation of transfected F11 cells was induced solely by removing the serum. All the groups of F11 clones, transfected or non-transfected, were thus treated the same way. Periodic treatment with HAT medium (100 μm hypoxanthine, 0.4 μm aminopterin and 16 μm thymidine) prevented the growth of any cells that were reverting to the parental N18TG2 neuroblastoma phenotype (Greene et al. 1975).
Stable transfection of F11 cells
F11 cells were maintained in DMEM medium (with 4.5 mg l−1d-glucose) as described above. The 1.9 kb BamH I–Xba I fragment of the rat 5-HT1A receptor (wild-type receptor clones, designated wt-1) or receptor mutants (T149A, called i2 clones or T229A-S253G-T343A, called i3 clones) was subcloned into BamH I–Xba I-cut pcDNA3 (Invitrogen) to generate p3-DBX or mutants. For transfection, F11 cells were plated at 5 × 106 cells per 10 cm dish and incubated overnight. Calcium phosphate precipitate (2 ml per dish) containing 20 μg p3-DBX or mutant plasmid and 1 μg pPGK-puro (a gift of Dr M. McBurney, University of Ottawa), was overlaid with 8 ml per dish DMEM, 10% FCS and 20 mm Hepes, pH 7.0, and incubated for 4–6 h as described above. The calcium phosphate was then aspirated and replaced with 8 ml per dish fresh medium for incubation overnight as described above. The cells were selected in DMEM plus 10 %FCS and 10 μg ml−1 puromycin (Sigma, St Louis, MO, USA) for 2 weeks until colonies appeared. Northern blot analysis of mRNA extracted from individual colonies identified positive clones expressing 5-HT1A (wild-type or mutant) receptors. For the i2–1 clone, RT-PCR analysis of RNA with 5-HT1A-specific primers followed by DNA sequence analysis demonstrated that the mutation (T149A) was exclusively present in 5-HT1A RNA from these cells. The cells were propagated for 1 month in 10 μg ml−1 puromycin and subsequently maintained in 1–10 μg ml−1 puromycin.
Ligand binding assay
Membranes were prepared from transfected F11 cells as previously described (Lembo et al. 1997) and stored as pellets at −80 °C. Briefly, the cells were re-suspended in hyposmotic TME (1:5 dilution of TME, (mm): Tris 75, pH 7.4, 12.5 mm MgCl2, 1 mm EDTA) on ice for 10 min, lysed by passage through a 25 gauge needle and centrifuged at 1000 g for 5 min at 4 °C to remove nuclei and debris. The supernatant was then centrifuged at 12 000 g for 30 min at 4 °C to pellet membranes. For the binding assay membrane pellets were re-suspended in TME, aliquotted (100 μg in 50 μl) into tubes containing 200 μl TME and 1–20 nm 8-hydroxy-[3H]2-(dipropylamino)-tetralin (8-OH-[3H]DPAT, Amersham, Quebec, Canada) and incubated at room temperature for 40 min. The binding reaction mixtures were applied to Whatman GF/C filters under vacuum and rinsed three times with 3 ml of ice-cold 50 mm Tris, pH 7.4. The filters were then placed in 3 ml of scintillation fluid (Ultima-Gold, Packard) and counted for 2 min using the direct-dpm program of the Tricarb TR2100 counter (Packard). 5-HT (10 μm) was used to define non-specific binding.
cAMP assay
Cells were plated in 6-well dishes and 24 h later were rinsed with DMEM and incubated at 37 °C in 1 ml per well DMEM, 0.01% bovine serum albumin, 20 mm Hepes, pH 7.0, and 100 μm 3-isobutyl 1-methyl xanthine (IBMX) for 15–20 min. Experimental compounds were added to triplicate wells. The medium was recovered and centrifuged at 13 000 g for 30 s to remove floating cells and the supernatants were stored at −20 °C. Attached cells were extracted using reporter lysis buffer (Promega), centrifuged for 10 min at 4 °C and stored at −20 °C. The medium was assayed for cAMP by a specific radioimmunoassay, as previously described (Lembo et al. 1997). Statistical analyses of the data were done using Graphpad Prism (San Diego, CA, USA) software and groups were compared using Student's unpaired t test with Welsh's correction to determine significance (P < 0.05).
Whole-cell recording
Experiments designed to measure Ca2+ currents used as a pipette solution: CsCl 110 mm, Hepes 40 mm, EGTA 10 mm, Mg-ATP 4 mm, MgCl2 1 mm, GTP 300 μm, phosphocreatine 14 mm, pH adjusted to 7.3 with CsOH. The extracellular solution was continually perfused at a rate of about 2 ml min−1 into a bath containing about 1 ml of recording solution. In order to eliminate the contribution of Na+ ions to the inward current, we added 0.1 μm TTX to all recording solutions. We routinely used the following solution to establish seals for whole-cell recording (mm): NaCl 134, Hepes 20, glucose 10, sucrose 20, CaCl2 2, KCl 2.5, MgCl2 2. The external recording solution, designed to isolate calcium channel currents (carried by Ba2+), contained (mm): TEA-Cl 140, BaCl2 30, Hepes 10, sucrose 20, pH adjusted to 7.3 with TEA-OH. Recordings were carried out at room temperature (22–25 °C). The osmolarity of the external solution was adjusted with sucrose such that the pipette solution was 20 mosmol l−1 hyposmotic compared to the recording solution (300 mosmol l−1). This usually prevented cell swelling. Drugs that dissolved in the extracellular solution were added to the perfusate.
An Axopatch 200A patch-clamp amplifier (Axon Instruments) was used to voltage-clamp neurons with truncated dendrites and a cell soma with one dimension of at least 20 μm, using the whole-cell configuration. Electrodes, pulled from soda-lime glass capillary tubes, were regularly coated with Sylgard and ranged in resistance from 1.8 to 2.5 MΩ. The series resistance circuit of the amplifier was used to compensate 80% of the apparent series resistance. Clamp settling time was typically less than 300 μs. When measuring Ca2+ currents in TEA, the seal resistance is often greater than 5 GΩ. Subtraction of the leak and capacitance from the current records was done using the Axobasic software system (Axon Instruments). During the experiment, at regular periods, we obtained leak sweeps. Leak sweeps consisted of 16 averaged hyperpolarizing steps of 10 mV. The leak sweep currents were scaled to the appropriate size and then subtracted from the individual current records. The voltage-clamp data (measurement of Ca2+ current) were filtered at 2 kHz then digitized at 100 μs per point. Voltage protocols were generated and analysed by an IBM PC Pentium clone using the Axobasic 1 patch-clamp software and the resultant data were written to disk for analysis off-line.
It was ascertained that addition of 10 or 500 nm PMA had little effect on baseline voltage-dependent Ca2+ current in these cells. Furthermore, in five wt-1 cells in which 500 nm PMA was tested, the final concentration of vehicle in the external solution (1:10 000 DMSO) caused no change in the average baseline current (685.0 ± 166.8 versus 588.4 ± 79.7 pA with DMSO). In all the other transfected cells, the effect of 500 nm PMA on the baseline current was not distinguishable from the normal rate of Ca2+ current rundown. This was measured in six wt-1 cells in which the control Ca2+ current was 1148.5 ± 75.2 pA. After 200 s during the fast phase of rundown the current fell to 935.0 ± 78.8 pA, a rundown of almost 19%. The response to 5-HT in these cells, however, was 49.4 ± 4.1% at time zero and 53.4 ± 4.9% after the baseline current had run down (no significant difference by Student's paired t test), thus rundown does not lead to a reduction in the effect of 5-HT. The measurements of Ca2+ current are expressed as means ± s.e.m., unless noted otherwise and in some cases the means were tested for equality using Student's t test or paired t analysis. Comparisons of multiple groups were done using ANOVA followed by Student-Newman-Keuls post hoc test or Duncan's multiple range test.
There was a large group of non-responders in both groups (70 wt-1 and 108 i2-1 cells, Table 1). Of these non-responders there was only one response measuring 7%; all the others were scored as ‘0’. The criterion that we used to define a non-responder was the lack of any perceptible inhibition when the data were plotted as a peak current versus time plot. The data were inspected to see whether the points showed a trend towards inhibition that reversed when the 5-HT was removed. If there was a hint of inhibition it was measured.
Table 1.
5-HT1A receptor binding and inhibition of adenylyl cyclase and calcium channels in F11 clones
| Cell line | Specific binding (fmol(mg protein)−1) | cAMP response (% of forskolin level) | Calcium channel inhibition (% of cells responding (n)) |
|---|---|---|---|
| F11 | 0 | 112.0 ± 4.0 | 0(0/9) |
| wt-1 | 14.0 ± 0.8 | 50.0 ± 6.9** | 38.1 (43/113) |
| i2-1 | 119.2 ± 6.2 | 69.7 ± 9.6** | 31.6 (50/158) |
| i2-6 | 92.8 ± 6.3 | 79.3 ± 4.4* | 4.8 (1/21) |
| i2-8 | 201.1 ± 6.5 | 58.0 ± 5.4** | 0 (0/23) |
| i3-2 | 416.9 ± 24.2 | 47.7 ± 5.5** | 68 (34/50) |
| i3-8 | 573.4 ± 49.3 | 15.3 ± 4.4** | 100 (65/65) |
The level of 5-HT1A receptor was measured in triplicate using a saturating concentration of the 5-HT1A-selective agonist 8-OH-[3H]DPAT. 8-OH-[3H]DPAT (2.5μM)-induced inhibition of forskolin (1μM)-induced adenylyl cyclase is shown as percentage inhibition and was measured in triplicate in 3 independent experiments. 8-OH-[3H]DPAT had no effect on basal cAMP accumulation. Data are tabulated as means ± s.e.m. For each clone, the percentage of cells responding to 1 mM 5-HT is shown for the number of cells tested (n). Asterisks denote a significant difference compared with F11 control cells
P < 0.01
P < 0.001; by ANOVA with a Newman-Keuls post hoc test).
RESULTS
The results of whole-cell patch-clamp recording from F11 cells revealed that they had Cd2+-sensitive Ca2+ currents that averaged 604.0 ± 51.4 pA, when recorded using 30 mm external Ba2+ (n = 162). In nine control (non-transfected F11) cells, 5-HT applied at 1 μm had no effect on ICa (Table 1). F11 cells were also demonstrated by Banerjee et al. (1993), and in the present study, to lack binding sites for the selective 5-HT1A receptor agonist 8-OH-[3H]DPAT and failed to exhibit inhibition of forskolin-stimulated cyclase activity upon application of 8-OH-[3H]DPAT (Table 1).
Calcium channels expressed in F11 cells
F11 cell Ca2+ current was suppressed by 60.4 ± 5.0 % (in wt-1 and i3–8 cells, n = 6) by ω-conotoxin GVIA (1 μm), a selective N-type Ca2+ channel blocker, with no further effect when this was followed by an application of 10 μm ω-conotoxin GVIA. These experiments were done using 10 mm external Ba2+ as the charge carrier because higher [Ba2+] decreases ω-conotoxin binding (Abe et al. 1986). On average about 40 % of the total current appeared not to be N-type, as determined by sensitivity to ω-conotoxin (Penington & Fox, 1995). The Ca2+ current was inhibited by 57.0 ± 3.2% (n = 4) by 5-HT treatment but after ω-conotoxin application 5-HT inhibited the current by only 27.2 ± 12.6%, suggesting that it acted mainly to inhibit N-type currents. The response to 5-HT was further evaluated in the presence of two specific Ca2+ channel blockers, nifedipine (an L-type channel blocker) and ω-agatoxin IVA (a P/Q-type channel blocker). Nifedipine (1 μm) was applied to the i3-8 clonal cells using a holding potential of −40 mV to increase the binding affinity of the drug (Bean, 1984). In four cells the Ca2+ current was decreased by 17.2 ± 7.5% after nifedipine treatment. If the holding potential had been −80 mV, the L-type current would have made up a smaller proportion of the total current due to the removal of N-type channel inactivation. The response to 5-HT was not decreased significantly by pre-treatment with nifedipine (reduced, on average, by 19.3 ± 15.0%, n = 4). In five i3-8 cells, 100 nm ω-agatoxin decreased the Ca2+ current by only 3.7 ± 4.1% and on average the response to 5-HT was not significantly decreased (reduced by 13.4 ± 8.8%, n = 5). These results indicated that 5-HT1A receptor activation preferentially inhibits N-type calcium channels in F11 cells.
In F11 cells transfected with the wild-type 5-HT1A receptor (wt-1 clone), specific binding of 8-OH-[3H]DPAT and inhibition of forskolin-stimulated cAMP formation were detected (Table 1). The percentage inhibition of baseline current by 5-HT remained constant (in the absence of PMA), irrespective of the absolute size of the baseline current. In the presence of PMA, a PKC activator, the inhibitory effect of 5-HT on calcium current was reduced (Fig. 1). In 10 cells, the baseline Ca2+ current was not altered by 10 nm PMA (805.0 ± 103.8 vs. 697.0 ± 117.2 pA; P > 0.05, Student's paired t test). However, addition of PMA (10 nm) did reduce the effect of 5-HT on Ca2+ current inhibition by 28.6 ± 6.8% (Fig. 1 and Table 2). PMA (500 nm) was applied to five wt-1 cells and an even greater reduction of 44.3% in the effect of 5-HT was obtained. Thus the activation of PKC partially uncoupled the 5-HT1A receptor from inhibition of calcium currents in F11 cells.
Figure 1. The phorbol ester PMA reduces the inhibitory effect of 5-HT on Ca2+ current in F11 cells transfected with the wild-type 5-HT1A receptor (wt-1 clone).
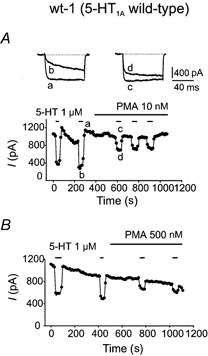
In this, and all other Ca2+ current figures, the holding potential was −80 mV with a step to 0 mV every 10 s. All Ca2+ currents have been leakage subtracted. A, application of 1 μm 5-HT from a sewer pipe arrangement inhibited the Ca2+ current. a refers to control Ca2+ current and b is Ca2+ current in the presence of 5-HT. c shows control current with 10 nm PMA and d the current obtained in the presence of 5-HT and PMA. Addition of 10 nm PMA to the bath decreased the effectiveness of 5-HT irreversibly. B, the effect of 500 nm PMA on 1 μm 5-HT-induced calcium current inhibition. In these cells neither concentration of PMA had any effect on baseline Ca2+ current.
Table 2.
Quantitative analysis of the action of PMA on 5-HT-induced calcium channel inhibition in wild-type and mutant 5-HT1A receptor clones
| 10 nM PMA treatment | 500 nM PMA treatment | |||||||
|---|---|---|---|---|---|---|---|---|
| n | Inhibition before (%) | Inhibition after (%) | Uncoupling (%) | n | Inhibition before (%) | Inhibition after (%) | Uncoupling (%) | |
| wt-1 | 10 | 57.6 ± 5.7 | 42.4 ± 6.6 | 28.6 ± 6.8 | 5 | 61.7 ± 4.5 | 34.7 ± 4.6 | 44.3 ± 4.7 |
| i3-2 | 8 | 66.2 ± 2.0 | 48.3 ± 4.1 | 26.9 ± 5.7 | 8 | 53.5 ± 4.1 | 35.7 ± 6.1 | 33.0 ± 10.2 |
| i3-8 | 16 | 58.1 ± 2.0 | 46.6 ± 2.5 | 19.6 ± 3.4 | 11 | 53.5 ± 3.9 | 37.3 ± 4.0 | 29.7 ± 6.0 |
| i2-1 | 16 | 42.2 ± 4.5* | 37.4 ± 4.6 | 13.4 ± 3.4* | 14 | 40.6 ± 4.0* | 26.4 ± 4.9 | 34.0 ± 11.1 |
Shown are the means ± s.e.m. of the indicated number (n) of independent measurements for the various F11 clones expressing wild-type (wt-1), i2 mutant (i2-1) or i3 mutant (i3-2 and i3-8) 5-HT1A receptors. All groups were compared by ANOVA as described in the Methods.
Significantly different from wild-type (P < 0.05). Cells that showed no response to 5-HT were excluded from these data.
High but not low concentrations of PMA reduce the effect of GTP-γ-S
The inhibition of calcium current by 5-HT can be divided into two components, one voltage dependent and the other voltage independent. A depolarizing prepulse to +80 mV partially reversed the voltage-dependent component of modulation by GTP-γ-S and the resulting facilitation ratio (defined as: current after pre-pulse in GTP-γ-S/current in GTP-γ-S) estimates the voltage-dependent component of inhibition (Ikeda, 1991; Jones & Elmslie, 1997). To address whether PMA acted downstream of the 5-HT1A receptor, GTP-γ-S was used to bypass the receptor and directly activate G protein-induced coupling to calcium channels as measured by prepulse facilitation. We chose the i3-8 clones for this experiment as nearly 100% of the cells responded to 5-HT (Table 1), they were more likely to be homogeneous with our population of wt-1 and i2-1 responding clones, with respect to post-receptor machinery, and they responded normally to PMA.
GTP-γ-S (100 μm) was perfused into the cells from the patch pipette until a stable Ca2+ current was recorded. Prepulse facilitation was elicited following depolarization to +80 mV and was measured as an index of the effect of GTP-γ-S (Fig. 2). In 15 control cells the facilitation measured 57.0 ± 8.4%. When the cells were pretreated with 500 nm PMA for 5 min before they were patched with GTP-γ-S in the pipette, the facilitation measured 21.6 ± 7.2% (n = 6). This was a significantly smaller effect of GTP-γ-S (P < 0.05). However, when the cells were pretreated with 10 nm PMA for the same amount of time before patching them with GTP-γ-S in the pipette, the facilitation measured 50.6 ± 9.4% (n = 5), a value that was not different from the control (ANOVA using Duncan's multiple range test). Thus at high, but not low, levels of activation, PKC appeared to uncouple the G protein-mediated modulation of channel opening. A similar result was obtained in a wt-1 cell that was not pretreated with PMA. Infusion of GTP-γ-S yielded a facilitation of 89.9 %; after the addition of 10 nm PMA it was only slightly changed, measuring 80.6 %, but after addition of 500 nm PMA the facilitation measured only 49.5%.
Figure 2. High but not low concentrations of PMA inhibit GTP-γ-S-induced prepulse facilitation.
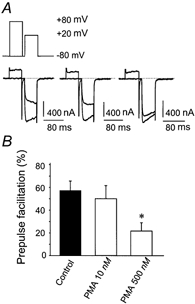
A shows the effect on calcium current of loading i3-8 cells (an F11 clone expressing 5-HT1A receptor) with GTP-γ-S (100 μm). The current was elicited from −80 mV with a step to +20 mV, with or without a prepulse to +80 mV that temporarily reversed the effect of the GTP-γ-S. A gap of 10 ms between the prepulse and the test pulse was sufficient to allow the tail current to decay. Pretreatment of other cells with 10 nm PMA did not affect prepulse facilitation (n = 5) but pretreatment with 500 nm PMA (n = 6) significantly decreased prepulse facilitation compared to that in control cells (n = 15, P < 0.05). B, bar graphs showing the mean ± s.e.m. for each condition. * Significantly different from Control (P < 0.05).
Reduced sensitivity to PKC of 5-HT1A receptor mutants
To identify the site of action of PKC, the 5-HT1A receptor was mutated to eliminate PKC substrate sites located in the second and third intracellular loops (i2 and i3) and the action of PMA on the response to 5-HT was examined in stably transfected F11 clones. Specific binding of 8-OH-[3H]DPAT was 5- to 10-fold greater for clones expressing mutant receptors than for the wt-1 clone. All clones displayed 8-OH-[3H]DPAT-induced inhibition of forskolin-stimulated adenylyl cyclase (Table 1), suggesting functional expression of the transfected receptors at the membrane. Previous studies in stably transfected Ltk− fibroblasts have shown that these mutations do not significantly alter binding affinity and that coupling to adenylyl cyclase is retained (Lembo & Albert, 1995; Lembo et al. 1997). Both clones that expressed the i3 mutant 5-HT1A receptor (i3-2 and i3-8) also mediated the 5-HT-induced reduction in calcium currents, and this was not significantly different from that seen in the wt-1 clone (Table 2). However, F11 cells that were transfected with the 5-HT1A receptor containing the i2 mutation T149A generally did not mediate the 5-HT-induced inhibition of calcium currents (Table 1). In one clonal line (i2-1), about 30% of the cells responded with a statistically significant 20% reduction in the 5-HT-induced response compared to wt-1, i3-2 or i3-8 clones (P < 0.05, n = 50 i2-1 cells, ANOVA and Duncan's multiple range test), which might be attributed to the effect of the T149A mutation. The integrity of the T149A mutation in the 5-HT1A receptor RNA from i2-1 cells was confirmed by DNA sequence analysis of RT-PCR products generated using receptor-specific oligonucleotide primers (data not shown).
Swartz (1993) found that application of PMA to several types of neuronal cell (with the notable exception of dorsal root ganglion neurons) caused an increase in the baseline Ca2+ current, perhaps by interfering with tonic G protein-mediated current inhibition. Therefore we examined the action of PMA on baseline calcium currents in our F11 clones. We found that F11 clones containing the wild-type (wt-1) 5-HT1A receptors exhibited a negligible increase in baseline Ca2+ current after 500 nm PMA treatment (Fig. 3 and Fig. 4). In addition, there was no measurable increase in baseline current after 10 nm PMA treatment. These findings fit well with the observation that there was only a weak facilitation current, measuring 13–15 %, in i3-8 and i2-8 cells in response to a depolarizing pulse to +80 mV without 5-HT, suggesting that these cells had little tonic G protein activity.
Figure 3. Relative insensitivity of a T149A 5-HT1A mutant F11 clone to low concentrations of PMA.
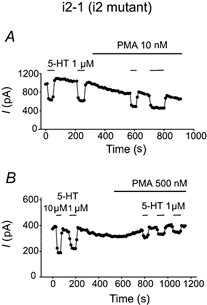
In i2-1 clone cells that responded to 5-HT, the i2 mutants were insensitive to 10 nm PMA-mediated uncoupling of the effect of 5-HT. A, 10 nm PMA had little obvious effect until data from 10 cells were averaged. However, 500 nm PMA was capable, on average, of inhibiting the response to 5-HT as effectively as in clonal lines expressing wild-type or i3 triple mutant receptors (B).
Figure 4. Responsiveness of i3 triple mutant F11 clones to high and low concentrations of PMA.
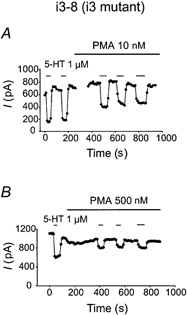
As seen with the wild-type 5-HT1A receptor, PMA at both 10 (A) and 500 nm (B) reduced the inhibitory effect of 5-HT on ICa in F11 cells expressing the triple mutant of PKC consensus phosphorylation sites in the i3 loop of the 5-HT1A receptor.
The sensitivity to PMA of 5-HT-induced inhibition of calcium currents in the i2 clones was examined. In 16 i2-1 clone cells that responded to 5-HT, addition of 10 nm PMA slightly (13%) reduced 5-HT-induced Ca2+ current inhibition, an effect that was significantly smaller than that seen with 10 nm PMA in the wt-1 clone (P < 0.05, Student's unpaired t test; see Table 2 and Fig. 3). When comparing the means of the four groups (wt, i2 and the two i3 clones) by one way ANOVA and Duncan's multiple range test, only the i2-1 clone response to 10 nm PMA was significantly different from that of the wt-1 clone (P < 0.05). By contrast, no significant difference in sensitivity to 500 nm PMA was observed.
The effect of 10 and 500 nm PMA was tested in cells containing receptors with a triple mutation of the i3 loop of the 5-HT1A receptor (i3-2 and i3-8 clones, Fig. 4). The response of the i3-2 clonal cells to PMA was not significantly different to that of the wt-1 clones at either concentration of PMA (Table 2). Similar data were obtained with the triple mutant clonal line i3-8 in which sensitivity to PMA was not different from that observed in i3-2 cells or the wt-1 clone (Table 2 and Fig. 4). A high concentration of PMA (500 nm) was equally effective in uncoupling the inhibitory response to 5-HT in all of these cells, including the i2-1 clone. Sensitivity to strong activation of PKC suggests that a lower-affinity target of phosphorylation exists further downstream from the receptor that mediates PKC-induced uncoupling of the 5-HT1A receptor from calcium current inhibition.
This result may indicate that the T149 residue is a critical target for relatively low levels of PKC stimulation. The resistance to PKC in the i2-1 clone provided evidence that in F11 cells, in some circumstances, the crucial action of PKC at low levels of stimulation is on the receptor. It is interesting that uncoupling of the receptor from the channel is observed (in i2-6, i2-8 and 62 % of i2-1 clones, Table 1) whether T149 is mutated or (presumably) phosphorylated by PKC (Table 2). Thus modification of T149 appears to have a crucial action to reduce receptor coupling to the channel.
Comparison of the effect of 10 nm PMA on opiate responses in i2-1 and wt-1 cells
As the i2-1 cell line was significantly less sensitive to 10 nm PMA than the other clones we reasoned that a good control would be to examine the sensitivity to 10 nm PMA of these cells in the response to the δ-opioid receptor-selective agonist d-alanine-5-leucine-enkephalin (DADLE, 100 nm). Despite the general lack of response to 5-HT in the i2 clones, all the clones of F11 cells always responded to 1 μm DADLE with a large reversible inhibition of Ca2+ current. DADLE inhibited the Ca2+ current in all the clones by a similar amount: wt-1 clones by 44.2 ± 12.2% (n = 4), i3-8 clones by 59.6 ± 3.4% (n = 5) and i2-1 clones by 55.3 ± 4.5% (n = 5).
Like the 5-HT1A receptor, the Gi/Go-coupled δ-opioid receptor is uncoupled from calcium mobilization by PKC activation (Ueda et al. 1995; Yoon et al. 1998), an action that appears to be mediated by receptor phosphorylation at S344, located at the receptor C-terminal tail domain (Xiang et al. 2001). In six i2-1 cells, 10 nm PMA decreased the response to DADLE by 29.4 ± 8.1% whilst in five wt-1 cells the response to DADLE was inhibited by 29.7 ± 15.1% and so the effect of the 5-HT1A receptor mutation was specific for the effect of PMA on the response to 5-HT.
Pharmacological inhibition of the action of PMA using a PKC blocker
To assess the role of PKC in the actions of PMA, the selective PKC inhibitor Go6976 (200 nm) was tested on F11 wt-1 cells (Fig. 5A). First, cells expressing the wild-type receptor were treated with PMA to reduce the effect of 5-HT. Before 10 nm PMA treatment, 5-HT inhibited the Ca2+ current by on average 58.7%; after PMA treatment 5-HT inhibited the current by 37.9%. This represented an irreversible (in the time course of our recordings) reduction of 35% in the effect of 5-HT. Go6976 was dissolved in DMSO (at 5000 times dilution) and was then applied for approximately 4 min. It was found that addition of Go6976 after PMA application allowed the response to 5-HT to recover to 53% (n = 3), which was quite close to the control level of inhibition. The i3-8 cells were also tested to confirm whether there was any effect of Go6976, alone, on the response to 5-HT. The inhibition of the Ca2+ current by 5-HT was measured under three conditions (Fig. 5B): before addition of Go6976, after addition of 200 nm Go6976 and after addition of Go6976 plus 10 nm PMA. With this protocol, 5-HT inhibited the Ca2+ current by 61.9 ± 6.7, 72.4 ± 2.9 and 75.4 ± 5.4%, respectively. On average, 5-HT inhibition of calcium currents was not significantly changed after application of Go6976 with or without 10 nm PMA (P > 0.05, n = 6; Student's paired t test).
Figure 5. The effects of both 10 and 500 nm PMA were reversed or prevented by the PKC inhibitor Go6976.
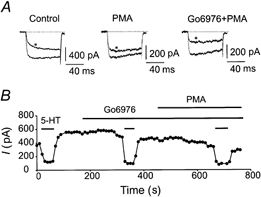
A, calcium currents elicited from a holding potential of −80 mV, with a step to +20 mV, recorded from an F11 cell stably expressing wt-1 5-HT1A receptors. * Current in the presence of 1 μm 5-HT. The centre panel shows that PMA (10 nm) decreased the effect of 5-HT, normally in an irreversible fashion; however, addition of 200 nm Go6976 reversed the effect of PMA. B, current vs. time plot obtained from an F11 cell stably expressing the i3-8 5-HT1A receptor. Go6976 did not affect the average size of the baseline Ca2+ current or the percentage inhibition by 5-HT. Addition of PMA (10 nm) had little effect on the percentage inhibition by 5-HT after pretreatment of the cell with Go6976.
DISCUSSION
Differential action of low and high concentrations of PMA
As observed in a variety of acutely isolated neurons (Golard et al. 1993; Swartz, 1993; Zhu & Ikeda, 1994; Chen & Penington, 1996) the application of the phorbol ester PMA decreased the ability of 5-HT to inhibit Ca2+ current in cultured F11 cells transfected with the 5-HT1A receptor. At low concentrations, 10 nm PMA produced a smaller uncoupling of 5-HT-induced calcium current modulation and appeared to activate a smaller amount of PKC, which may preferentially target a ‘high affinity’ site on the 5-HT1A receptor. When the T149 residue in the second intracellular loop of the 5-HT1A receptor was mutated to alanine, 10 nm PMA had a reduced effect on the 5-HT1A receptor-channel coupling, compared to its effect on the wild-type receptor. The uncoupling by PMA application of a similar response resulting from endogenous opiate receptor activation was not altered in the 5-HT1A receptor (T149A) mutation-containing clone. This indicates that the observed reduction in the sensitivity to 10 nm PMA of responses to 5-HT was due to the mutation in the 5-HT1A receptor and not to alterations in PKC activity.
Further evidence in support of a selective action of low doses of PMA on the 5-HT1A receptor was obtained when the effect of different concentrations of PMA on the prepulse facilitation of calcium current was studied in GTP-γ-S-loaded F11 i3-8 cells. In these experiments, the G protein was activated directly and the receptors were bypassed. High concentrations of PMA (500 nm) displayed full effectiveness in uncoupling direct G protein-mediated regulation of N-type calcium channels. In contrast, low concentrations of PMA (10 nm) did not significantly affect prepulse facilitation in GTP-γ-S-loaded cells, despite the fact that 10 nm PMA uncoupled the effect of 5-HT1A receptor activation. It remains a possibility that 100 μm GTP-γ-S may have released more Gβγ than 1 μm 5-HT, despite the similar degree of inhibition of Ca2+ current, thus providing more competition for PKC at a potential site of phosphorylation on the Ca2+ channel.
The 5-HT1A receptor was not the only receptor in F11 cells that was responsive to low concentrations of PMA. DADLE-induced inhibition of calcium current mediated by endogenous δ-opioid receptors was also inhibited by 10 nm PMA to a similar extent as the response to 5-HT. This suggests that a variety of receptors can serve as high affinity sites for the action of PKC. In the case of the δ-opioid receptor, mutation of the S344 PKC site located in the C-terminal tail of the receptor blocked PMA-induced receptor phosphorylation and internalization processes (Xiang et al. 2001).
The precise mechanism for the greater PMA sensitivity of the 5-HT1A receptor T149 site than of downstream sites is unclear. The response to PMA was blocked by Go6976, which preferentially targets PKC α, β and γ subtypes, and this suggests a role for these subtypes in receptor phosphorylation. These kinases have high sensitivities to PMA, whereas other PKC subtypes perhaps targeting downstream PKC sites on the 5-HT1A receptor might have a lower affinity for PMA. Alternatively, PKC may be anchored in close proximity to the receptor by scaffolding proteins (e.g. protein kinase A anchoring proteins, AKAPs; Edwards & Scott, 2000) allowing for greater sensitivity to PMA. Specific interactions between the AKAP family and G protein-coupled receptors have not, however, been demonstrated. Finally the T149 site may provide a better substrate (having a lower Km) for PKC than other sites.
Influence of the 5-HT1A T149 site on receptor coupling
Mutation of the 5-HT1A receptor at PKC sites revealed that the i3 sites play no significant role in influencing the sensitivity of 5-HT-induced inhibition of calcium channels to low or high PMA concentrations. This is in contrast to the > 70% resistance to 100 nm PMA-induced uncoupling of 5-HT1A-mediated calcium mobilization in Ltk− fibroblast cells, a response mediated by Gβγ-induced phospholipase C-β activation (Lembo & Albert, 1995). These results suggest that PKC sites on the receptor differentially modulate distinct Gβγ signalling pathways. The T149A mutation resulted in uncoupling of the 5-HT1A receptor from inhibition of calcium channels in most clones, even though the i2 mutant retained coupling to Gαi-mediated inhibition of adenylyl cyclase. Previously we have observed that the T149A mutant is also uncoupled from other Gβγ-mediated pathways, including 5-HT-induced calcium mobilization and activation of adenylyl cyclase type II, but remains coupled to inhibition of forskolin-stimulated adenylyl cyclase (Lembo et al. 1997; Albert et al. 1999). The wt-1 clones responded inconsistently due to their lower level of receptor expression. Curiously, the i2-containing cells as a group, especially the i2-6 and i2-8 clones, did not respond or responded with a 5-HT-induced Ca2+ current inhibition of significantly lower frequency (i2-1, 32 % response rate) and efficacy (20 % lower response magnitude). In preliminary studies we have observed increased Gα protein expression in the i2 clones that lack 5-HT1A coupling to ion channels (i.e. i2-6 and i2-8); no increase in Gα proteins was detected in the i2-1 clone (N. Kushwaha & P. Albert, unpublished observations). These data suggest that an increase in the level of G proteins in these cells correlates inversely with the ability of the i2 5-HT1A mutant to couple to the calcium channel. We hypothesize that increased expression of Gα subunits, and their association with the mutant 5-HT1A receptor, may prevent mobilization of Gβγ subunits hence preventing 5-HT1A receptor coupling to calcium currents in the i2-6 and i2-8 clones. This specific effect of Gα in association with the T149A mutant receptor would not alter the effect of Gαi on adenylyl cyclase (Liu et al. 1999). It should also be pointed out that coupling to inhibition of forskolin-induced adenylyl cyclase seemed to be partially impaired in the i2 clones and was not as robust in some clones as in the wt-1 or i3 mutant. It does not appear that changes in G proteins in the i2 clones render the channel, rather than the receptor, resistant to PKC. If a general insensitivity to PKC occurred, then δ-opioid receptor coupling would also be resistant to a low concentration (10 nm) of PMA, which was not the case. Furthermore, in the i2-1 clone, which displays resistance of the 5-HT1A response to PKC, there was no measurable change in the G protein levels. Thus the alterations in G proteins in some clones may regulate coupling, but do not appear to affect the inherent PKC sensitivity of the i2 mutant 5-HT1A receptor. Our interpretation of the specific uncoupling of the 5-HT1A receptor from inhibition of calcium channels is that it is indirectly the result of the interaction of the T149A mutation with an elevated level of Gα.
Receptors as sites for high sensitivity to PMA
A major finding reported in this paper is that the T149A mutation results in a cell in which the Ca2+ channel modulation pathway is significantly less sensitive to low concentrations of PMA, but larger concentrations of PMA were as effective as in the i3-2, i3-8 and wt-1 cell groups. We interpret this finding to indicate that lower degrees of PKC activation might exert their effects on the PKC consensus phosphorylation site T149 in the i2 loop of the receptor. On the other hand, higher concentrations of PMA might have effects on other consensus sites for PKC-mediated phosphorylation at other points in the transduction pathway.
The response to 10 nm PMA in the wild-type clone was an inhibition of the effect of 5-HT that measured 28.6%, but only 13.4% inhibition was seen in the i2-1 clones. At low concentrations of PMA (10 nm), 53% of the effect appears to be mediated by receptor phosphorylation of the i2 loop. The partial response to PMA of the i2 clone could be mediated by PKC-induced phosphorylation of other sites at downstream targets (e.g. the G protein or channel).
It is known that PMA activates PKC and this has been suggested to alter receptor–channel coupling by acting on phosphorylation targets downstream of receptor- G protein coupling (Swartz, 1993; Zhu & Ikeda, 1994; Zamponi et al. 1997). Very little is known, however, about the direct effects of PKC on receptors that couple to various types of ion channel. Some evidence indicates that PKC may phosphorylate receptors and consequently block modulation of calcium channels by neurotransmitters (Golard et al. 1993; Garcia et al. 1998). Chen & Penington (1997) found that pretreatment with 1 μm PMA partially blocked the facilitation of GTP-γ-S-inhibited Ca2+ current in serotonergic neurons. However, activation of PKC after intracellular application of GTP-γ-S did not block the facilitation of Ca2+ current under any circumstances. This result suggests that PKC might be acting upstream of the Ca2+ channel in these cells. Garcia et al. (1998) found that PKC disrupts the inhibitory actions of cannabinoid agonists on P/Q-type calcium channels by phosphorylation of the CB1 receptor. Their results showed that mutation of the third intracellular loop of the CB1 receptor did not affect the ability of cannabinoid agonists to modulate P/Q-type calcium channels, but the effect of PKC on the CB1 receptor was eliminated. By contrast, we find that the i3 PKC sites of the 5-HT1A receptor are dispensable, whereas the i2 site (T149) is important for PMA-induced uncoupling of the receptor from N-type calcium channels.
The present communication is not the first observation of a critical role for the i2 loop of seven transmembrane G protein coupled receptors in the action of PKC. Francesconi & Duvoisin (2000) found that a modification at T695 in the second intracellular loop selectively disrupts the mGluR1-Gq/11 interaction without affecting signalling through Gs. When T695 was replaced with E in the mGluR1 message injected into oocytes, the receptor stimulation-induced opening of Ca2+-activated Cl− channels appeared smaller and slower. As glutamate itself is negatively charged, it mimics modulation by PKC phosphorylation. The current was ‘constitutively desensitized’ in this mutant preparation. Thus PKC-induced desensitization was eliminated.
N-type calcium channels as a target for PKC
Recent studies of PKC phosphorylation sites on N-type Ca2+ channels indicate that there are two sites, T422 and S425, on the I-II loop of the α1B subunit. One (T422) appears to be the site at which PKC mediates upregulation of the calcium current and antagonizes Gβγ coupling (see below). The other (S425) appears to regulate current upregulation but has no effect on G protein modulation (Zamponi et al. 1997; Hamid et al. 1999). This suggestion does not fully explain data obtained in other studies. In one case, the action of PKC produced only the upregulation of Ca2+ current (Yang & Tsien, 1993) and it did not reduce G protein modulation. In other studies, activation of M2 muscarinic receptors occluded D2 modulation, suggesting a shared signalling element. However, activation of PKC attenuated the M2 modulation without significantly affecting the D2 modulation. Thus PKC disrupted one receptor-mediated mechanism of N-type calcium current modulation but not another in the same cells (Yan et al. 1997), suggesting that calcium channels may not be the exclusive targets of PKC action. Another possibility is that different receptors use different combinations of Gβγ (Kleuss et al. 1993) or that these subunits bind to different sites on the Ca2+ channel. Either of these schemes could explain why, in a given neuron, the phosphorylation of T422 and S425 on the I–II loop of the α1B subunit might have no effect on the response to a certain transmitter. In these cells the effect of PKC might be mediated, instead, at a point higher up in the signalling cascade. In this regard a more recent paper by the Zamponi group (Cooper et al. 2000) has presented evidence that opiate receptor-mediated inhibition is affected to a lesser extent than are somatostatin-mediated responses. Their data showed that phosphorylation or mutation of residue 422 antagonized only the effects of Gβ1 whereas the effect of over-expressing the other Gβ subunits was not reduced. Thus the cross-talk between N-type Ca2+ channel phosphorylation and G proteins is dependent on the use of the Gβ1 subunit by the neurotransmitter in question. The potency of PMA to phosphorylate and regulate the N-type channel relative to its potency at receptors such as the 5-HT1A receptor remains incompletely addressed. Our results suggest that the receptor is the site of highest sensitivity to PKC activation, while the channel or G protein appears to respond only at high levels of PKC activation.
Conclusion
Taken together, our data indicate that the action of PKC to uncouple the 5-HT1A receptor from inhibition of N-type calcium channels depends in part on the presence of a PKC site located in the i2 loop of the receptor. However, since PMA-induced uncoupling persists upon addition of higher concentrations of PMA, even after elimination of this site, we hypothesize that a downstream action of PKC on G proteins and/or channel subunits is involved in this. Thus, the T149 residue phosphorylation site on the second intracellular loop of the 5-HT1A receptor may be a critical site for the coupling between the receptor and G protein. Mutation of this site reduces the disruptive effect of PKC on 5-HT1A receptor coupling to calcium channels.
Acknowledgments
This work was supported by National Institute of Mental Health award no. MH5504101 to N.J.P. and a Canadian Institute of Health Research grant to P.R.A.
REFERENCES
- Abe T, Koyano K, Saisu H, Nishiuchi Y, Sakakibara S. Binding of ω-conotoxin to receptor sites associated with the voltage-sensitive calcium channel. Neuroscience Letters. 1986;71:203–208. doi: 10.1016/0304-3940(86)90559-8. [DOI] [PubMed] [Google Scholar]
- Ahlijanian MK, Striessnig J, Catterall WA. Phosphorylation of an α1-like subunit of an ω-conotoxin-sensitive brain calcium channel by cAMP-dependent protein kinase and protein kinase C. Journal of Biological Chemistry. 1991;266:20192–20197. [PubMed] [Google Scholar]
- Albert PR, Sajedi N, Lemonde S, Ghahremani MH. Constitutive G(i2)-dependent activation of adenylyl cyclase type II by the 5-HT1A receptor. Inhibition by anxiolytic partial agonists. Journal of Biological Chemistry. 1999;274:35469–35474. doi: 10.1074/jbc.274.50.35469. [DOI] [PubMed] [Google Scholar]
- Banerjee P, Berry-Kravis E, Bonafede-Chhabra D, Dawson G. Heterologous expression of the serotonin 5-HT1A receptor in neural and non-neural cell lines. Biochemical and Biophysical Research Communications. 1993;192:104–110. doi: 10.1006/bbrc.1993.1387. [DOI] [PubMed] [Google Scholar]
- Bean BP. Nitrendipine block of cardiac calcium channels: high affinity binding to the inactivated state. Proceedings of the National Academy of Sciences of the USA. 1984;81:6388–6392. doi: 10.1073/pnas.81.20.6388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Penington NJ. Differential effects of PKC activation on 5-HT1A receptor coupling to Ca2+ and K+ currents in rat serotonergic neurones. Journal of Physiology. 1996;496:129–137. doi: 10.1113/jphysiol.1996.sp021670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Penington NJ. Order of application determines the interaction between phorbol esters and GTP- γ-S in dorsal raphe neurons: evidence that the effect of 5-HT is modified upstream of the G protein Ca channel interaction. Journal of Neurophysiology. 1997;77:2697–2703. doi: 10.1152/jn.1997.77.5.2697. [DOI] [PubMed] [Google Scholar]
- Cooper CB, Arnot MI, Feng Z-P, Jarvis SE, Hamid J, Zamponi GW. Cross-talk between G-protein and protein kinase C modulation of N-type calcium channels is dependent on the G-protein β subunit isoform. Journal of Biological Chemistry. 2000;275:40777–40781. doi: 10.1074/jbc.C000673200. [DOI] [PubMed] [Google Scholar]
- Edwards AS, Scott JD. A-kinase anchoring proteins: protein kinase A and beyond. Current Opinion in Cell Biology. 2000;12:217–221. doi: 10.1016/s0955-0674(99)00085-x. [DOI] [PubMed] [Google Scholar]
- Fan GH, Zhao J, Wu YL, Lou LG, Zhang Z, Jing Q, Ma L, Pei G. N-methyl-d-aspartate attenuates opioid receptor-mediated G protein activation and this process involves protein kinase C. Molecular Pharmacology. 1998;53:684–690. doi: 10.1124/mol.53.4.684. [DOI] [PubMed] [Google Scholar]
- Francesconi A, Duvoisin RM. Opposing effects of protein kinase C and protein kinase A on metabotropic glutamate receptor signaling: selective desensitization of the inositol triphosphate/Ca2+ pathway by phosphorylation of the receptor-G protein-coupling domain. Proceedings of the National Academy of Sciences of the USA. 2000;97:6185–6190. doi: 10.1073/pnas.97.11.6185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia DE, Brown S, Hille B, Mackie K. Protein kinase C disrupts cannabinoid actions by phosphorylation of the CB1 cannabinoid receptor. Journal of Neuroscience. 1998;18:2834–2841. doi: 10.1523/JNEUROSCI.18-08-02834.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golard A, Role LW, Siegelbaum SA. Protein kinase C blocks somatostatin-induced modulation of calcium current in chick sympathetic neurons. Journal of Neurophysiology. 1993;70:1639–1643. doi: 10.1152/jn.1993.70.4.1639. [DOI] [PubMed] [Google Scholar]
- Greene LA, Shain W, Chalazonitis A, Breakfield X, Minna J, Coon HG, Nirenberg M. Neuronal properties of hybrid neuroblastoma X sympathetic ganglion cells. Proceedings of the National Academy of Sciences of the USA. 1975;72:4923–4927. doi: 10.1073/pnas.72.12.4923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamid J, Nelson D, Spaetgens R, Dubel SJ, Snutch TP, Zamponi GW. Identification of an integration center for cross-talk between protein kinase C and G protein modulation of N-type calcium channels. Journal of Biological Chemistry. 1999;274:6195–6202. doi: 10.1074/jbc.274.10.6195. [DOI] [PubMed] [Google Scholar]
- Ikeda S. Double-pulse calcium channel current facilitation in adult rat sympathetic neurones. Journal of Physiology. 1991;439:181–214. doi: 10.1113/jphysiol.1991.sp018663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda SR, Dunlap K. Voltage-dependent modulation of N-type calcium channels: role of G protein subunits. Advances in Second Messenger and Phosphoprotein Research. 1999;33:131–151. doi: 10.1016/s1040-7952(99)80008-1. [DOI] [PubMed] [Google Scholar]
- Jones SW, Elmslie KS. Transmitter modulation of neuronal calcium channels. Journal of Membrane Biology. 1997;155:1–10. doi: 10.1007/s002329900153. [DOI] [PubMed] [Google Scholar]
- Kleuss C, Scherubl H, Hescheler J, Schultz G, Wittig B. Selectivity in signal transduction determined by gamma subunits of heterotrimeric G proteins. Science. 1993;259:832–834. doi: 10.1126/science.8094261. [DOI] [PubMed] [Google Scholar]
- Kozasa T, Gilman AG. Protein kinase C phosphorylates G12α and inhibits its interaction with Gβγ. Journal of Biological Chemistry. 1996;271:12562–12567. doi: 10.1074/jbc.271.21.12562. [DOI] [PubMed] [Google Scholar]
- Lembo PM, Albert PR. Multiple phosphorylation sites are required for pathway selective uncoupling of the 5-hydroxytryptamine1A receptor by protein kinase C. Molecular Pharmacology. 1995;48:1024–1029. [PubMed] [Google Scholar]
- Lembo PM, Ghahremani MH, Morris SJ, Albert PR. A conserved threonine residue in the second intracellular loop of the 5-hydroxytryptamine 1A receptor directs signaling specificity. Molecular Pharmacology. 1997;52:164–171. doi: 10.1124/mol.52.1.164. [DOI] [PubMed] [Google Scholar]
- Liu YF, Ghahremani MH, Rasenick MM, Jakobs KH, Albert PR. Stimulation of cAMP Synthesis by Gi-coupled receptors upon ablation of distinct Gαi protein expression: Gi subtype specificity of the 5-HT1A receptor. Journal of Biological Chemistry. 1999;274:16444–16450. doi: 10.1074/jbc.274.23.16444. [DOI] [PubMed] [Google Scholar]
- Macek TA, Schaffhauser H, Conn PJ. Protein kinase C and A3 adenosine receptor activation inhibit presynaptic metabotropic glutamate receptor (mGluR) function and uncouple mGluRs from GTP-binding proteins. Journal of Neuroscience. 1998;18:6138–6146. doi: 10.1523/JNEUROSCI.18-16-06138.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishizuka Y. Protein kinase C and lipid signaling for sustained cellular responses. FASEB Journal. 1995;9:484–496. [PubMed] [Google Scholar]
- Penington NJ, Fox AP. Toxin insensitive Ca current in dorsal raphe neurons. Journal of Neuroscience. 1995;15:5719–5726. doi: 10.1523/JNEUROSCI.15-08-05719.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platika D, Boulos MH, Baizer L, Fishman MC. Neuronal traits of clonal cell lines derived by fusion of dorsal root ganglion neurons with neuroblastoma cells. Proceedings of the National Academy of Sciences of the USA. 1985;82:3499–3503. doi: 10.1073/pnas.82.10.3499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymond JR. Protein kinase C induces phosphorylation and desensitization of the human 5-HT1A receptor. Journal of Biological Chemistry. 1991;266:14747–14753. [PubMed] [Google Scholar]
- Strassheim D, Malbon CC. Phosphorylation of Gi2 attenuates inhibitory adenyl cyclase in neuroblastoma/glioma (NG 108–15) cells. Journal of Biological Chemistry. 1994;269:14307–14313. [PubMed] [Google Scholar]
- Swartz KJ. Modulation of Ca2+ channels by protein kinase C in rat central and peripheral neurons: disruption of G protein mediated inhibition. Neuron. 1993;11:305–320. doi: 10.1016/0896-6273(93)90186-u. [DOI] [PubMed] [Google Scholar]
- Ueda H, Miyamae T, Hayashi C, Watanabe S, Fukushima N, Sasaki Y, Iwamura T, Misu Y. Protein kinase C involvement in homologous desensitization of δ-opioid receptor coupled to Gi1-phospholipase C activation in Xenopus oocytes. Journal of Neuroscience. 1995;15:7485–7499. doi: 10.1523/JNEUROSCI.15-11-07485.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang B, Yu GH, Guo J, Chen L, Hu W, Pei G, Ma L. Heterologous activation of protein kinase C stimulates phosphorylation of δ-opioid receptor at serine 344, resulting in β-arrestin- and clathrin-mediated receptor internalization. Journal of Biological Chemistry. 2001;276:4709–4716. doi: 10.1074/jbc.M006187200. [DOI] [PubMed] [Google Scholar]
- Yan Z, Song W-J, Surmeier DJ. D2 dopamine receptors reduce N-type Ca2+ currents in rat neostriatal cholinergic interneurons through a membrane-delimited, protein-kinase-C-insensitive pathway. Journal of Neurophysiology. 1997;77:1003–1015. doi: 10.1152/jn.1997.77.2.1003. [DOI] [PubMed] [Google Scholar]
- Yang J, Tsien RW. Enhancement of N- and L-type calcium channel currents by protein kinase C in frog sympathetic neurons. Neuron. 1993;10:127–136. doi: 10.1016/0896-6273(93)90305-b. [DOI] [PubMed] [Google Scholar]
- Yoon SH, Jin W, Spencer RJ, Loh HH, Thayer SA. Desensitization of δ-opioid-induced mobilization of Ca2+ stores in NG108–15 cells. Brain Research. 1998;802:9–18. doi: 10.1016/s0006-8993(98)00531-9. [DOI] [PubMed] [Google Scholar]
- Zamponi GW, Bourinet E, Nelson D, Nargeot J, Snutch TP. Crosstalk between G proteins and protein kinase C mediated by the calcium channel α1 subunit. Nature. 1997;385:442–446. doi: 10.1038/385442a0. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Ikeda S. Modulation of Ca2+-channel currents by protein kinase C in adult rat sympathetic neurons. Journal of Neurophysiology. 1994;72:1549–1560. doi: 10.1152/jn.1994.72.4.1549. [DOI] [PubMed] [Google Scholar]