

Abstract
Inhibition of cAMP-dependent stimulation of vectorial fluid transport across the alveolar epithelium following haemorrhagic shock is mediated by reactive nitrogen species released within the airspaces of the lung. We tested here the hypothesis that the prior activation of the cellular heat shock or stress response, via exposure to either heat or geldanamycin, would attenuate the release of airspace nitric oxide (NO) responsible for the shock-mediated failure of the alveolar epithelium to respond to catecholamines in rats. Rats were haemorrhaged to a mean arterial pressure of 30–35 mmHg for 60 min, and then resuscitated with a 4 % albumin solution. Alveolar fluid clearance was measured by change in concentration of a protein solution instilled into the airspaces 5 h after the onset of haemorrhage. Stress preconditioning restored the cAMP-mediated upregulation of alveolar liquid clearance after haemorrhage. The protective effect of stress preconditioning was mediated in part by a decrease in the expression of iNOS in the lung. Specifically, stress preconditioning decreased the production of nitrite by endotoxin-stimulated alveolar macrophages removed from haemorrhaged rats or by A549 and rat alveolar epithelial type II cell monolayers stimulated with cytomix (a mixture of TNF-α, IL-1β and IFN-γ) for 24 h. In summary, these results provide the first in vivo evidence that stress preconditioning restores a normal fluid transport capacity of the alveolar epithelium in the early phase following haemorrhagic shock by attenuating NO-mediated oxidative stress to the lung epithelium.
Major trauma associated with haemorrhagic shock is one of the most important causes of acute lung injury and pulmonary oedema in critically ill patients (Hudson et al. 1995). Upregulation of alveolar epithelial fluid transport by endogenous catecholamines is a major mechanism that prevents alveolar flooding following septic or haemorrhagic shock (Pittet et al. 1994, 1996; Modelska et al. 1997). However, after severe haemorrhage, this protective mechanism is compromised by the development of an inflammatory response mediated by neutrophils that accumulate in the lung (Modelska et al. 1999). Sequestration of neutrophils in the lung causes the release of oxygen radicals, proinflammatory cytokines (tumour necrosis factor-α (TNF-α) and interleukin-1β (IL-1β)) and nitric oxide (NO) through the activation of inducible NO synthase (iNOS) (Pittet et al. 2001). The production of a large amount of NO during sustained haemorrhagic shock may contribute to lung damage via two different pathways: (a) NO may further activate the inflammatory cascade via the activation of nuclear factor κB (NF-κB) that stimulates recruitment and sequestration of more neutrophils to the lung (Hierholzer et al. 1998); (b) NO may inhibit β-adrenergic receptor and ion channel activities in the lung epithelium through the production of peroxynitrite, a strong oxidant that arises due to the reaction between NO and superoxide (Pittet et al. 2001). Thus NO released within the airspaces secondary to the NF-κB-dependent activation of iNOS is one of the major proximal mediators of the oxidant-mediated decrease in the fluid transport capacity of the alveolar epithelium after prolonged haemorrhagic shock.
The heat shock or stress response is a highly conserved defence mechanism characterized by the increased expression of stress proteins that provide cellular protection via increased chaperone activity (Morimoto et al. 1990; Minowada & Welch, 1995). Activation of the stress response by mild hyperthermia allows cells to withstand a subsequent metabolic insult that would otherwise be lethal, a phenomenon referred to as ‘thermotolerance’ or ‘preconditioning’. Recent experimental work indicates that an important feature of the stress response in the lung is the modulation of the inflammatory response by transient inhibition of the expression of pro-inflammatory mediators. For example, activation of the stress response inhibited cytokine-mediated iNOS expression in rat pulmonary smooth muscle cells (Wong et al. 1996) and murine lung epithelium without affecting the expression of constitutively expressed genes, such as those of surfactant proteins (Wong et al. 1997a). Therefore we hypothesized that the activation of the stress response could decrease the release of NO within the airspaces of the lung following haemorrhagic shock. This is turn would lead to a decrease in the oxidative stress experienced by the alveolar epithelium and potentially restore a physiological response of this lung barrier to catecholamines following haemorrhage.
Our first objective was to determine whether stress preconditioning with whole body hyperthermia or geldanamycin, a benzoquinone ansamycin that activates stress protein expression (Hedge et al. 1995), would prevent the shock-mediated failure of the alveolar epithelium to respond to catecholamines. Stress preconditioning with whole body hyperthermia or geldanamycin did indeed help to restore the cAMP-mediated upregulation of alveolar liquid clearance after haemorrhage.
We have recently reported that the inability to increase alveolar epithelial fluid transport in response to catecholamines was mediated by the release of NO within the airspaces of the lung after haemorrhagic shock in rats (Pittet et al. 2001). In addition, both alveolar macrophages and alveolar epithelial cells have been shown to be a major source of NO production within the airspaces of the lung (Warner et al. 1995). Thus the second objective was to test the hypothesis that stress preconditioning would attenuate the production of NO both in vivo and in vitro. Stress preconditioning decreased the production of nitrite by endotoxin-stimulated alveolar macrophages removed from haemorrhaged rats or by A549 and rat alveolar epithelial type II cell monolayers stimulated with cytomix (a mixture of TNF-α, IL-1β and IFN-γ) for 24 h. These results provide the first in vivo evidence that stress preconditioning restores a normal fluid transport capacity of the alveolar epithelium in the early phase following haemorrhagic shock by attenuating NO-mediated oxidative stress to the lung epithelium.
METHODS
The protocol for these studies was approved by the University of California at San Francisco (UCSF) Animal Research Committee.
Lung barrier function studies
Surgical preparation
Male Sprague-Dawley rats weighing 300–350 g were anaesthetized with pentobarbitone sodium (60 mg kg−1i.p.), and anaesthesia was maintained with 30 mg kg−1 of pentobarbitone sodium i.p. every 2 h. An endotracheal tube (PE 220) was inserted through a tracheostomy. The rats were maintained in lateral decubitus position and were mechanically ventilated with a constant-volume pump (Harvard, Millis, MA, USA) throughout the experiment. The rats were ventilated with an inspired oxygen fraction of 1.0, peak airway pressure of 8–12 cmH2O supplemented with a positive end-expiratory pressure of 3 cmH2O. The respiratory rate was adjusted to maintain a Pa,CO2 between 35 and 40 mmHg. During the experiment, the level of anaesthesia was constantly monitored for signs of distress, response to noxious pinch, arterial hypertension and changes in arterial blood gases to determine whether additional doses of anaesthetic were necessary, as done previously (Modelska et al. 1997, 1999). At the end of the experiment, an additional dose of anaesthetic was given and the animals were killed by transection of the abdominal aorta.
Vascular and alveolar protein tracers, 131I- and 125I-labelled human albumin, were used as previously described (Modelska et al. 1997, 1999). At the end of the experiment, an alveolar fluid sample from the distal airspaces (0.1–0.2 ml) was obtained by gently passing the sampling catheter (PE-50 catheter, 0.5 mm i.d.) into a wedged position in the instilled area of the left lower lobe. After centrifugation, the total protein concentration and the radioactivity of the liquid sampled were measured. The right and left lungs were homogenized separately for water: dry weight ratio measurements and radioactivity counts.
Specific protocols
Group 1. Effect of prolonged haemorrhagic shock and fluid resuscitation (n = 20)
The rats were haemorrhaged and resuscitated in order to determine the effect of haemorrhagic shock and fluid resuscitation on fluid transport across the lung epithelium. Five hours after the onset of haemorrhagic shock, 3 ml kg−1 of the 5 % bovine albumin solution with 1 μCi of 125I-labelled albumin were instilled into the left lung (n = 6). Since there was no upregulation of alveolar fluid clearance after haemorrhagic shock, adrenaline was added to the protein solution instilled into the distal airspaces at a concentration of 10−5m(n = 5). This concentration of intra-alveolar adrenaline has previously been shown to be sufficient to upregulate alveolar fluid clearance in pilot experiments. The first series of control studies included rats that underwent the same surgical preparation, were studied for the same period of time, but were neither haemorrhaged nor fluid resuscitated (n = 6). The second series of control studies included rats that were not haemorrhaged nor fluid resuscitated, but had their distal airspaces instilled with an albumin solution containing 10−5m adrenaline for the last hour of the experiment (n = 3).
Group 2. Stress preconditioning with heat (n = 15)
Stress preconditioning was induced with whole body hyperthermia by slowly increasing the rectal temperature of anaesthetized (i.p. pentobarbitone) rats to 42 °C with heating lamps and heating pads 16 h before onset of haemorrhage. The rectal temperature was maintained at 42 °C for 20 min, then the rats were slowly cooled to a physiological body temperature. Haemorrhagic shock and fluid resuscitation were performed as described previously (n = 5). Controls studies included rats that were neither haemorrhaged nor fluid resuscitated, but were preconditioned with whole body hyperthermia and were instilled with a protein solution containing adrenaline (10−5m) (n = 4). In pilot experiments (n = 6), we found that activation of the stress response with whole body hyperthermia did not affect basal alveolar fluid clearance in rats.
Group 3. Stress preconditioning with geldanamycin (n = 14)
Stress preconditioning was induced with geldanamycin (1 mg kg−1i.p. 48 and 24 h before onset of haemorrhage). Haemorrhagic shock and fluid resuscitation were performed as described previously (n = 4). Controls studies included rats that were neither haemorrhaged nor fluid resuscitated, but were preconditioned with geldanamycin and were instilled with a protein solution containing adrenaline (10−5m) (n = 4). In pilot experiments (n = 6), we found that activation of the stress response with geldanamycin did not affect basal alveolar fluid clearance in rats.
Measurements
Haemodynamics, pulmonary gas exchange and protein concentration
Systemic arterial, central venous and airway pressures were continuously measured. Arterial blood gases were measured at 1 h intervals. Samples from the instilled protein solution, from final distal airspace fluid, and from initial and final blood were collected to measure total protein concentration with an automated analyser (AA2 Technicon, Tarrytown, NY, USA).
Albumin flux across endothelial and epithelial barriers
Two different methods were used to measure the flux of albumin across the lung endothelial and epithelial barriers, as done previously (Pittet et al. 1994; Modelska et al. 1997, 1999). The first method measures residual 125I-labelled albumin (the airspace protein tracer) in the lungs as well as accumulation of 125I-labelled albumin in plasma. The second method measures 131I-labelled albumin (the vascular protein tracer) in the extravascular space of the lungs (Pittet et al. 1994; Modelska et al. 1997, 1999).
Alveolar fluid clearance
Changes in the concentration of the non-labelled bovine albumin and the instilled 125I-labelled albumin over the study period (1 h) were used to measure fluid clearance from the distal airspaces, as done previously (Pittet et al. 1994; Modelska et al. 1997, 1999). There is a good correlation between the changes in the concentration of instilled non-labelled bovine albumin and 125I-labelled albumin.
Tracer binding measurement
To determine 125I binding to albumin, trichloracetic acid (20 %) was added to all tubes, which were then centrifuged to obtain the supernatant for measurement of free 125I radioactivity. The results are expressed as a percentage of the unbound 125I radioactivity to the total amount of 125I-labelled albumin radioactivity instilled. These fluid samples always had less than 1 % of unbound iodine present.
Production of nitrite by alveolar macrophages
To determine the effect of stress preconditioning on the production of nitrite by alveolar macrophages after prolonged shock, a first series of experiments included rats (n = 4) that were haemorrhaged and fluid resuscitated as described above. Then the rats were exsanguinated and their lungs inflated to total lung capacity with sterile phosphate-buffered saline (PBS, 10 ml) and lavaged once.
A second series of experiments included rats that were preconditioned with whole body hyperthermia (n = 4) 16 h before they underwent prolonged haemorrhagic shock and fluid resuscitation using the protocol described above. At the end of the experiments, the lungs were inflated to total lung capacity with sterile PBS (10 ml) and lavaged once.
Control studies included: (1) rats (n = 4) that were neither haemorrhaged nor fluid resuscitated and had their airspaces lavaged once at the end of the studies with sterile PBS (10 ml); (2) rats that were preconditioned with whole body hyperthermia but were neither haemorrhaged nor fluid resuscitated and had their airspaces lavaged once at the end of the studies with sterile PBS (10 ml).
In all experiments, the lavage samples were then centrifuged at 800 g for 10 min at 4 °C to remove cells, and quickly frozen at −70 °C. The cell pellet was resuspended in 1 ml of sterile red blood cell lysis buffer (Sigma, USA) for 10 min at room temperature. The cells were again recovered by centrifugation at 800 g for 10 min at 4 °C. Then viable cells were counted and a small aliquot was removed for cytospin and cell differential staining (Diff-Quick, Dade Diagnostics, Aguada, PR, USA). Cell concentration was adjusted by dilution in serum-free RPMI-1640 medium (UCSF Cell Culture Facility) to 5 × 106 cells ml−1 and equal numbers of cells (1 × 106) were plated on 96-well tissue culture plates (Falcon, Lincoln Park, NJ, USA). Cells were treated with a range of lipopolysaccharide concentrations from 0 to 10 ng ml−1 (E. coli 055:B5, Sigma). The alveolar macrophages were then incubated at 37 °C (5 % CO2) for 24 h and cell supernatants were collected. Nitrite production was quantified in the harvested supernatant using a modified Griess Reagent (Sigma) with measurement of visible light absorption at 540 nm. Results are expressed as nitrite production (μm) over 24 h × 105 cells.
Production of nitrite by A549 and alveolar epithelial type II cell monolayers
A549 cells, an alveolar epithelial cell line, were plated at a concentration of 1.0 × 106 cells cm−2 and maintained in a room air-5 % CO2 incubator at 37 °C using DMEM-H21 medium containing 10 % fetal calf serum and penicillin/streptomycin (Gibco BRL). Rat alveolar epithelial type II (ATII) cells were isolated by elastase digestion and selective adhesion plating on rat immunoglobulin G, as done previously (Folkesson et al. 1996). The cells were plated onto tissue culture-treated wells at a concentration of 1.0 × 106 cells cm−2 in DMEM-H21 medium with 10 % fetal calf serum and penicillin/streptomycin (Gibco BRL). Twenty-four hours later, A549 and ATII cell monolayers were stress preconditioned with heat (43 °C for 60 min for A549 cells and 30 min in for ATII cells). In each series of experiments, we verified the activation of the stress response by measuring the expression of the heat shock protein Hsp72 in cell monolayers by Western blotting. Cell monolayers recovered for 12 h at 37 °C, then were stimulated by a mixture of pro-inflammatory cytokines (cytomix) (IL-1β, TNF-α, IFN-γ) at a concentration of 10 ng ml−1 for 24 h (Boehringer Mannheim, Indianapolis, IN, USA). This concentration of cytomix induced a significant increase in the production of nitrite by ATII cell monolayers without increasing cell death measured by Trypan Blue exclusion assay or dimethylthiazol-diphenyl tetrazolium bromide (MTT) assay (Gutierrez et al. 1995). Nitrite production was quantified in the harvested supernatant using a modified Griess Reagent (Sigma) with measurement of visible light absorption at 540 nm. Results are expressed as nitrite production (μm) over 24 h × 106 cells.
Transient transfection and luciferase assay
To determine whether stress preconditioning would affect NF-κB regulation in response to pro-inflammatory stimuli, A549 or ATII cells were transiently transfected with a plasmid in which the luciferase gene was driven by three tandem NF-κB binding motifs followed by a minimal interferon-γ promoter (3NF-κB-Luc, a kind gift from Dr H. Wong, University of Cincinnati, Cincinnati, OH, USA). This plasmid has previously been demonstrated to be a sensitive tool for specifically evaluating NF-κB activation (Wong et al. 1997).
Cells were transfected in triplicate, in 6-well plates, at a density of 300 000 cells per well by incubation with cationic liposomes (Fugene, Roche Biochemicals, Indianapolis, IN, USA) for 48 h in DMEM. Then the cells were exposed to a mixture of pro-inflammatory cytokines (TNF-α, IL-1β, IFN-γ; Boehringer Mannheim) or its vehicle at a concentration of 10 ng ml−1. A group of cells were stress preconditioned with heat (43 °C for 60 min for A549 cells and 30 min for ATII cells) 12 h before exposure to pro-inflammatory cytokines. Twenty-four hours later, cellular proteins were extracted and analysed for luciferase activity according to the manufacturer's instructions (Promega) using a luminometer. Luciferase activity was corrected for total cellular protein and reported as fold-induction over the control cells (cells that were transfected and treated with cytokine vehicle alone).
Western blot measurements (iNOS, Hsp72)
Lungs from haemorrhaged and control rats that underwent or did not undergo stress preconditioning with either whole body hyperthermia or geldanamycin (n = 5 in each group) were thawed in a tissue homogenization buffer, homogenized and the samples were immediately boiled for 5 min at 95–100 °C. Then the samples were centrifuged at 14 000 g at 4 °C for 30 min and the supernatant was quickly frozen at −70 °C.
A549 or ATII cell monolayers were lysed, centrifuged at 14 000 g at 4 °C for 30 min and the supernatant was quickly frozen at −70 °C. The protein concentration of lung tissue and cell lysates was measured using the bicinchoninic acid assay kit with bovine serum albumin as the standard (Pierce Chemical Co., Rockford, IL, USA). A portion of the lung tissue or cell lysate was then boiled in Laemmli sample buffer for 5 min at 95–100 °C prior to loading 5 μg (Hsp72) or 25 μg (iNOS) of total protein per lane on a 10 % Tris-glycine SDS-polyacrylamide gel. The electrophoretically separated proteins were subsequently transferred to nitrocellulose membranes, which were blocked with 5 % milk protein in PBS for 120 min. To detect iNOS protein, nitrocellulose membranes were immunoblotted using a commercially available 1 : 500 or 1 : 1000 dilution of a monoclonal anti-mouse iNOS antibody overnight at 4 °C (Transduction Laboratories, Lexington, KY, USA). The blot was then incubated with horseradish peroxidase-labelled goat anti-mouse immunoglobulin (dilution 1 : 2000). To detect Hsp72 protein, nitrocellulose membranes were immunoblotted using a 1 : 1500 dilution of a monoclonal anti-mouse Hsp72 antibody for 1 h (gift from W.J. Welch, UCSF). The blot was then incubated with horseradish peroxidase-labelled goat anti-rabbit immunoglobulin (dilution 1 : 2000). Protein bands were visualized using a chemiluminescence method (SuperSignal, Pierce Chemical Co.) and quantified using a digital image analysis system (ChemiImager, Alpha Innotech).
Myeloperoxidase activity assay
Myeloperoxidase (MPO) activity in the lung was measured using a modification of the method described by Mullane et al. (1985) as done previously (Modelska et al. 1997).
Statistics
All data are summarized as means ± s.e.m. One-way analysis of variance and the Fisher's exact t test were used to compare experimental and control groups. A P value of < 0.05 was considered statistically significant.
RESULTS
Haemorrhagic shock decreases alveolar epithelial fluid transport
In order to induce haemorrhagic shock, 39 ± 2 % of the blood volume was withdrawn (9.8 ± 0.6 ml of blood), causing systemic arterial hypotension (Table 1) and metabolic acidosis (Table 2) at the end of the ischaemic phase of shock. In addition, there was a significant 3-fold increase in the lung MPO activity in rats that were haemorrhaged and fluid resuscitated compared with controls (Fig. 2). In contrast, arterial PO2 and PCO2 were not statistically different between the experimental groups throughout the study (data not shown).
Table 1.
Effect of preconditioning either with heat or geldanamycin on changes in mean systemic arterial pressure in control and haemorrhaged rats
| Experimental conditions | n | Baseline (mmHg) | Before shock (mmHg) | End of shock (mmHg) | Before instillation (mmHg) | End of experiment (mmHg) |
|---|---|---|---|---|---|---|
| No preconditioning | ||||||
| Controls – no shock | 6 | 101 ± 7 | 106 ± 7 | 100 ± 4 | 99 ± 4 | 103 ± 5 |
| Controls + alveolar adrenaline | 3 | 105 ± 6 | 102 ± 6 | 108 ± 4 | 100 ± 5 | 108 ± 7 |
| Haemorrhagic shock | 6 | 110 ± 5 | 108 ± 3 | 35 ± 4* | 103 ± 6 | 102 ± 9 |
| Haemorrhagic shock + alveolar adrenaline | 5 | 108 ± 5 | 105 ± 6 | 36 ± 4* | 97 ± 5 | 101 ± 5 |
| Stress preconditioning with heat | ||||||
| Controls + alveolar adrenaline | 4 | 105 ± 5 | 108 ± 6 | 104 ± 4 | 98 ± 5 | 102 ± 12 |
| Haemorrhagic shock + alveolar adrenaline | 5 | 108 ± 7 | 102 ± 4 | 37 ± 5* | 99 ± 4 | 99 ± 5 |
| Stress preconditioning with geldanamycin | ||||||
| Controls + alveolar adrenaline | 4 | 114 ± 5 | 97 ± 4 | 106 ± 3 | 100 ± 3 | 101 ± 4 |
| Haemorrhagic shock + alveolar adrenaline | 4 | 114 ± 8 | 103 ± 8 | 36 ± 2* | 102 ± 5 | 107 ± 5 |
Data are given as means ± s.e.m.
P < 0.05 vs. baseline.
Table 2.
Effect of preconditioning either with heat or geldanamycin on changes in arterial pH and calculated base deficit in control and haemorrhaged rats
| Baseline | End of haemorrhagic shock | End of experiment | |||||
|---|---|---|---|---|---|---|---|
| Experimental conditions | n | pH | BE | pH | BE | pH | BE |
| No preconditioning | |||||||
| Controls – no shock | 6 | 7.41 ± 0.02 | 1.1 ± 0.6 | 7.39 ± 0.02 | 0.9 ± 0.7 | 7.38 ± 0.01 | 1.8 ± 0.8 |
| Controls + alveolar adrenaline | 3 | 7.42 ± 0.03 | 0.5 ± 0.3 | 7.41 ± 0.03 | 1.0 ± 0.4 | 7.39 ± 0.02 | 2.0 ± 1.0 |
| Haemorrhagic shock | 6 | 7.44 ± 0.02 | 1.2 ± 0.6 | 7.24 ± 0.01* | −13.3 ± 1.0* | 7.39 ± 0.05 | 2.4 ± 1.3 |
| Haemorrhagic shock + alveolar adrenaline | 5 | 7.41 ± 0.02 | 1.0 ± 0.5 | 7.25 ± 0.02* | −12.3 ± 1.2* | 7.40 ± 0.03 | 2.0 ± 1.2 |
| Stress preconditioning with heat | |||||||
| Controls + alveolar adrenaline | 4 | 7.43 ± 0.02 | 0.5 ± 0.8 | 7.40 ± 0.03 | 0.7 ± 0.7 | 7.39 ± 0.02 | 0.5 ± 1.4 |
| Haemorrhagic shock + alveolar adrenaline | 5 | 7.42 ± 0.04 | 0.6 ± 0.5 | 7.26 ± 0.02* | −12.0 ± 1.4* | 7.40 ± 0.02 | 1.3 ± 0.8 |
| Stress preconditioning with geldanamycin | |||||||
| Controls + alveolar adrenaline | 4 | 7.41 ± 0.01 | 0.8 ± 0.5 | 7.41 ± 0.04 | 0.9 ± 0.4 | 7.38 ± 0.02 | 1.3 ± 1.1 |
| Haemorrhagic shock + alveolar adrenaline | 4 | 7.43 ± 0.02 | 0.7 ± 0.5 | 7.23 ± 0.04* | −13.2 ± 1.2* | 7.40 ± 0.02 | 1.3 ± 1.1 |
Data are given as means ± s.e.m.
P < 0.05 vs. controls. BE, base excess.
Figure 2. Increase in the lung myeloperoxidase (MPO) activity following haemorrhage is significantly reduced by stress preconditioning with heat.
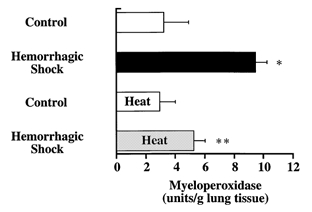
MPO activity (mean ± s.e.m.) is shown for control (n = 4) and haemorrhaged rats (n = 4) that were or were not stress preconditioned with heat (42 °C for 20 min, 16 h before haemorrhage). *P < 0.05 from control rats; **P < 0.05 from haemorrhaged and resuscitated rats that were not stress preconditioned with heat.
Haemorrhagic shock was associated with a failure of the alveolar epithelium to increase vectorial fluid transport in response to catecholamines, despite the absence of any increase in the alveolar epithelial permeability to protein. Protein flux (125I-labelled albumin) from the airspaces to the plasma was comparable in haemorrhaged and control rats (0.5 ± 0.1 vs. 0.6 ± 0.2 %, n.s.). The final-to-initial distal airspace unlabelled protein concentration did not increase in haemorrhaged rats, despite adding adrenaline to the instilled protein solution in order to maximize the response of β-adrenergic receptors (Table 3). In contrast, the expected normal increase in airspace unlabelled protein concentration did occur in control rats instilled with a protein solution containing adrenaline (Table 3). In response to catecholamines, alveolar fluid clearance significantly increased in control rats, but not in haemorrhaged rats (Fig. 3A). Comparable values were obtained when alveolar fluid clearance was measured with the 125I-labelled albumin (control rats: 31 ± 2 to 43 ± 1 %, P < 0.05; haemorrhaged rats: 34 ± 2 to 32 ± 3 %, n.s.). Finally, haemorrhagic shock was associated with a small increase in lung endothelial permeability to protein, as indicated by the significant increase in extravascular plasma equivalents of non-instilled lung in haemorrhaged rats compared with controls (Table 4).
Table 3.
Effect of preconditioning either with heat or geldanamycin on alveolar fluid protein concentration after haemorrhagic shock
| Alveolar protein concentration (g (100 ml)−1) | ||||
|---|---|---|---|---|
| Experimental conditions | n | Initial | Final | F/I ratio |
| No preconditioning | ||||
| Controls — no shock | 6 | 4.50 ± 0.12 | 6.45 ± 0.22 | 1.37 ± 0.05 |
| Controls + alveolar adrenaline | 3 | 4.40 ± 0.30 | 7.05 ± 0.30 | 1.59 ± 0.07* |
| Haemorrhagic shock | 6 | 4.42 ± 0.07 | 6.02 ± 0.15 | 1.34 ± 0.02 |
| Haemorrhagic shock + alveolar adrenaline | 5 | 4.44 ± 0.04 | 5.98 ± 0.12 | 1.32 ± 0.03 |
| Stress preconditioning with heat | ||||
| Controls + alveolar adrenaline | 4 | 4.42 ± 0.12 | 6.99 ± 0.15 | 1.58 ± 0.03* |
| Haemorrhagic shock + alveolar adrenaline | 5 | 4.39 ± 0.02 | 6.45 ± 0.13 | 1.48 ± 0.02*† |
| Stress preconditioning with geldanamycin | ||||
| Controls + alveolar adrenaline | 4 | 4.37 ± 0.12 | 6.63 ± 0.15 | 1.54 ± 0.02* |
| Haemorrhagic shock + alveolar adrenaline | 4 | 4.40 ± 0.07 | 6.52 ± 0.11 | 1.49 ± 0.03*† |
Data are given as means ± s.e.m.; F/I ratio: final over initial alveolar protein concentration ratio
P < 0.05 vs. controls
P < 0.05 vs. shock.
Figure 3. Stress preconditioning restores the ability of alveolar epithelium to respond to catecholamines by upregulating alveolar fluid clearance after haemorrhage.
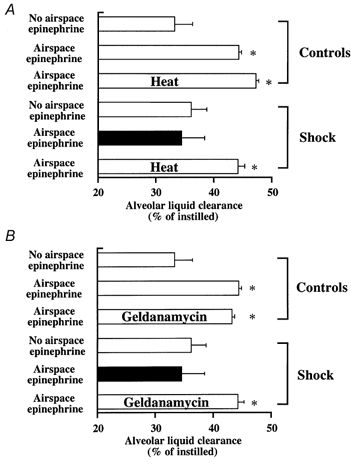
Rats were anaesthetized and then either subjected to whole body hyperthermia (42 °C for 20 min 12–16 h before onset of haemorrhage) or injected with geldanamycin (i.p., 1 mg kg−1, 48 and 24 h before onset of haemorrhage). Half of the animals were left untreated while the other half were subjected to haemorrhagic shock as described in Methods. Measurement of alveolar liquid clearance was performed as described in Methods. A, alveolar fluid clearance (% of instilled liquid) (mean ± s.e.m.) is shown for control and haemorrhaged rats that were or were not pretreated with whole body hyperthermia; B, alveolar fluid clearance (% of instilled liquid) (mean ± s.e.m.) is shown for control and haemorrhaged rats that were or were not pretreated with geldanamycin; *P < 0.05 from haemorrhaged and resuscitated rats that did not receive any adrenaline in their airspaces.
Table 4.
Preconditioning either with heat or geldanamycin attenuates lung endothelial permeability to protein after haemorrhagic shock
| Experimental condition | n | Extravascular PE (ml) |
|---|---|---|
| No preconditioning | ||
| Controls — no shock | 6 | 0.07 ± 0.01 |
| Controls + alveolar adrenaline | 3 | 0.08 ± 0.01 |
| Haemorrhagic shock | 6 | 0.30 ± 0.08* |
| Haemorrhagic shock + alveolar adrenaline | 5 | 0.28 ± 0.07* |
| Stress preconditioning with heat | ||
| Controls + alveolar adrenaline | 4 | 0.06 ± 0.02 |
| Haemorrhagic shock + alveolar adrenaline | 5 | 0.10 ± 0.03 |
| Stress preconditioning with geldanamycin | ||
| Controls + alveolar adrenaline | 4 | 0.07 ± 0.03 |
| Haemorrhagic shock + alveolar adrenaline | 4 | 0.11 ± 0.02 |
Data are given as means ± s.e.m. Extravascular PE, extravascular plasma equivalents.
P < 0.05 from controls. Lung endothelial permeability to protein was measured as accumulation of the vascular tracer 131I-labelled albumin inextravascular spaces of the non-instilled lung, and was expressed as extravascular plasma equivalents.
Stress preconditioning with heat or geldanamycin restores normal alveolar epithelial fluid transport after haemorrhagic shock
Recent work indicates that activation of the cellular stress response in the lung results in the modulation of the inflammatory response (Wong et al. 1997a–d). Thus our second series of experiments was designed to determine whether stress preconditioning, either with whole body hyperthermia or via pharmacological activation of the stress response (geldanamycin), would attenuate the shock-mediated failure of the alveolar epithelium to respond to catecholamines. Rats preconditioned with heat or geldanamycin (as described in Fig. 1) underwent a haemorrhagic shock similar to that of non-pretreated rats. The quantity of blood removed, systemic arterial hypotension, metabolic acidosis and calculated base deficit of the arterial blood at the end of the ischaemic phase of haemorrhagic shock were comparable in all experimental groups (Tables 1 and 2). However, stress preconditioning with heat was associated with a significant reduction in the lung MPO activity compared with haemorrhaged, but non-preconditioned rats (Fig. 2).
Figure 1. Schematics outlining the general experimental protocols used in the studies.
See Methods section for further explanation.
Stress preconditioning with heat or geldanamycin was associated with a significant increase in the expression of Hsp72 protein in the lung (Fig. 4 and Fig. 5). Vectorial fluid transport across the alveolar epithelium was not affected by stress preconditioning in control non-haemorrhaged rats (data not shown). Moreover, stress preconditioning preserved the ability of the alveolar epithelium to upregulate vectorial fluid transport in response to catecholamines in control rats (Table 3, Fig. 3A and B). Comparable values were found when alveolar fluid clearance was measured with 125I-labelled albumin (data not shown).
Figure 4. Stress preconditioning via whole body hyperthermia leads to increased expression of Hsp72 protein in the lung.
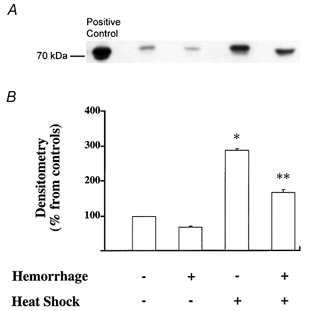
Rats were left untreated or preconditioned via whole body hyperthermia. Some of the animals were then subjected to haemorrhagic shock. Animals were killed, the lungs removed and then examined for their expression of Hsp72 by Western blotting. A, Western blot analysis for Hsp72. One representative lung is shown for each experimental condition. Three additional experiments gave comparable results. B, densitometry analysis of four comparable Western blots for detection of Hsp72 protein showed a significant increase in the expression of Hsp72 protein in lung homogenate from rats that underwent whole body hyperthermia compared with non-pretreated rats; *P < 0.05 from control rats that did not receive any pretreatment; ** P < 0.05 from haemorrhaged and resuscitated rats that did not receive any pretreatment.
Figure 5. Stress preconditioning using geldanamycin leads to increased expression of Hsp72 in the lung.
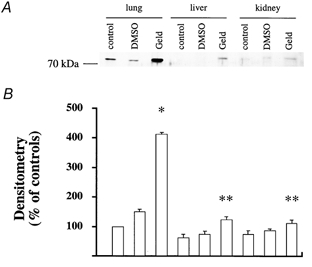
Rats were left untreated or injected with geldanamycin. Then the animals were killed and the lungs, liver and kidneys were removed and examined for their expression of Hsp72 by Western blotting. Equal amounts of total protein were applied to the gel. A, Western blot analysis for Hsp72 protein in control rats that have or have not been pretreated with geldanamycin. One representative experiment is shown for each experimental condition. Three additional experiments gave comparable results. B, densitometry analysis of four comparable Western blots for detection of Hsp72 protein showed a significant increase in the expression of Hsp72 protein in lung homogenate from rats that were pretreated with geldanamycin compared with non-pretreated rats; *P < 0.05 from control rats that did not receive any pretreatment; **P < 0.05 from the values measured in the lung.
Activation of the cellular stress response restored the normal catecholamine-mediated upregulation of alveolar liquid clearance in haemorrhaged rats (Table 3, Fig. 3A and B). Comparable values were found when alveolar fluid clearance was measured with 125I-labelled albumin (whole body hyperthermia: 33 ± 2 to 42 ± 3 %; geldanamycin: 33 ± 2 to 41 ± 3 %). This protective effect was not observed when haemorrhaged rats were pretreated with i.p. DMSO, the vehicle by which geldanamycin was delivered (data not shown). Finally, stress preconditioning with heat or geldanamycin also significantly attenuated the increase in lung endothelial permeability to protein in haemorrhaged rats (Table 4).
Stress preconditioning attenuates the release of NO by alveolar macrophages and alveolar epithelial cells
We have recently reported that the inability to increase alveolar epithelial fluid transport in response to a cAMP-dependent stimulation was mediated by the release of NO within the airspaces of the lung after haemorrhage in rats (Pittet, 2001). Therefore, our third series of experiments was to test the hypothesis that stress preconditioning with heat or geldanamycin would attenuate the shock-mediated expression of iNOS protein in the lung. As is shown in Fig. 6, stress preconditioning with either whole body hyperthermia or geldanamycin did indeed significantly reduce the expression of iNOS protein in the lung of haemorrhaged rats. Hence a restoration of the cAMP-dependent upregulation of alveolar fluid clearance in haemorrhaged rats that have been stress preconditioned with whole body hyperthermia or geldanamycin may be due, in part, to reduced iNOS expression.
Figure 6. Stress preconditioning with whole body hyperthermia or geldanamycin results in an attenuation of iNOS protein levels in the lung.
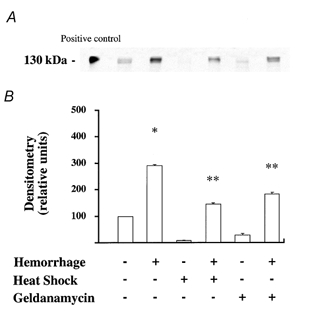
Rats were left untreated or preconditioned via whole body hyperthermia or geldanamycin. Some of the animals were then subjected to haemorrhagic shock. Animals were killed, the lungs removed and then examined for their expression of iNOS by Western blotting. A, Western blot analysis for iNOS protein in control and haemorrhaged rats that have or have not been pretreated with whole body hyperthermia or geldanamycin. One representative experiment is shown for each experimental condition. Four additional experiments gave comparable results. B, densitometry analysis of five comparable Western blots for detection of iNOS protein showed a significant reduction in the expression of iNOS protein in lung homogenate from stress preconditioned and haemorrhaged rats compared with non-pretreated haemorrhaged rats; *P < 0.05 from control rats that did not receive any pretreatment; ** P < 0.05 from haemorrhaged and resuscitated rats that did not receive any pretreatment.
Alveolar macrophages and alveolar epithelial cells have both been shown to be a major source of NO production within the airspaces of the lung (Warner et al. 1995). Thus in our fourth series of experiments we examined whether stress preconditioning would attenuate the production of NO in either alveolar macrophages or rat alveolar epithelial type II cells.
Rats were left untreated or preconditioned via whole body hyperthermia as described above. Some of the animals were then subjected to haemorrhagic shock. Animals were killed, alveolar macrophages were recovered from bronchoalveolar lavage (BAL) fluid and were cultured ex vivo for 24 h with or without stimulation with lipopolysaccharide (LPS; 0, 0.01, 0.1, 1 and 10 ng ml−1). There was a significant increase in the production of nitrite, one of the major end-products of NO metabolism, by alveolar macrophages removed from haemorrhaged rats 6 h after onset of haemorrhagic shock and then cultured ex vivo for 24 h in the presence of increasing concentrations of LPS. Stress preconditioning significantly decreased this LPS-induced production of nitrite by the alveolar macrophages (Fig. 7).
Figure 7. Stress preconditioning with whole body hyperthermia attenuates the increase in nitrite production following endotoxin stimulation of alveolar macrophages from haemorrhaged rats.
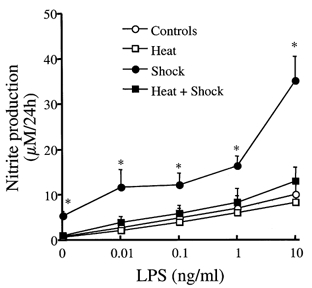
Rats were left untreated or preconditioned via whole body hyperthermia. Some of the animals were then subjected to haemorrhagic shock. Animals were killed, alveolar macrophages were recovered from BAL fluid and were cultured ex vivo for 24 h with or without stimulation with LPS (0, 0.01, 0.1, 1 and 10 ng ml−1). Nitrite production (μm per 24 h) (mean ± s.e.m.) is shown for control (○) and haemorrhaged and resuscitated rats (•), and control (□) and haemorrhaged and resuscitated rats (▪) pretreated with whole body hyperthermia; *P < 0.05 from control rats.
In order to determine if stress preconditioning attenuates NO production in alveolar epithelial cells, we utilized A549 cells, a human alveolar epithelial cell line, as well as primary cultures of rat ATII cells. A549 and ATII cell monolayers were stimulated for 24 h with cytomix (a mixture of TNF-α, interferon-γ and IL-1β), an in vitro model representative of the oxidative stress to the alveolar epithelium observed after haemorrhagic shock. There was a 5- to 6-fold increase in the production of nitrite, a stable end-product of NO, in the medium of both cell monolayers 24 h after stimulation with cytomix (Fig. 8A and B). Under these conditions, cell death measured by Trypan Blue exclusion was less than 2 %. Stress preconditioning with heat reduced, by 50 and 70 %, respectively, the production of nitrite by A549 and ATII cell monolayers (Fig. 8A and B). That cell exposure to heat was effective in eliciting a stress response was confirmed by an increased expression of Hsp72 protein in both A549 and ATII cell monolayers (Fig. 9A and B). The reduction in the production of nitrite in thermotolerant cells was not associated with any significant change in cell death as measured by Trypan Blue exclusion (less than 2 %).
Figure 8. Stress preconditioning reduces cytokine-stimulated nitrite production by A549 or primary cultures of rat alveolar epithelial type II cell monolayers.
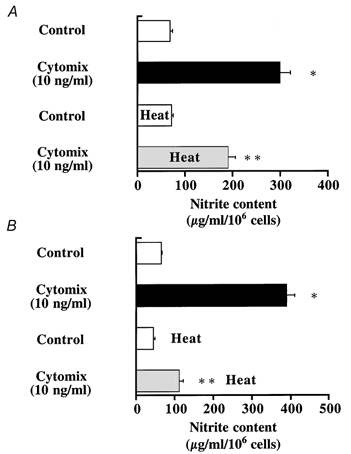
Control cells were maintained in basal growth media. Cytokine-treated cells were exposed to a mixture of pro-inflammatory cytokines (TNF-α, IL-1β, IFN-γ) for 24 h at a concentration of 10 ng ml−1. A group of cells were stress preconditioned with heat (43 °C for 60 min for A549 cells and 30 min for ATII cells) 12 h before exposure to pro-inflammatory cytokines. Nitrite production in the cell culture medium 24 h after exposure to cytomix or its vehicle is shown for cell monolayers that were (n = 4) or were not stress preconditioned (n = 4) with heat. *P < 0.05 from controls; ** P < 0.05 from cell monolayers that were exposed to cytomix, but not stress preconditioned with heat.
Figure 9. Increased expression of Hsp72 protein production by A549 or primary cultures of rat ATII cell monolayers after stress preconditioning with heat.
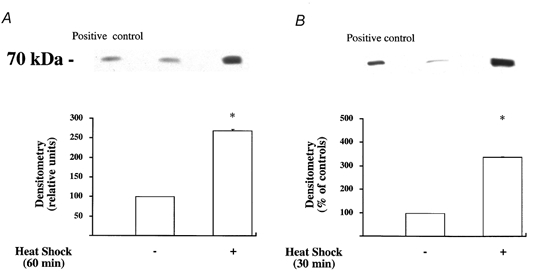
Control cells were maintained at 37 °C. A group of cells were stress preconditioned with heat (43 °C for 60 min for A549 cells and 30 min for ATII cells) 12 h before being harvested. A, Western blot analysis for Hsp72 protein expression in A549 cell monolayers that were or were not stress preconditioned with heat. One representative experiment is shown for each experimental condition. Three additional experiments gave comparable results. B, Western blot analysis for Hsp72 protein expression in primary cultures of rat ATII cell monolayers that were or were not stress preconditioned with heat. One representative experiment is shown for each experimental condition. Three additional experiments gave comparable results; *P < 0.05 from cell monolayers that were not stress preconditioned with heat.
Activation of the pro-inflammatory transcriptional factor, NF-κB, occurs early (15 min) after the onset of septic (Blackwell et al. 1996) and haemorrhagic shock (Shenkar et al. 1996). In vivo inhibition of NF-κB activation prevents endotoxin-dependent iNOS expression in the lung (Liu et al. 1997). Thus the fifth series of experiments was designed to determine whether the attenuation of NO release by thermotolerant cells would be mediated by an inhibition of cytokine-induced activation of NF-κB pathway. Exposure to pro-inflammatory cytokines significantly increased luciferase activity in A549 and ATII cells transiently transfected with a κ-dependent luciferase reporter construct (3IgκBLuc) (Fig. 10A and B). In contrast, stress preconditioning with heat significantly reduced NF-κB activation of this κ-dependent luciferase reporter construct in both A549 and ATII cell monolayers (Fig. 10A and B).
Figure 10. Stress preconditioning with heat attenuates cytokine-mediated activation of NF-κB in A549 (A) or primary cultures of rat ATII cell monolayers (B).
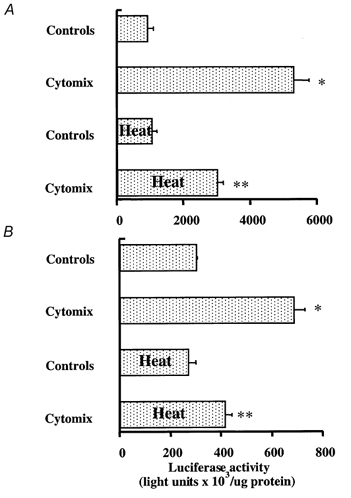
Cells were transiently transfected with luciferase reporter plasmid containing three tandem NF-κB motifs. Control cells were maintained in basal growth media. Cytokine-treated cells were exposed to a mixture of proinflammatory cytokines (TNF-α, IL-1β, IFN-γ) for 24 h at a concentration of 10 ng ml−1. A group of cells were stress preconditioned with heat 12 h before exposure to pro-inflammatory cytokines. Luciferase activity was corrected for total cellular protein and reported as fold-induction over the control cells (cells that were transfected and treated with cytokine vehicle alone). Data represent the means ± s.e.m. of four separate experiments with each condition carried out in triplicate; *P < 0.05 from control cells not treated with cytokine; ** P < 0.05 from cytokine-treated cells that were not stress preconditioned with heat.
DISCUSSION
Because the preservation of the capacity of the alveolar epithelium to actively remove fluid from the airspaces is critical for the survival of patients with acute lung injury (Matthay & Wiener-Kronish, 1990; Ware & Matthay, 2001), we developed an experimental animal model to investigate the effect of haemorrhagic shock on the vectorial fluid transport across the alveolar epithelium. Under pathological conditions that predispose to the development of increased permeability pulmonary oedema (septic or haemorrhagic shock), upregulation of alveolar epithelial fluid transport by endogenous catecholamines is a major mechanism that prevents alveolar flooding (Pittet et al. 1994, 1996; Modelska et al. 1997). After severe haemorrhage, however, this protective mechanism is abolished by the development of an inflammatory response mediated by neutrophils that accumulate in the airspaces of the lung (Laffon et al. 1999; Modelska et al. 1999). The primary objective of these studies, therefore, was to determine whether stress preconditioning would restore the catecholamine-mediated upregulation of alveolar liquid clearance after haemorrhage, one of the common causes of acute lung injury and pulmonary oedema in humans (Hudson et al. 1995).
The heat shock or stress response is a highly conserved cellular defence mechanism that is characterized by the increased expression of heat shock or stress proteins. Increased levels of the stress proteins, for example by mild hyperthermia, allow cells to withstand a subsequent thermal insult that would otherwise be lethal, a phenomenon referred to as ‘thermotolerance’ or ‘preconditioning’. Non-thermal forms of cellular stress, such as exposure to heavy metals or geldanamycin, also increase heat shock protein expression and thereby confer upon cells a thermotolerant phenotype (Li, 1983; Hedge et al. 1995; Zou et al. 1998). Perhaps most relevant for our studies described here are previous observations showing that increased stress protein expression imparts protection against other injuries, including oxidant and endotoxin exposure (Bellmann et al. 1995). We therefore hypothesized that the activation of the stress response could decrease the oxidative stress to the alveolar epithelium and help to restore a normal physiological response of the lung to catecholamines following haemorrhage.
As we have shown here, activation of the stress response, either by whole body hyperthermia or geldanamycin, resulted in a substantial increase in the expression of Hsp72 protein in the lung. Normally present in cells at relatively low levels, increased expression and accumulation of Hsp72 is a reliable indicator that activation of the cellular stress response has occurred. Moreover and consistent with our hypothesis, stress preconditioning did in fact help to restore cAMP-dependent upregulation of vectorial fluid transport across the alveolar epithelium in haemorrhaged rats.
Our work adds to a growing number of studies showing that stress preconditioning mediates cytoprotection in cell and animal models of acute lung injury. For example, prior activation of the stress response protected rats against a subsequent and acute lung injury caused by intratracheal administration of phospholipase A1 (Villar et al. 1993) or by systemic administration of endotoxin (Villar et al. 1994). Induction of the stress response was shown to protect lung grafts from reperfusion injury after transplantation in rats (Hiratsuka et al. 1999). Increased expression of stress proteins, either by heat or sodium arsenite treatment, attenuated endotoxin-mediated injury to cultured sheep pulmonary artery endothelial cells (Wong et al. 1996). Finally, in cultured murine lung epithelium and bovine pulmonary endothelial cells, induction of the stress response was shown to confer added protection against oxidant injury (Wong et al. 1997a,b). Results presented herein provide the first experimental evidence that stress preconditioning can have an effect on ion transport activities across the alveolar epithelium and thereby help to restore the ability of this barrier to upregulate fluid transport in response to cAMP-dependent stimulation after severe haemorrhage. Thus stress preconditioning appears to provide protection to the lung against various forms of acute injury.
The mechanisms by which activation of the stress response confers cytoprotection in the lung are likely to be multi-fold. First, in their role as molecular chaperones, stress proteins help stabilize and/or refold proteins damaged as a consequence of the particular stress event. In addition, when present at higher levels, the stress proteins collectively facilitate the synthesis and maturation of new proteins needed to replace those irreparably damaged by the earlier metabolic insult. Finally, a specific role for the most highly stress inducible protein, Hsp72, in cellular protection has been suggested from experiments utilizing molecular means to increase its intracellular levels. For example, overexpression of Hsp72 was demonstrated to protect cultured lung cells against NO- (Wong et al. 1997b), endotoxin- (Wong et al. 1996) or hyperoxia-mediated cell death (Wong et al. 1998).
Recent data indicate that another important feature of the stress response in the lung is a corresponding decrease in the inflammatory response. For example, activation of the stress response has been reported to inhibit endotoxin-mediated expression of TNF-α and IL-1β in cultured mononuclear cells (Schmidt & Abdulla, 1988) and in vivo in the lung (Ribeiro et al. 1996). Furthermore, inhibition of cytokine-mediated iNOS expression and the subsequent release of NO in either rat pulmonary smooth muscle cells (Wong et al. 1995) or in murine lung epithelium (Wong et al. 1997a) following stress preconditioning has been reported. Recent work from our laboratory has provided in vivo evidence that NO is the proximal mediator of the inflammatory response that limits the rate of alveolar epithelial transport after prolonged haemorrhagic shock. Increased levels of NO appeared to directly alter the function of membrane proteins involved in β-adrenergic receptor-cAMP signalling pathways (Pittet et al. 2001). Consistent with our previous observations, we have shown in the present study that stress preconditioning, using either whole body hyperthermia or administration of geldanamycin, was associated with a significant attenuation in the shock-mediated expression of iNOS and subsequent release of NO in the lung. There are several mechanisms that may explain why attenuation of NO induction helps to restore the response of the alveolar epithelium to β-adrenergic agonists after prolonged haemorrhagic shock. First, abnormally high levels of NO may result in the inhibition of cation channels present on rat alveolar type II cells (DuVall et al. 1998; Guo et al. 1998). Secondly, high concentrations of NO may inhibit membrane proteins involved in the β-adrenergic receptor-cAMP signalling pathway of the alveolar epithelium (Pittet et al. 2001). Finally, increased levels of NO could lead to an increase in chloride secretion by lung epithelial cells. Consistent with this idea are studies showing that NO can activate non-cystic fibrosis transmembrane regulator (CFTR) chloride channels in human lung epithelial cells by a cGMP-dependent mechanism (Kamosinska et al. 1997). Thus the release of induced NO within the airspaces of haemorrhaged rats could result in an unopposed chloride secretion by the alveolar epithelium in the event that sodium absorptive pathways are perturbed by the effect of NO on the apical alveolar epithelial cation channels. Whatever the exact mechanism(s), stress preconditioning may provide protection via an impact on some aspect of the immune response leading to lower NO levels.
That this may be the case was supported by our in vitro studies examining alveolar macrophages and alveolar epithelial cells, two types of cell known to be a major source of NO production within the airspaces of the lung (Warner et al. 1995). Our results showed that stress preconditioning with heat did in fact cause a significant decrease in the production of NO by alveolar macrophages removed from the airspaces of preconditioned and haemorrhaged rats when compared with the control, haemorrhaged animals not provided with a pre-conditioning treatment. Similarly, stress preconditioning resulted in a significant decrease in the production of NO by either A549 cells, an alveolar epithelial cell line, or primary cultures of rat ATII cells upon their subsequent exposure to pro-inflammatory cytokines. These results are in line with prior studies showing that induction of the heat shock response inhibited cytokine-mediated expression of iNOS in pulmonary artery smooth muscle cells (Wong et al. 1995) and lung epithelial cells (Wong et al. 1997a). Moreover, stress preconditioning has also been reported to inhibit the expression of a variety of other pro-inflammatory genes including IL-8 (Thomas et al. 1998), RANTES (Ayad et al. 1998), TNF-α (Snyder et al. 1992) and IL-1β (Schmidt & Abdulla, 1988). This effect may be rather specific to pro-inflammatory genes since the expression of other products, such as the surfactant proteins, was largely unaffected (Wong et al. 1997a). Thus activation of the cellular stress response may provide protection in the lung via its attenuation of pro-inflammatory mediators, explaining (at least in part) why the stress preconditioning protects against various forms of acute lung injury.
Exactly how stress preconditioning might inhibit cellular inflammatory responses is still under investigation. One study reported that the transcription factor controlling the expression of the heat shock genes (HSF) might act as a repressor, binding to regulatory sequences of the promoter of the genes encoding certain proinflammatory cytokines, such as IL-1β (Cahil et al. 1996). Yet other studies have suggested a more general mechanism, involving the inhibition of the whole NF-κB pathway (Feinstein et al. 1996; Wong et al. 1997d; Pritts et al. 2000). For example, it has been reported that the heat shock-mediated inhibition of NF-κB activation was associated with a decrease in the phosphorylation and ubiquitination of IκBα, thereby leading to the stabilization of this NF-κB inhibitor (Wong et al. 1997c,d; Pritts et al. 2000). In addition, activation of the stress response itself was suggested to result in an increase in the expression of the IκB gene and a corresponding increase in the level of the IκB protein (Wong et al. 1997d; Thomas et al. 1998; Wong et al. 1999). It is important to note, however, that these effects of heat on the NF-κB pathway appear to be restricted to only the early time periods following the heating regimen. Thus how NF-κB activities might be attenuated in cells preconditioned via heat shock treatment 12–24 h prior to cytokine stimulation is still not clear and therefore represents a major focus of our ongoing work.
In summary, our studies provide in vivo evidence that stress preconditioning, by whole body hyperthermia or geldanamycin exposure, attenuates NO-mediated oxidative stress to the alveolar epithelium following haemorrhagic shock. This protective effect, at least in part, was mediated by an attenuation of the NF-κB pathway and resulted in a restoration of normal fluid transport capacity of the alveolar epithelium in the early phases following haemorrhagic shock. These results are of potential clinical importance. Results of a recent study in critically ill patients revealed an increased production of reactive oxygen-nitrogen intermediates in the distal airspace of patients with acute lung injury. In turn these changes were associated with impaired alveolar fluid clearance (Zhu et al. 2001), an early predictor of prolonged duration of mechanical ventilation and higher hospital mortality in patients with acute lung injury (Ware & Matthay, 2001). Whether we can exploit the protective effects of cellular stress preconditioning to attenuate these sorts of changes in the lung remains an important but still unanswered question.
Acknowledgments
This work was primarily supported by NIH grants GM62188 (to J. F. Pittet) and HL51854 (to M. A. Matthay). W. J. W. acknowledges the continued support of the NIH (GM33551).
REFERENCES
- Ayad O, Stark JM, Fiedler MA, Menendez IY, Ryan MA, Wong HR. The heat shock response inhibits RANTES gene expression on cultured lung epithelium. Journal of Immunology. 1998;161:2594–2599. [PubMed] [Google Scholar]
- Bellmann K, Wenz A, Radons J, Burkart V, Leemann R, Kolb H. Heat shock induces resistance in rat pancreatic islet cells against nitric oxide, oxygen radicals and streptozotocin. Journal of Clinical Investigation. 1995;95:2840–2845. doi: 10.1172/JCI117989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackwell TS, Blackwell TR, Holden EP, Christman BW, Christman JW. In vivo antioxidant treatment suppresses nuclear factor κB activation and neutrophilic lung inflammation. Journal of Immunology. 1996;157:1630–1637. [PubMed] [Google Scholar]
- Cahil CM, Waterman WR, Xie Y, Auron PE, Calderwood SK. Transcriptional repression of the prointerleukin-1β gene by heat shock factor 1. Journal of Biological Chemistry. 1996;271:24874–24879. [PubMed] [Google Scholar]
- DuVall MD, Zhu S, Fuller M, Matalon S. Peroxynitrite inhibits amiloride-sensitive sodium currents in Xenopus oocytes expressing αβγ-rENaC. American Journal of Physiology. 1998;274:C1417–1423. doi: 10.1152/ajpcell.1998.274.5.C1417. [DOI] [PubMed] [Google Scholar]
- Feinstein DL, Galea E, Aquino DA, Li GC, Xu H, Reis DJ. Heat shock protein 70 suppresses astroglial-inducible nitric oxide synthase expression by decreasing NF-κB expression. Journal of Biological Chemistry. 1996;271:17224–17232. doi: 10.1074/jbc.271.30.17724. [DOI] [PubMed] [Google Scholar]
- Folkesson HG, Pittet JF, Nitenberg G, Matthay MA. Transforming growth factor-α increases alveolar liquid clearance in anesthetized, ventilated rats. American Journal of Physiology. 1996;271:L236–244. doi: 10.1152/ajplung.1996.271.2.L236. [DOI] [PubMed] [Google Scholar]
- Guo Y, DuVall MD, Crow JP, Matalon S. Nitric oxide inhibits sodium absorption across cultured alveolar type II cell monolayers. American Journal of Physiology. 1998;274:L369–377. doi: 10.1152/ajplung.1998.274.3.L369. [DOI] [PubMed] [Google Scholar]
- Gutierrez HH, Pitt BR, Schwartz M, Watkins SC, Lowenstein C, Caniggia I, Chumley P, Freeman BA. Pulmonary alveolar epithelial inducible NO synthase gene expression: regulation by inflammatory mediators. American Journal of Physiology. 1995;268:L501–508. doi: 10.1152/ajplung.1995.268.3.L501. [DOI] [PubMed] [Google Scholar]
- Hedge RS, Zuo J, Voellmy R, Welch WJ. Short-circuiting stress protein expression via tyrosine inhibitor, herbimycin. Journal of Cellular Physiology. 1995;165:186–200. doi: 10.1002/jcp.1041650122. [DOI] [PubMed] [Google Scholar]
- Hierholzer C, Harbrecht B, Menezes JM, Kane J, Macmicking J, Nathan CF, Peitzman AB, Billiar TR, Tweardy DJ. Essential role of induced nitric oxide in the initiation of the inflammatory response after hemorrhagic shock. Journal of Experimental Medicine. 1998;187:917–928. doi: 10.1084/jem.187.6.917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiratsuka M, Mora BN, Yano M, Mohanakumar T, Patterson A. Gene transfer of heat shock protein 70 protects lung grafts from ischemia-reperfusion injury. Annals of Thoracic Surgery. 1999;67:1421–1427. doi: 10.1016/s0003-4975(99)00164-2. [DOI] [PubMed] [Google Scholar]
- Hudson LD, Milberg JA, Anardi D, Maunder R. Clinical risks for development of the acute respiratory distress syndrome. American Journal of Respiratory and Critical Care Medicine. 1995;151:293–301. doi: 10.1164/ajrccm.151.2.7842182. [DOI] [PubMed] [Google Scholar]
- Kamosinska B, Radomski MW, Duszyk M, Radomski A, Man SFP. Nitric oxide activates chloride currents in human lung epithelial cells. American Journal of Physiology. 1997;272:L1098–1104. doi: 10.1152/ajplung.1997.272.6.L1098. [DOI] [PubMed] [Google Scholar]
- Laffon M, Lu LN, Modelska K, Matthay MA, Pittet JF. α-Adrenergic blockade restores the normal fluid transport capacity of the alveolar epithelium after hemorrhagic shock. American Journal of Physiology. 1999;277:L760–768. doi: 10.1152/ajplung.1999.277.4.L760. [DOI] [PubMed] [Google Scholar]
- Li GC. Induction of thermotolerance and enhanced heat shock protein synthesis in Chinese hamster fibroblasts by sodium arsenite and ethanol. Journal of Cellular Physiology. 1983;115:116–122. doi: 10.1002/jcp.1041150203. [DOI] [PubMed] [Google Scholar]
- Liu SF, Ye X, Malik AB. In vivo inhibition of nuclear factor κB activation prevents inducible nitric oxide synthase expression and systemic hypotension in a rat model of septic shock. Journal of Immunology. 1997;159:3976–3983. [PubMed] [Google Scholar]
- Matthay MA, Wiener-Kronish JP. Intact epithelial barrier function is critical for the resolution of alveolar edema in humans. American Review of Respiratory Diseases. 1990;142:1250–1257. doi: 10.1164/ajrccm/142.6_Pt_1.1250. [DOI] [PubMed] [Google Scholar]
- Minowada G, Welch WJ. Clinical implications of the stress response. Journal of Clinical Investigation. 1995;95:3–12. doi: 10.1172/JCI117655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modelska KM, Matthay MA, Brown LAS, Deusch E, Lu LN, Pittet JF. Inhibition of β-adrenergic-dependent alveolar epithelial clearance by oxidant mechanisms after hemorrhagic shock. American Journal of Physiology. 1999;276:844–857. doi: 10.1152/ajplung.1999.276.5.L844. [DOI] [PubMed] [Google Scholar]
- Modelska KM, Matthay MA, McElroy MC, Pittet JF. Upregulation of alveolar liquid clearance after fluid resuscitation for hemorrhagic shock in rats. American Journal of Physiology. 1997;273:L305–314. doi: 10.1152/ajplung.1997.273.2.L305. [DOI] [PubMed] [Google Scholar]
- Morimoto RI, Tissieres A, Geogopoulos C. Stress Proteins in Biology and Medicine. Cold Spring Harbor, NY, USA: Cold Spring Harbor Laboratories; 1990. [Google Scholar]
- Mullane KM, Kraemer E, Smith B. Myeloperoxidase activity as a quantitative assessment of neutrophil infiltration into ischemic myocardium. Journal of Pharmacological Methods. 1985;14:157–167. doi: 10.1016/0160-5402(85)90029-4. [DOI] [PubMed] [Google Scholar]
- Pittet JF, Brenner TB, Modelska K, Matthay MA. Alveolar liquid clearance is increased by endogenous catecholamines in hemorrhagic shock in rats. Journal of Applied Physiology. 1996;81:830–837. doi: 10.1152/jappl.1996.81.2.830. [DOI] [PubMed] [Google Scholar]
- Pittet JF, Lu LN, Morris DG, Modelska K, Welch WJ, Carey HV, Roux J, Matthay MA. Reactive nitrogen species inhibit alveolar epithelial fluid transport by NF-κB dependent mechanisms after hemorrhagic shock in rats. Journal of Immunology. 2001;166:6301–6310. doi: 10.4049/jimmunol.166.10.6301. [DOI] [PubMed] [Google Scholar]
- Pittet JF, Wiener-Kronish JP, McElroy MC, Folkesson HG, Matthay MA. Stimulation of alveolar epithelial liquid clearance by endogenous release of catecholamines in septic shock. Journal of Clinical Investigation. 1994;94:663–671. doi: 10.1172/JCI117383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritts TA, Want Q, Sun X, Moon MR, Fisher DR, Fisher JE, Wong HR, Hasselgren PO. Induction of the stress response in vivo decreases NF-κB activity in jejunal mucosa of endotoxemic mice. Archives of Surgery. 2000;135:860–866. doi: 10.1001/archsurg.135.7.860. [DOI] [PubMed] [Google Scholar]
- Ribeiro SP, Villar J, Downey GP, Edelson JE, Slutsky AS. Effect of the stress response in septic rats and LPS-stimulated alveolar macrophages: evidence for TNF-α post-translational regulation. American Journal of Respiratory and Critical Care Medicine. 1996;154:1843–1850. doi: 10.1164/ajrccm.154.6.8970379. [DOI] [PubMed] [Google Scholar]
- Schmidt JA, Abdulla E. Down-regulation of IL-β biosynthesis by inducers of the heat shock response. Journal of Immunology. 1988;141:2027–2034. [PubMed] [Google Scholar]
- Shenkar R, Schwartz MD, Terada LS, Repine JE, McCord J, Abraham E. Hemorrhage activates NF-κB in murine lung mononuclear cells in vivo. American Journal of Physiology. 1996;270:L729–735. doi: 10.1152/ajplung.1996.270.5.L729. [DOI] [PubMed] [Google Scholar]
- Snyder YL, Guthrie L, Evans GF, Zuckerman S. Transcriptional inhibition of endotoxin-induced monokine synthesis following heat shock in murine peritoneal macrophages. Journal of Leukocyte Biology. 1992;51:181–187. doi: 10.1002/jlb.51.2.181. [DOI] [PubMed] [Google Scholar]
- Thomas SC, Ryan MA, Shanley TP, Wong HR. Induction of the stress response with prostaglandin-A increases I-κB gene expression. FASEB Journal. 1998;12:1371–1378. doi: 10.1096/fasebj.12.13.1371. [DOI] [PubMed] [Google Scholar]
- Villar J, Edelson JD, Post M, Mullen JB, Slutsky AS. Induction of heat stress proteins is associated with decreased mortality in an animal model of acute lung injury. American Journal of Respiratory and Critical Care Medicine. 1993;147:177–181. doi: 10.1164/ajrccm/147.1.177. [DOI] [PubMed] [Google Scholar]
- Villar J, Ribeiro SP, Mullen JB, Kuliszewski M, Post M, Slutsky AS. Induction of heat shock response reduces mortality rate and organ damage in a sepsis-induced acute lung injury model. Critical Care Medicine. 1994;22:914–922. [PubMed] [Google Scholar]
- Ware LB, Matthay MA. Alveolar epithelial fluid clearance is impaired in the majority of patients with acute lung injury and the acute respiratory distress syndrome. American Journal of Respiratory and Critical Care Medicine. 2001;163:1376–1383. doi: 10.1164/ajrccm.163.6.2004035. [DOI] [PubMed] [Google Scholar]
- Warner RL, Paine R, Christensen PJ, Marletta MA, Richards MK, Wilcoxen SE, Ward PA. Lung sources and cytokine requirements for in vivo expression of inducible nitric oxide synthase. American Journal of Respiratory Cell and Molecular Biology. 1995;12:649–661. doi: 10.1165/ajrcmb.12.6.7539274. [DOI] [PubMed] [Google Scholar]
- Wong HR, Finder JD, Wasserloos K, Pitt BR. Expression of inducible nitric oxide synthase in cultured rat pulmonary artery smooth cells is inhibited by the heat shock response. American Journal of Physiology. 1995;269:L843–848. doi: 10.1152/ajplung.1995.269.6.L843. [DOI] [PubMed] [Google Scholar]
- Wong HR, Mannix J, Rusnak JM, Boota A, Zar H, Watkins SC, Lazo JS, Pitt BR. The heat shock response attenuates lipopolysaccharide-mediated apoptosis in cultured sheep pulmonary artery endothelial cells. American Journal of Respiratory Cell and Molecular Biology. 1996;15:745–751. doi: 10.1165/ajrcmb.15.6.8969269. [DOI] [PubMed] [Google Scholar]
- Wong HR, Menendez IY, Ryan M, Denenberg AG, Wispe JR. Increased expression of heat shock protein 70 protects A549 cells against hyperoxia. American Journal of Physiology. 1998;275:L836–841. doi: 10.1152/ajplung.1998.275.4.L836. [DOI] [PubMed] [Google Scholar]
- Wong HR, Ryan M, Gebb S, Wispe JR. Selective and transient in vitro effects of heat shock on alveolar type II cell gene expression. American Journal of Physiology. 1997a;272:L132–138. doi: 10.1152/ajplung.1997.272.1.L132. [DOI] [PubMed] [Google Scholar]
- Wong HR, Ryan M, Menendez IY, Denenberg A, Wispe JR. Heat shock protein induction protects human respiratory epithelium against nitric oxide-mediated cytotoxicity. Shock. 1997b;8:213–218. doi: 10.1097/00024382-199709000-00010. [DOI] [PubMed] [Google Scholar]
- Wong HR, Ryan M, Menendez I, Wispe JR. Heat shock activates the I-κBa promoter and increases I-κBa mRNA expression. Cell Stress and Chaperones. 1999;4:1–7. doi: 10.1006/csac.1998.0123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong HR, Ryan M, Wispe JR. The heat shock response inhibits activation of inducible nitric oxide synthase gene expression by blocking I-κB degradation and NF-κB nuclear translocation. Biochemical and Biophysical Research Communications. 1997c;231:257–263. doi: 10.1006/bbrc.1997.6076. [DOI] [PubMed] [Google Scholar]
- Wong HR, Ryan M, Wispe JR. Stress response decreases NF-kB nuclear translocation and increases I-κBa expression in A549 cells. Journal of Clinical Investigation. 1997d;99:2423–2428. doi: 10.1172/JCI119425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu S, Ware LB, Geiser T, Matthay MA, Matalon S. Increased levels of nitrate and surfactant protein A nitration in the pulmonary edema fluid of patients with acute lung injury. American Journal of Respiratory and Critical Care Medicine. 2001;163:166–172. doi: 10.1164/ajrccm.163.1.2005068. [DOI] [PubMed] [Google Scholar]
- Zou J, Guo Y, Guettouche T, Smith DF, Voellmy R. Repression of heat shock transcription HSF1 activation by Hsp 90 (Hsp 90 complex) that forms a stress-sensitive complex with HSF1. Cell. 1998;94:471–480. doi: 10.1016/s0092-8674(00)81588-3. [DOI] [PubMed] [Google Scholar]