

Abstract
Stimulation of ovine airway epithelial cells with 10 μm ATP for 1 min at 25 °C transiently increased both cytoplasmic calcium (fura-2 epifluorescence microscopy) and ciliary beat frequency (CBF; differential interference contrast microscopy) with a similar time course. Identical purinergic stimulation of human airway epithelial cells at 25 or 35 °C, however, lead to an increase in CBF that outlasted the calcium transient at least 20 min. While a nitric oxide synthase inhibitor had no effect, pre-treatment of human cells with inhibitors of cAMP-dependent kinase (PKA), 10 μm myristoylated PKA-inhibitory peptide and 1 μm KT-5720, as well as an inhibitor of adenylyl cyclase, 1 mm SQ22536, blocked the prolonged, but not calcium-coupled CBF increase. Addition of PKA inhibitors after purinergic stimulation only partially reduced CBF from its elevated plateau. Prolonged CBF increases did not depend on adenosine production as 10 μm UTP had an effect similar to ATP and 8-sulphophenyl-theophylline did not block them. After increasing human CBF in a PKA-dependent manner to a stable plateau with forskolin (10 μm), ATP caused only a transient, calcium-coupled CBF increase. Calcium transients were necessary for both short-term and prolonged CBF changes as ATP failed to produce CBF increases after emptying calcium stores with 1 μm thapsigargin. These data suggest that in human, but not ovine airway epithelial cells, ATP-induced calcium transients activate a signalling cascade including adenylyl cyclase and PKA. The resulting prolonged CBF stimulation does not rely only on PKA activity, suggesting that the decay of CBF is influenced by ciliary phosphatase activity.
The regulation of ciliary beat frequency (CBF) has been studied in a variety of species, ranging from single cell organisms like Paramecium to mammalian cells, including those from humans. Although the general microtubular ultrastructure of cilia is preserved throughout all species (Fawcett & Porter, 1954; Rhodin & Dalhamn, 1956; Gibbons, 1961; Rhodin, 1966), the regulation of ciliary beating varies between organisms. These differences are probably the consequence of particular functions that cilia fulfil in a variety of cells. For example, in Paramecium, cilia provide locomotion whereas cilia on epithelial cells in the mammalian tracheobronchial tree contribute to mucociliary clearance, the most important host defense mechanism of the airways.
In mammals, there seem to exist at least three partially independent mechanisms to increase CBF: (1) cAMP-dependent phosphorylation (Di Benedetto et al. 1991; Salathe et al. 1993; Wyatt et al. 1998); (2) calcium-dependent NO/cGMP-dependent phosphorylation (Jain et al. 1993; Wyatt et al. 1998; Uzlaner & Priel, 1999); and (3) direct Ca2+ action (e.g. Salathe & Bookman, 1999). While it may be tempting to speculate that all mammalian species regulate CBF in a similar way, recent evidence contradicts this notion. For instance, species differences were found between CBF responses to sole cGMP exposure: in bovine and human ciliated cells, CBF increases (Geary et al. 1995; Wyatt et al. 1998), whereas in rabbit and sheep, CBF remains stable (Uzlaner & Priel, 1999; Salathe et al. 2000). In one paper, cGMP production was even associated with a decrease in CBF (Tamaoki et al. 1991). Therefore, the regulation of CBF by these different mechanisms may vary widely depending on the species studied.
Signal transduction with respect to CBF regulation in mammalian cells has been specifically investigated through a variety of G protein-coupled receptors using muscarinic, purinergic and β-adrenergic stimuli to increase CBF (Sanderson & Dirksen, 1989; Di Benedetto et al. 1991; Kelsen et al. 1995; Korngreen & Priel, 1996; Yang et al. 1996; Salathe et al. 1997; Uzlaner & Priel, 1999; Morse et al. 2001). While such approaches were used in an effort to pinpoint the mechanisms of CBF increases through the intracellular second messengers mentioned above, they also indicated that some of the regulatory pathways might interact. For instance, β-adrenergic stimulation of rabbit tracheal explants increased CBF both in an initial calcium-coupled and prolonged cAMP-dependent fashion (Lansley et al. 1992). Since β-adrenergic agonists stimulate adenylyl cyclase, these results indicated a cAMP-dependent calcium release mechanism in these cells. Similar results were obtained in a non-mammalian species, namely the frog oesophagus (Braiman et al. 1998).
Purinergic activation of the P2Y receptor is one of the strongest and most consistent stimulus to increase CBF in a variety of mammalian species (Wong & Yeates, 1992; Gheber et al. 1995; Korngreen & Priel, 1996; Korngreen et al. 1998; Evans & Sanderson, 1999; Morse et al. 2001). P2Y receptors couple to phospholipase C to produce inositol 1,4,5-trisphosphate (IP3) that releases calcium from internal stores. There is considerable disagreement over how increasing calcium concentrations stimulate mammalian CBF; both a direct action of calcium on dynein arms as well as phosphorylation cascades have been implicated (Korngreen & Priel, 1994, 1996; Evans & Sanderson, 1999; Lansley & Sanderson, 1999; Salathe & Bookman, 1999). But purinergic stimulation may also increase CBF through a cAMP-dependent pathway as shown by Morse et al. (2001) as ATP can be hydrolysed extracellularly and stimulate adenosine receptors. We provide here evidence that an additional cAMP-dependent pathway, not dependent on the activation of adenosine receptors, can be activated by purinergic stimulation of human airway epithelial cells.
METHODS
Chemicals
Dulbecco's modified Eagle's medium (DMEM), Ham's F-12 nutrient, and Hanks’ balanced salt solution (HBSS) were purchased from Gibco, Life Technologies (Grand Island, NY, USA). Forskolin, ATP, UTP, and 8-sulphophenyl-theophylline were from Sigma (St Louis, MO, USA). Fura-2 AM was from Molecular Probes (Eugene, OR, USA). Thapsigargin, myristoylated protein kinase A inhibitory peptide 14–22 (PKI14–22), KT-5720, KT-5823, SQ22536, and 3-bromo-7-nitroindazole were from Calbiochem (La Jolla, CA, USA).
Preparation of submerged tracheal epithelial cultures
Primary cultures of tracheal epithelial cells were prepared as previously described (Salathe & Bookman, 1995). Briefly, the mucosa from freshly obtained tracheas was dissected from the underlying cartilage under sterile conditions and incubated in 0.05 % protease (Sigma, Type 14) in DMEM overnight at 4 °C. Ovine tissue was obtained from adult ewes killed by intravenous injection of 30 mg kg−1 pentobarbital according to NIH and local animal care use committee-approved protocols. Human tissue was obtained from organ donors whose lungs were not suited for transplant through the organ procurement team at the University of Miami, appropriate consent was obtained as approved by the local human subjects committee.
After protease treatment, epithelial cells were released by vigorous shaking and harvested by centrifugation. They were plated on collagen-coated glass coverslips (human placental collagen, Sigma) at a density of 0.5 × 106 cells cm−2 in a minimal volume of 100 μl cm−2 (= 1 ml per 35 mm dish). The culture media consisted of 50 % DMEM, 50 % Ham's F-12 medium supplemented with insulin (10 μg ml−1), transferrin (5 μg ml−1), hydrocortisone (0.36 μg ml−1), triiodothyronine (20 ng ml−1), endothelial cell growth supplement (7.5 μg ml−1), penicillin (100 μg ml−1), and streptomycin 100 μg ml−1). Media were exchanged every other day. Cells from these cultures were used for physiological measurements 3–14 days after plating.
Preparation of air liquid interface cultures of tracheal epithelium
Human and ovine air-liquid interface (ALI) cultures were prepared according to published methods (Adler et al. 1990; Matsui et al. 1998a,b; Bernacki et al. 1999). Briefly, tracheal epithelial cells were harvested as described above. They were plated on collagen-coated plastic dishes, grown to confluence and passaged after enzyme dissociation with trypsin yielding undifferentiated airway epithelial cells. Cells from passage 1 or 2 were plated onto Transwell collagen-coated inserts with a diameter of 24 mm (Corning Costar Corporation, Cambridge, MA, USA). Cells were grown in an incubator at 37 °C in ambient air supplemented with 5 % CO2 until fully redifferentiated (about 4 weeks) and then used for measurements.
Measurement of CBF
Cells grown on coverslips in submerged cultures were mounted at room temperature onto the stage of a Nikon Eclipse E600FN upright water-immersion lens microscope in a Warner Instrument RC-25F perfusion chamber with a 150 μl working volume and perfused constantly. Ciliated cells were imaged with infrared differential interference contrast (DIC) optics with an optical gain of × 600. For online CBF measurements, the light path was directed to a CCD video camera (CCD 100, Dage MIT, Michigan City, IN, USA) and a box of 3 × 3 pixels from the live, digitized, contrast-enhanced video image was selected (where one pixel samples an area of 180 nm × 180 nm). The magnitude spectra from a fast Fourier transform (FFT) of each of the pixel's intensity signals were computed online and displayed on the monitor for immediate adjustments. The intensity signals were recorded and later used for analysis according to published methods (Salathe & Bookman, 1999) using a sliding FFT window approach (128 frames per FFT, sliding the FFT window through the data set by 100 frames at a time), providing a frequency resolution of at least 0.23 Hz and a time resolution of ∼3 s. The individual FFT magnitude spectra were peak extracted for graphing (Salathe & Bookman, 1999).
For a few experiments with cells in submerged culture, the microscope was encased in an environmentally controlled chamber heated to 35 °C. These CBF measurements were obtained with a special CCD camera (Sony XC-7500), acquiring the images at 60 Hz rather than the usual 30 Hz, providing an upper frequency detection limit of 30 Hz. All solutions for these experiments were preheated to 35 °C as well.
In order to measure CBF in cells grown at the air-liquid interface (but imaged with the apical surface ‘submerged’), we used a holding chamber for the Transwell membranes allowing selective perfusion of the apical or basolateral sides of the cells. For these experiments, the basolateral side was filled with buffer but not perfused, while the apical side was perfused to allow drug additions and imaging through a water-immersion lens (Nikon) at a total magnification of × 600. Data acquisition and processing was identical to the one described for submerged cultures.
Measurement of [Ca2+]i
Incubation protocols
Coverslips were rinsed 3 times with HBSS buffered with 10 mm Hepes, pH 7.40 (HBSS-Hepes), and loaded at room temperature with 4 μm fura-2 AM in HBSS-Hepes containing 2.5 % fetal calf serum (Hyclone, UT, USA) for 60 min on a rocking table. The dishes were washed 3 times with HBSS-Hepes and used for measurements after a minimum of 30 min.
Imaging hardware and software
For [Ca2+]i measurements with fura-2, a lambda DG4 excitation system (Sutter, Novato, CA, USA) was used with 10 nm wide excitation filters centred on 340 and 380 nm (Chroma Technology Corp., Brattleboro, VT, USA). ‘Ratio-tool’ software from Inovision (Durham, NC, USA) controlled the output of the lambda DG4. Ratiometric calcium estimates were made by capturing the light (510–600 nm) emitted from the cells excited with wavelengths of 340 and 380 nm for 600 ms through a × 60 water immersion objective (Nikon Inc.) and directing it to a cooled CCD camera (Quantix, Photometrics, Tucson, AZ, USA). Using Inovision's ‘ratio-tool’ software, individual cells were identified as regions of interest (ROIs). The fura-2 ratio within each ROI was computed on a pixel by pixel basis (after background fluorescence correction). Ratios were computed every 20 s, or more frequently as needed (every 10 s during the time of exposure to different stimuli). Average fura-2 ratio values for each ROI were written to disk for later analysis and graphing.
Simultaneous measurement of CBF and [Ca2+]i
Both the CBF analysis software as well as ‘ratio-tool’ were running on an SGI O2 workstation (Silicon Graphics, Mountain View, CA, USA). By using a dual-image module and guiding the infrared signal for CBF measurements to the CCD camera while sending all fluorescence signals (< 600 nm) to the cooled CCD camera, we were able to measure recordings of CBF and calcium of the same single cell simultaneously.
Expression of [Ca2+]i data
Since we were not able to perform reliable calibrations of the calcium indicator dye fura-2 intracellularly in ciliated cells (cells exposed to ionomycin did not change their intracellular calcium concentration when exposed to different extracellular calcium concentrations), conversions of the fura-2 ratio data into [Ca2+]i using in vitro calibration procedures would have given rough, but not accurate estimates of the true calcium values. We therefore decided against conversions and report here the original ratiometric fura-2 data. Rough estimates indicated the baseline [Ca2+]i in ciliated cells from both human and sheep tracheas to be between 70 and 100 nm and peak responses to be between 200 and 350 nm.
Incubation protocols / experimental procedures
PKA was inhibited with the cell membrane-permeant, myristoylated protein kinase A inhibitory peptide 14–22 (PKI14–22) (Muniz et al. 1997; Rimon & Rubin, 1998) and also with KT-5720 (Kase et al. 1987; Cabell & Audesirk, 1993). PKI14–22 was used at 10 μm (∼30-fold excess of estimated Ki) and KT-5720 at 1 μm (Ki = 56 nm) in HBSS-Hepes. PKG was inhibited with 1 μm KT-5823 (Ki = 234 nm) (Grider, 1993). Cells were pre-incubated with the inhibitors for 60 min at room temperature unless specified otherwise. 3-Bromo-7-nitroindazole was used at 5 μm (highest IC50 for any nitric oxide synthase isoform, 290 nm) during continuous perfusion for at least 5 min before adding ATP.
All baseline measurements were performed during continuous perfusion with HBSS-Hepes at a flow rate of 250 μl min−1. After recording baseline signals for 5–10 min, ATP was added for 1 min (2 min for cells cultured at the air-liquid interface) at a flow rate of 1000 μl min−1 and then washed away with buffer at flow rates of 1000 μl min−1 for 5 min. Flow rates were changed to accelerate the full exchange of the bathing solution during short-term agonist application (shown to be complete after 1 min). These changes in flow rates did neither influence CBF nor [Ca2+]i. Forskolin and thapsigargin were applied using continuous perfusion at the low flow rate of 250 μl min−1.
Statistics
Since some variations in the responses to agonists were encountered during the experimentations (see for instance Fig. 1 and Fig. 11), responses from cells were always compared to date and culture-matched controls. The statistical analysis used a one-way analysis of variance to compare the means of more than two groups (e.g. time courses) using JMP software from SAS Institute Inc. (Cary, NC, USA). If a significant difference was found, a group-by-group comparison was done using the Tukey-Kramer honestly significant difference test. Two groups were compared using Student's unpaired t test. A P < 0.05 was considered to be significant. Data were expressed as means ± s.e.m.
Figure 1. CBF and [Ca2+]i responses to short-term ATP exposure in ovine and to short-term ATP or UTP exposure in human tracheal epithelial cells.
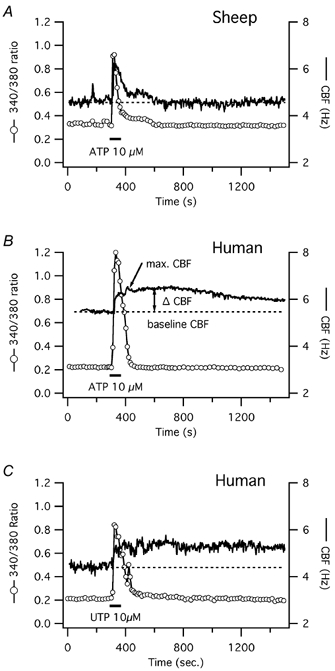
Simultaneous CBF and [Ca2+]i recordings of single tracheal epithelial cells loaded with fura-2. [Ca2+]i is represented by the original fura-2 ratiometric data obtained from its fluorescence emissions at excitations of 340 and 380 nm. A, representative recording from an ovine airway epithelial cell reveals a strictly calcium-coupled, transient increase in CBF upon short-term stimulation with ATP. B, recording from a representative human airway epithelial cell shows an initial calcium-coupled, and later prolonged increase in CBF upon short-term exposure to ATP. C, similar to ATP, a representative human airway epithelial cell responds with an initial calcium-coupled, and later prolonged increase in CBF upon short-term exposure to UTP.
Figure 11. Influence of late PKG inhibition with KT-5823 and late PKA inhibition with PKI14–22 on CBF and [Ca2+]i responses to short-term ATP exposure in human tracheal epithelial cells.
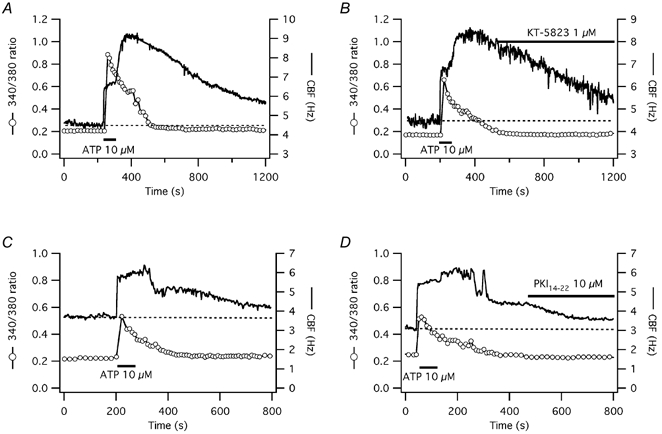
A and B, the [Ca2+]i and CBF responses to short-term ATP exposure of representative submerged human tracheal epithelial cells (date- and culture-matched) are shown. PKG inhibition during the prolonged CBF increase with 1 μm KT-5823 (B) did not significantly change the CBF decline towards baseline compared to control (A). C and D, in another pair of cells, PKA inhibition during the prolonged CBF increase with 10 μm PKI14–22 (D) significantly accelerated the CBF decline towards baseline compared with control (C), but CBF was still significantly higher than baseline at the end of the experiment.
RESULTS
CBF and [Ca2+]i response to extracellular ATP in human and ovine cells
Changes in CBF and [Ca2+]i were recorded simultaneously from single cells in submerged culture during ∼1 min exposures to 10 μm ATP. In both human (n = 12) and ovine (n = 7) cells, ATP caused a rapid increase in both [Ca2+]i and CBF (Fig. 1). Baseline CBF was 4.6 ± 0.5 Hz in human cells, a value significantly lower compared with ovine cells with a baseline CBF of 6.0 ± 0.6 Hz (P < 0.001). The maximal CBF increase in response to 10 μm ATP was similar in both species: frequency increased by 1.7 ± 0.2 Hz (or 37 ± 4 % above baseline) in human cells vs. 2.1 ± 0.3 Hz (or 35 ± 5 % above baseline) in ovine cells (P = 0.2 for both absolute and percentage changes). The maximal increase of the fura-2 ratio signal was also similar in both species: 0.53 ± 0.08 in human cells vs. 0.63 ± 0.10 in ovine cells (P = 0.6). In both species, the start of the [Ca2+]i and CBF increases occurred at the same time, i.e. within the time resolution of the CBF (3 s) and fura-2 ratio recordings (10 s). We and others have previously shown that this time resolution is insufficient to accurately describe the correlation between [Ca2+]i and CBF rises, which occur within 100–150 ms (Evans & Sanderson, 1999; Lansley & Sanderson, 1999; Salathe & Bookman, 1999). However, since the work described here concentrates on the decay of CBF back to baseline compared with the [Ca2+]i signal, we have not included any high resolution measurements.
The fura-2 ratio signal returned to baseline (± 0.03 of the original fura-2 ratio) within 205 ± 28 s in human cells and within 174 ± 36 s in ovine cells (P = 0.5). The duration of the CBF increases, however, was significantly different. In ovine cells, CBF declined back to baseline in parallel to the decline in [Ca2+]i; in fact, the time courses of these two signals were statistically indistinguishable considering the time resolution of the measurements (Fig. 1A). Again, this does not rule out some delay of the CBF signal compared to [Ca2+]i as described by us and others to be ∼8 s (Evans & Sanderson, 1999; Lansley & Sanderson, 1999; Salathe & Bookman, 1999). Higher concentrations of ATP (100 μm) did not change the relationship between CBF and [Ca2+]i seen with 10 μm ATP in these ovine cells (n = 5).
In human cells, on the other hand, CBF remained elevated 10 min after ATP stimulation despite the earlier decline of [Ca2+]i: CBF was still 1.0 ± 0.1 Hz (or 21 ± 3 %) above baseline (P = 0.01; Fig. 1B). Since most measurements were stopped within 20 min after ATP exposure and CBF remained elevated, the exact duration of the CBF elevation is unknown. To rule out a simultaneous activation of adenosine receptors due to ATP hydrolysis to adenosine (Morse et al. 2001), human cells were also exposed to UTP (Fig. 1C). Exposure to 10 μm UTP for 1 min resulted in a calcium and CBF response that was statistically indistinguishable to ATP exposure (n = 5). Thus, this significant (i.e. minutes and not seconds) ‘uncoupling’ of [Ca2+]i and CBF during the [Ca2+]i decrease in human cells in response to ATP or UTP suggests that the CBF changes are not solely due to a direct calcium effect on cilia in human cells.
To see whether long-term exposures to ATP and UTP yielded different responses compared with short-term exposures, human cells were treated with 100 μm ATP or UTP for the duration of the experiment (Fig. 2A and B). Statistically, long-term exposures to these purinergic agonists were indistinguishable from short-term exposures, both showing a slight decay of CBF towards baseline over time (each n = 5). The exposure to both 100 μm ATP plus 10 μm of the non-metabolizable adenosine analogue 5-(N-ethylcarboxamido)-adenosine (NECA), however, yielded a similar calcium response, but a different CBF response: the CBF increase was statistically higher compared with exposure to ATP alone (CBF increased by 2.8 ± 0.5 Hz above a baseline of 5.2 ± 0.1 Hz, n = 3, P = 0.04 compared with ATP alone) and CBF had a tendency to increase further after its peak and not to return to baseline (Fig. 2C). These results suggested that both short-term and long-term exposures to ATP or UTP stimulate CBF in a similar manner and that, at room temperature, adenosine receptor activation through ATP hydrolysis does not contribute significantly to these responses.
Figure 2. CBF and [Ca2+]i responses to long-term ATP, UTP or ATP plus NECA exposure in human tracheal epithelial cells.
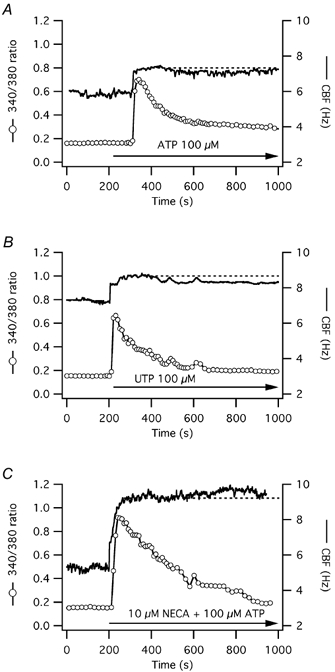
A, a representative human airway epithelial cell shows an initial calcium-coupled, and later prolonged increase in CBF upon long-term exposure to ATP, similar to the response upon short-term ATP exposure (as shown in Fig. 1B). B, another cell shows also an initial calcium-coupled, and later prolonged increase in CBF upon long-term exposure to UTP, similar to the response upon short-term UTP exposure (as shown in Fig. 1C). C, the exposure to both ATP plus 5-(N-ethylcarboxamido)-adenosine (NECA) yielded a calcium response similar to ATP alone. CBF, however, increased to a higher level and had a tendency to increase further after its initial peak as opposed to exposure to ATP alone, where CBF had a tendency to slowly decay towards baseline.
This finding was confirmed by testing the ability of 100 μm 8-sulphophenyl-theophylline (8-SPT), a broad adenosine receptor antagonist, to inhibit the prolonged CBF activation after short-term ATP exposure. Addition of 8-SPT 5 min after ATP stimulation (n = 4) had no influence on CBF compared to cells only exposed to ATP (Fig. 3; P > 0.05 compared with control cells). In fact, addition of 8-SPT lead, for unknown reasons, to a small increase in baseline [Ca2+]i with a concurrent, slight CBF increase. Thus, both the UTP and 8-SPT data confirm that the prolonged CBF elevation to short-term ATP exposure was not due to adenosine receptor stimulation.
Figure 3. Influence of adenosine receptor blockage on CBF and [Ca2+]i responses to short-term ATP exposure in human tracheal epithelial cells.
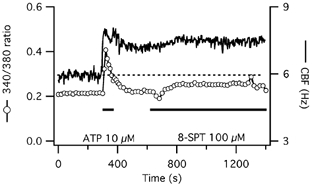
CBF and [Ca2+]i responses to short-term ATP exposure of a representative submerged human tracheal epithelial cell are shown. Blockage of adenosine receptors with 100 μm 8-sulphophenyl-theophylline (8-SPT) does not accelerate the decline of the prolonged CBF increase towards baseline but actually slightly increases both CBF and [Ca2+]i.
To make sure that the prolonged CBF response was not only seen in submerged cultures, CBF was also measured from cells grown and redifferentiated at the air-liquid interface (ALI). Calcium was not recorded since poor loading of fura-2 AM into these cells prevented reproducible epifluorescence measurements. Applied to the apical side of the cultures, higher concentrations of ATP (50 μm) were necessary to stimulate CBF, possibly because of mucus production in cultures grown at the ALI. The CBF responses were kinetically similar to those in submerged culture: after short ATP stimulation (2 min) of ovine cells, CBF (baseline 6.7 ± 1.4 Hz) increased transiently by 0.9 ± 0.2 Hz (or 14 ± 2 % above baseline). After 10 min, CBF had returned to baseline; in fact, CBF returned to baseline within 366 ± 33 s (n = 3, Fig. 4A). In human cells, a prolonged CBF response was seen after a 2 min exposure to 50 μm ATP: CBF (baseline 5.77 ± 0.67 Hz) increased by 1.7 ± 0.7 Hz (or 29 ± 11 % above baseline) and remained 1.2 ± 0.3 Hz above baseline (or 20 ± 6 %) 10 min after ATP stimulation (n = 6, Fig. 4B).
Figure 4. CBF responses to short-term ATP exposure in tracheal epithelial cells grown at the air-liquid interface.
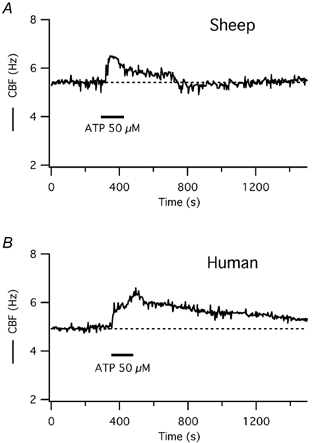
A, a representative ovine cell responds to 50 μm ATP, applied apically, with a transient increase in CBF, similar to ovine cells in submerged cultures (see Fig. 1A). B, in a representative human cell, on the other hand, CBF increases with two maxima and remains elevated above baseline for an extended period of time after short-term ATP exposure, similar to human cells in submerged culture (see Fig. 1B).
To rule out that the measurement temperature (25 °C) was responsible for the prolonged CBF increase to ATP in human cells, experiments were performed with submerged human cells at 35 °C. The principle of long CBF elevation (20 min) after short-term purinergic stimulation was confirmed (n = 3) at this temperature with statistically indistinguishable time courses compared with the room temperature measurements (Fig. 5A).
Figure 5. Neither physiological temperature nor a NOS inhibitor change CBF responses to short-term ATP exposure in human cells.
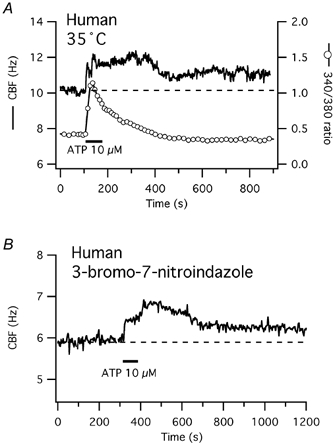
A, [Ca2+]i and CBF responses to short-term ATP exposure of a representative human cell measured at 35 °C. CBF reveals both a calcium-coupled and a prolonged increase after short-term ATP exposure, similar to cells measured at room temperature. B, representative human cell in submerged culture and measured at room temperature. Despite the presence of the NOS inhibitor 3-bromo-7-nitroindazole (5 μm), CBF shows an immediate rise and a delayed second peak, followed by a prolonged elevation of CBF, similar to control cells (Fig. 1B).
CBF response to ATP in the presence of a NOS inhibitor
Since rabbit cells showed a two-phased CBF increase to long-term ATP exposure (Uzlaner & Priel, 1999) similar to the one seen here in human cells, we examined whether the NO-cGMP-PKG pathway was responsible for the prolonged rise in human CBF. Nitric oxide synthase (NOS) was inhibited with 3-bromo-7-nitroindazole, a membrane-permeable inhibitor, at a concentration well above its highest Ki and known to inhibit nitric oxide synthase in other systems (e.g. Babbedge et al. 1993). The presence of 3-bromo-7-nitroindazole had no effect on the human CBF time course in response to short-term ATP exposure (n = 5): after a rapid rise of CBF, a delayed second peak was observed (Fig. 5B) and the elevation of CBF above baseline 10 min after ATP stimulation was not significantly different compared with control (see Fig. 1B).
CBF and [Ca2+]i responses to ATP after inhibition of PKA
To test the hypothesis that PKA was responsible for the prolonged CBF response to short-term purinergic stimulation of human cells, simultaneous measurements of CBF and [Ca2+]i in response to extracellular ATP were performed in human and ovine cells with and without inhibition of PKA.
In this series of experiments, baseline CBF of ovine cells was 6.0 ± 0.6 Hz (n = 7) in the control group and 7.0 ± 0.7 Hz in the group of cells pre-incubated with the PKA inhibitory peptide PKI14–22(n = 6; P = 0.3). The maximal CBF increase in response to ATP was 2.1 ± 0.3 Hz (or 35 ± 4 % above baseline) in controls and 2.3 ± 0.3 Hz (or 32 ± 4 % above baseline) in the PKI group (P = 0.7). The maximal increase in the fura-2 ratio signal was 0.77 ± 0.10 in the controls and 0.93 ± 0.11 in the PKI14–22 group (P = 0.3). PKA inhibition did not change the time course of the response to ATP and the coupling of CBF to calcium in ovine cells (Fig. 6A; P > 0.05 for all measurements in the PKI14–22 group compared to controls). Thus, the responses of ovine cells to ATP were analogous in the presence (Fig. 6A) or absence (compare Fig. 1A) of PKA inhibition with PKI14–22, suggesting that PKA activation was not involved in the purinergic stimulation of CBF in ovine cells and that cytoplasmic calcium itself played the major role as suggested previously for muscarinic activation (Salathe & Bookman, 1999).
Figure 6. CBF and [Ca2+]i responses to short-term ATP exposure after inhibition of PKA with PKI14–22 in ovine and human tracheal epithelial cells.
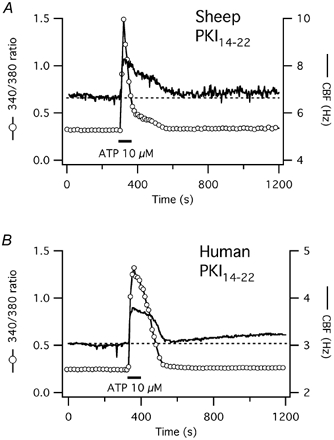
A, [Ca2+]i and CBF responses to short-term ATP exposure in a representative ovine tracheal epithelial cell after pre-treatment with 10 μm of the PKA inhibitory peptide PKI14–22 as outlined in Methods. The response of both signals and their coupling is not different from control cells (see Fig. 1A). B, in a representative human tracheal cell, however, the CBF response to short-term ATP exposure is now strictly calcium-coupled in contrast to control cells (see Fig. 1B).
In human cells, baseline CBF was 4.6 ± 0.5 Hz in the control group (n = 12) and 4.2 ± 0.5 Hz in cells pre-incubated with PKI14–22(n = 6; P = 0.6). The initial CBF response to ATP was similar in both groups: CBF increased by 1.7 ± 0.2 Hz (or 37 ± 4 % above baseline) in controls vs. 1.2 ± 0.3 Hz (or 28 ± 6 %) in the PKI group (P = 0.1). The initial increase in the fura-2 ratio signal was also similar in both groups: 0.53 ± 0.08 in the controls vs. 0.58 ± 0.11 in the PKI14–22 group (P = 0.7). The Ca2+ signal returned to baseline within 205 ± 28 s in the controls and within 257 ± 37 s in the PKI14–22 group (P = 0.7). However, CBF remained elevated in the control group but not in the PKI group (Fig. 6B). Ten minutes after ATP stimulation, when the fura-2 ratio signal had already returned to baseline (see above), CBF remained elevated in controls by 0.95 ± 0.13 Hz (or 20 ± 3 %) above baseline whereas CBF had returned to baseline (within the measurement error) in the PKI group (elevation was 0.15 ± 0.04 Hz or 4 ± 1 % above baseline; P < 0.001).
Since these results were obtained in cells from submerged cultures, they were confirmed in human cells redifferentiated at the ALI (n = 4). Cells were exposed to 50 μm ATP from the apical side for 2 min as described in Methods. After a 60 min pre-incubation with PKI14–22, baseline CBF was 5.0 ± 1 Hz (P = 0.6 compared with controls). The initial CBF response to 50 μm ATP was 0.75 ± 0.2 Hz, but there was no delayed second peak as seen in controls and no prolonged CBF activation: 10 min after ATP stimulation, CBF was only 0.15 ± 0.3 Hz (3 ± 6 %) above baseline, i.e. within the measurement error (P < 0.05; Fig. 7).
Figure 7. CBF response to short-term ATP exposure after inhibition of PKA with PKI14–22 in human tracheal epithelial cells grown at the air-liquid interface.
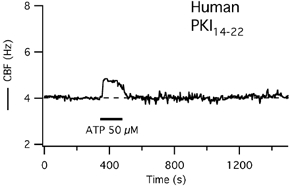
The CBF response to short-term ATP exposure is reduced to a transient increase in a representative human cell that has been pretreated with 10 μm of the PKA inhibitory peptide PKI14–22 as indicated in Methods.
Long-term incubation with PKI14–22 was not necessary: in three submerged cells tested, PKI14–22 addition as little as 5 min before exposure to 10 μm ATP was sufficient to inhibit the prolonged CBF increase. Thus, these findings suggest that PKA activation mediates the prolonged CBF rise seen after purinergic stimulation in human cells. These results were confirmed using a different PKA inhibitor, 1 μm KT-5720 (n = 4). In three of four cells pretreated with 1 μm KT-5720, the prolonged rise in CBF upon short-term ATP stimulation was blocked, similar to the results seen with PKI14–22: CBF returned to baseline with the calcium signal, i.e. within 197 ± 62 s (P > 0.05 for all measurements compared with cells pre-treated with PKI14–22 and P < 0.05 for CBF 10 min after ATP stimulation compared with control cells; Fig. 8A).
Figure 8. CBF and [Ca2+]i responses to short-term ATP exposure after inhibition of PKA with KT-5720, PKG with KT-5823, and adenylyl cyclase with SQ22536 in human tracheal epithelial cells.
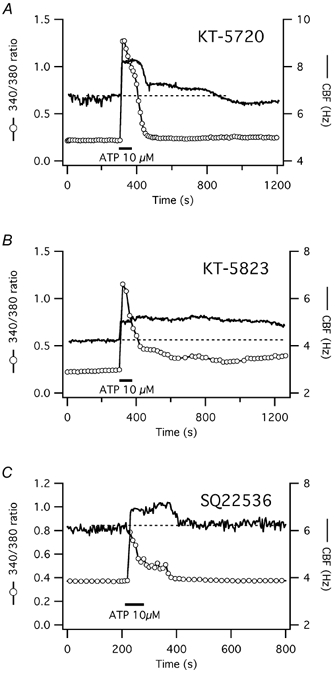
A, [Ca2+]i and CBF responses to short-term ATP exposure of a representative submerged human tracheal epithelial cell after pretreatment with 1 μm of the PKA inhibitor KT-5720. The CBF response to short-term ATP exposure is mainly calcium-coupled, similar to pretreatment with PKI14–22 (see Fig. 6B) and in contrast to control cells (see Fig. 1B). B, pretreatment with 1 μm of the PKG inhibitor KT-5823 has no effect on the prolonged CBF increase in response to short-term ATP exposure. Since both KT-5720 and KT-5823 are dissolved in DMSO, this solvent cannot explain the blocking ability of KT-5720. C, pretreatment with 1 mm of the adenylyl cyclase inhibitor SQ22536 inhibits the prolonged CBF increase upon short-tem exposure to ATP, similar to PKA inhibition.
Response to ATP after inhibition of PKG in human cells
cGMP-dependent protein kinase (PKG) has also been implicated in the regulation of CBF (Wyatt et al. 1998; Uzlaner & Priel, 1999). We therefore tested the effects of KT-5823, an inhibitor of PKG, on the CBF response to ATP of submerged human ciliated cells. Cells were pre-incubated for 60 min with 1 μm KT-5823 and then exposed to 10 μm ATP (n = 4). Baseline CBF was 4.8 ± 0.3 Hz and increased maximally by 2.0 ± 0.5 Hz (or 41 ± 9 % above baseline) in response to ATP (P > 0.05 for all measurements compared to control cells). Ten minutes after exposure to ATP, CBF was still elevated by 1.2 ± 0.3 Hz (or 26 ± 6 % above baseline), again not different from control cells. The fura-2 ratio values for these cells were also statistically indistinguishable from control cells at the peak responses; they reached baseline within 325 ± 29 s of ATP exposure (P = 0.1 compared with control). Thus, the prolonged CBF response of human ciliated cells to ATP was not blocked by PKG inhibition (Fig. 8B; compare with Fig. 1B).
Both KT-5720 and KT-5823 were dissolved in DMSO. Since KT-5823 did not change the [Ca2+]i or CBF signals compared with controls, DMSO itself cannot explain the blocking effect of KT-5720.
Response to ATP in human cells after inhibition of adenylyl cyclase with SQ22536
To see whether adenylyl cyclase activity was necessary for the prolonged CBF increase upon short-term ATP exposure, cells were pre-treated with 1 mm of the adenylyl cyclase inhibitor 9-(tetrahydro-2′-furyl)-adenine (SQ22536) (n = 6). Baseline CBF was 7.5 ± 0.6 Hz and increased maximally by 1.0 ± 0.1 Hz (or 14 ± 2 % above baseline) in response to ATP (increase was significantly lower compared with control cells). Ten minutes after exposure to ATP, CBF was back to baseline, again significantly different from control cells. The fura-2 ratio values for these cells, however, were statistically indistinguishable from control cells at the peak responses. Thus, the prolonged CBF response of human ciliated cells to ATP was blocked by adenylyl cyclase inhibition (Fig. 8C).
Response to ATP in human cells after stimulation of adenylyl cyclase with forskolin
Next, we stimulated PKA with 10 μm forskolin, an activator of adenylyl cyclase, previously known to stimulate CBF (Tamaoki et al. 1989a; Di Benedetto et al. 1991). Submerged human airway epithelial cells were exposed to 10 μm forskolin as described in Methods (n = 6). Forskolin increased CBF above the baseline of 5.2 ± 0.6 Hz by 1.3 ± 0.2 Hz (or 25 ± 4 %)(Fig. 9A). To show that forskolin also acts through PKA to stimulate CBF in human airway epithelial cells, PKA was inhibited with PKI14–22. Baseline CBF (6.1 ± 0.6 Hz) of cells pre-incubated with PKI14–22 was statistically indistinguishable from the forskolin only group (n = 6; P = 0.3). The CBF increase upon forskolin addition was blocked in the PKI14–22-treated group: ΔCBF was only 0.19 ± 0.2 Hz (3 ± 3 %, P < 0.01) or within the frequency resolution of the measurement (Fig. 9B). Thus, forskolin stimulated CBF in human airway epithelial cells through the activation of PKA.
Figure 9. CBF and [Ca2+]i responses to short-term ATP exposure in the presence of forskolin in human tracheal epithelial cells.
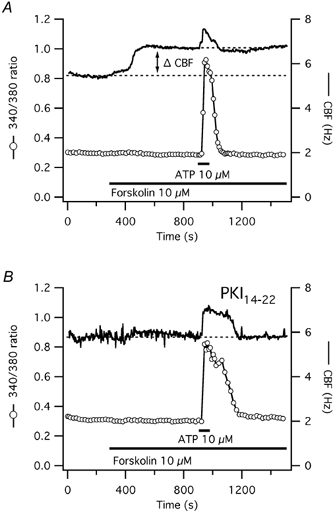
A, exposure of a representative human submerged tracheal epithelial cell to a direct activator of adenylyl cyclase, 10 μm forskolin, leads to a [Ca2+]i-independent rise of CBF to a stable plateau above the original baseline (ΔCBF). ATP stimulation in the presence of forskolin reveals only a short, calcium-coupled CBF increase. B, after PKA-inhibition with 10 μm PKI14–22, forskolin has no effect on CBF and ATP stimulation reveals again only a short, calcium-coupled rise in CBF.
We hypothesized that ATP would only stimulate a calcium-coupled CBF increase in cells pre-treated with forskolin since the cAMP-PKA pathway was already maximally stimulated. To test this hypothesis, controls and cells pre-incubated with PKI14–22 were stimulated with 10 μm ATP in the continued presence of forskolin. In the forskolin only group, 10 μm ATP caused a transient increase of the fura-2 ratio by 0.58 ± 0.13 and a CBF increase of 1.2 ± 0.65 Hz (or 19 ± 9 %) above the forskolin plateau of 6.5 ± 0.2 Hz. CBF and the fura-2 ratio returned to the pre-ATP plateau within 151 ± 15 s. In the forskolin plus PKI group, ATP caused a transient increase of the fura-2 ratio of 0.45 ± 0.13 (P = 0.5 compared with the forskolin group) and ΔCBF of 0.8 ± 0.2 Hz (or 14 ± 3 % above baseline; P = 0.3 compared to the forskolin group). CBF and the fura-2 ratio returned to the pre-ATP plateau within 198 ± 15 s (P = 0.1 compared with the forskolin group). Thus, in both groups, the fura-2 ratio and CBF rose and fell in parallel with each other (Fig. 9A, forskolin only and Fig. 9B, PKA inhibition). These results confirm our hypothesis that ATP leads to a strictly calcium-coupled transient CBF increase when PKA is inhibited or prestimulated and that ATP cannot increase CBF for an extended period of time beyond an already maximal PKA stimulation by forskolin.
Prolonged ATP-induced CBF increases require Ca2+ release from internal Ca2+ stores
Then we tested the hypothesis that the transient calcium increase stimulated the production of cAMP. We examined whether a prolonged CBF increase could be seen after short-term ATP exposure of human airway epithelial cells when the initial transient calcium increase was inhibited. Intracellular calcium stores were depleted by 1 μm thapsigargin (Salathe & Bookman, 1995), an inhibitor of the endoplasmic Ca2+-ATPase. In human ciliated cells (n = 3), the release of Ca2+ from intracellular stores was accompanied by a rise in CBF. Stimulation with 10 μm ATP after depletion of intracellular calcium stores lead to a short, barely detectable rise of [Ca2+]i, followed by a small drop of [Ca2+]i presumably due to uptake of calcium into mitochondria (Salathe et al. 2001). CBF also slightly decreased (Fig. 10A, arrow). Thus, the ATP-dependent activation of the PKA pathway to increase CBF requires a transient calcium increase.
Figure 10. Calcium requirements for prolonged CBF increases in response to short-term ATP exposure in human tracheal epithelial cells.
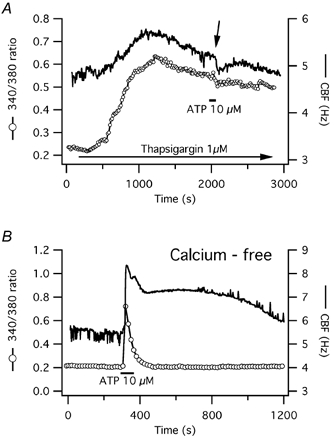
A, release of calcium from intracellular stores is required for prolonged CBF increases in human cells after short-term ATP exposure. CBF and [Ca2+]i responses to 1 μm thapsigargin, an inhibitor of the endoplasmic reticulum Ca2+-ATPase, which results in both a gradual calcium and CBF rise are shown. ATP exposure after calcium depletion elicits neither a calcium transient nor a CBF increase. In fact, both calcium and CBF decrease slightly (arrow). B, extracellular calcium is not required for extended CBF increases in response to short-term ATP exposure. The CBF and [Ca2+]i responses of a representative human tracheal cell exposed to ATP while bathed in calcium-free medium are shown.
Extracellular calcium was not required for the prolonged CBF stimulation, however. In the absence of calcium in the extracellular media (HBSS-Hepes without calcium plus 1 mm EGTA, restoring the Mg2+ concentration according to Fabiato & Fabiato, 1978), the [Ca2+]i and CBF responses to ATP were not different from the responses seen in the presence of extracellular Ca2+(n = 3; Fig. 10B). This finding rules out the possibility that that a local calcium influx could be responsible for the prolonged CBF rise.
Extended PKA activity is not solely responsible for prolonged ATP-induced CBF increases
To test whether prolonged PKA activity was responsible for the observed extended CBF increase in response to short-term ATP exposure in submerged human airway epithelial cells, we tested the ability of a PKA and a PKG inhibitor to hasten the decline of CBF back to baseline. Addition of 1 μm KT-5823 (PKG inhibitor) after initiation of the prolonged ATP-induced CBF increase did not reverse CBF back to baseline more rapidly compared with date and culture matched controls (n = 4; Fig. 11A and B). However, addition of 10 μm PKI14–22 (PKA inhibitor) decreased CBF from the elevated plateau statistically significantly compared with date- and culture-matched controls (n = 5; P = 0.01, Fig. 11C and D) but not back to baseline within 10 min of PKI14–22 addition (P = 0.01) in contrast to cells where PKA had been inhibited before ATP addition (each n = 5). These data suggest that continued PKA activity is not solely responsible for the prolonged CBF increase seen in human cells (Fig. 11).
DISCUSSION
Differences in signal transduction pathways to regulate ciliary beating have emerged between mammalian species over the last several years. As an example of such differences, we have explored the short-term purinergic stimulation of CBF in ovine and human airway epithelial cells. Purinergic stimulation of airway epithelial cells provides a strong signal to increase airway ciliary activity and since nucleotides can be locally produced and released into the airways (Watt et al. 1998; Dezaki et al. 2000; Homolya et al. 2000; Lazarowski et al. 2000), purinergic stimulation is likely one of the most important regulators of ciliary activity in vivo.
The P2Y2 receptor has been identified as the major purinoreceptor involved in ciliary regulation (Morse et al. 2001). In our experience and as shown here, short-term stimulation of ovine airway epithelial cells with ATP lead to a solely calcium-coupled, transient CBF increase similar to muscarinic stimulation (Salathe & Bookman, 1999). Transient calcium increases are typical after activation of G protein receptors that couple to phospholipase C as P2Y2 receptors do. To our surprise, however, short-term purinergic stimulation of human airway epithelial cells, in contrast to ovine cells, lead to a prolonged CBF elevation that outlasted the transient calcium increase for over 20 min.
There have been previous reports of prolonged ciliary stimulation with ATP in both rabbit airway (Korngreen & Priel, 1996; Korngreen et al. 1998; Uzlaner & Priel, 1999) and human nasal epithelial cells (Morse et al. 2001) using long-term exposures to ATP. In rabbit airway epithelial cells, prolonged ATP exposure has been shown to gate a P2X receptor, possibly leading to local calcium influx that is not detected by whole-cell calcium measurements (Korngreen et al. 1998). Such influx was considered to contribute to the extended CBF increase. In a follow-up paper, the same group showed that the ATP-induced transient calcium elevation (and possibly locally sustained calcium increase due to influx) stimulated NOS and subsequently PKG, which was responsible for prolonged CBF elevation (Uzlaner & Priel, 1999). Both of these mechanisms, however, did not seem to play a role in our experiments. First, ovine airway epithelial cells did not reveal any prolonged CBF activation after short-term ATP exposure. Second, human airway epithelial cells depicted a long-term CBF stimulation after short-term ATP exposure both at room temperature and at 35 °C, but neither inhibition of NOS, PKG or extracellular calcium influx was capable of blocking the prolonged CBF response in these cells. Therefore, species differences are likely to account for some of the differences seen between rabbit, human and ovine cells in addition to the different ATP exposure protocols.
Recently, another group showed that in human nasal epithelial cells, prolonged ATP exposure is associated with stimulation of an adenosine receptor due to ecto-hydrolysis of ATP to adenosine (Morse et al. 2001). Adenosine receptor activation is known to produce cAMP thereby stimulating CBF through a PKA-dependent pathway (Tamaoki et al. 1989b). In our experiments with human airway epithelial cells, both short- and long-term exposures to UTP (not expected to yield adenosine) and ATP were causing a prolonged, PKA-dependent CBF increase (see below). Together with the 8-SPT data (a broad adenosine receptor antagonist; Fig. 3), however, our results suggest that PKA activation was not due to adenosine receptor activation.
Our data provide clear evidence that the prolonged activation of CBF after short-term extracellular purinergic stimulation in human airway epithelial cells was due to activation of PKA. First, two inhibitors of PKA (PKI14–22 and KT-5720) blocked the prolonged CBF increase to ATP, an effect not seen with the PKG inhibitor KT-5823. Second, a direct activator of adenylyl cyclase, forskolin, increased CBF before ATP addition in a PKA-dependent manner. Subsequent ATP exposure produced only a short, strictly calcium-coupled rise in CBF but failed to produce a prolonged CBF increase above the forskolin/PKA-dependent plateau. In addition, PKA-inhibitors blocked both the forskolin effect on CBF as well as the prolonged CBF increase after short-term ATP exposure in both control cells and cells pre-exposed to forskolin. Finally, an inhibitor of adenylyl cyclase prevented a prolonged CBF increase after short-term ATP stimulation. Taken together, these results suggest that a signalling cascade likely involving adenylyl cyclase, cAMP and PKA is responsible for the prolonged ATP-dependent CBF elevation in human cells seen in our studies.
Several lines of experimentation suggest that the initial transient calcium increase was responsible for the PKA-dependent activation of CBF in human cells exposed short-term to ATP. After depletion of intracellular Ca2+ stores with thapsigargin, ATP stimulation led to a slight drop in [Ca2+]i (possibly due to calcium uptake from the cytoplasm into mitochondria as suggested in Salathe et al. 2001). The prolonged rise of CBF that would have been expected if PKA activation was calcium independent was not seen; on the contrary, CBF also decreased. In addition, prolonged calcium influx was unlikely to occur since calcium-free extracellular fluid had no influence on the prolonged CBF response. Thus, PKA activation required an initial transient calcium increase through release of calcium from intracellular stores upon ATP exposure.
These data are interesting given recent publications implicating that the calcium-sensitive adenylyl cyclases type I and VIII are mainly activated by capacitative calcium influx (Fagan et al. 1996). However, the same publication shows that adenylyl cyclase type VIII was stimulated by carbachol 70 % above baseline in the absence of extracellular calcium and that this effect was abolished by thapsigargin treatment. Although capacitative calcium influx increased cAMP production much more than intracellular calcium release, it is quite possible that the amount of cAMP produced after intracellular calcium release is sufficient to stimulate CBF maximally. Published data on adenylyl cyclase activation by calcium are also complicated by the fact that most experiments pre-stimulate adenylyl cyclase with forskolin (Fagan et al. 1996). However, forskolin pretreatment of our cells already stimulated CBF to an extent that could not be potentiated by calcium-induced cAMP production. Thus, our data are consistent with the published literature and it is possible that human airway epithelial cells express an adenylyl cyclase that can be activated by calcium, such as type VIII.
The prolonged CBF rise in our human cells after short-term purinergic stimulation did not rely entirely on sustained PKA activity. Thus, the activity of phosphatases in cilia may be critical in the determination of the prolonged frequency response if cells are only exposed to ATP for a brief period of time. On the other hand, a longer exposure to ATP, through ATP hydrolysis to adenosine, may stimulate additional cAMP production through adenosine receptors (Morse et al. 2001) and therefore counterbalance any increase in phosphatase activity in vivo.
Together, the results of this study show that the CBF response to short-term purinergic stimulation is distinctively different in ovine and human cells. PKA stimulation through calcium-induced activation of adenylyl cyclase and the subsequent production of cAMP is probably the mechanism involved in the prolonged CBF response to short-term ATP exposure seen in human cells. Other mechanisms to maintain increased CBF in human cells upon long-term ATP exposure likely involve adenosine receptors as reported by others (Morse et al. 2001). From a physiological point of view, prolonged CBF increases in the range 15–30 min could have a biological role, considering the fact that the transport of mucus from the carina to the larynx in humans requires about 10–15 min (Sackner et al. 1973; Yeates et al. 1975; Serafini et al. 1976). An increase in CBF will probably shorten this time and may assure that noxious substances that trigger the ciliary response are transported out of the airways.
Acknowledgments
The technical assistance of Sara Donoghue and Marie-Christine Nlend, the elegant programming of Nenad Amodaj, as well as the guidance of Gregory E. Conner and Adam Wanner are gratefully acknowledged. Supported in part by grants from NIH (HL-55341 to R.J.B.; HL-60644 & HL-67206 to M.S.).
REFERENCES
- Adler KB, Cheng PW, Kim KC. Characterization of guinea pig tracheal epithelial cells maintained in biphasic organotypic culture: cellular composition and biochemical analysis of released glycoconjugates. American Journal of Respiratory Cell and Molecular Biology. 1990;2:145–154. doi: 10.1165/ajrcmb/2.2.145. [DOI] [PubMed] [Google Scholar]
- Babbedge RC, Bland-Ward PA, Hart SL, Moore PK. Inhibition of rat cerebellar nitric oxide synthase by 7-nitro indazole and related substituted indazoles. British Journal of Pharmacology. 1993;110:225–228. doi: 10.1111/j.1476-5381.1993.tb13796.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernacki SH, Nelson AL, Abdullah L, Sheehan JK, Harris A, William Davis C, Randell SH. Mucin gene expression during differentiation of human airway epithelia in vitro. Muc4 and muc5b are strongly induced. American Journal of Respiratory Cell and Molecular Biology. 1999;20:595–604. doi: 10.1165/ajrcmb.20.4.3442. [DOI] [PubMed] [Google Scholar]
- Braiman A, Zagoory O, Priel Z. PKA induces Ca2+ release and enhances ciliary beat frequency in a Ca2+-dependent and -independent manner. American Journal of Physiology. 1998;275:C790–797. doi: 10.1152/ajpcell.1998.275.3.C790. [DOI] [PubMed] [Google Scholar]
- Cabell L, Audesirk G. Effects of selective inhibition of protein kinase C, cyclic AMP-dependent protein kinase, and Ca2+-calmodulin-dependent protein kinase on neurite development in cultured rat hippocampal neurons. International Journal Developmental Neuroscience. 1993;11:357–368. doi: 10.1016/0736-5748(93)90007-z. [DOI] [PubMed] [Google Scholar]
- Dezaki K, Tsumura T, Maeno E, Okada Y. Receptor-mediated facilitation of cell volume regulation by swelling-induced ATP release in human epithelial cells. Japanese Journal of Physiology. 2000;50:235–241. doi: 10.2170/jjphysiol.50.235. [DOI] [PubMed] [Google Scholar]
- Di Benedetto G, Manara-Shediac FS, Mehta A. Effect of cyclic AMP on ciliary activity of human respiratory epithelium. European Respiratory Journal. 1991;4:789–795. [PubMed] [Google Scholar]
- Evans JH, Sanderson MJ. Intracellular calcium oscillations regulate ciliary beat frequency of airway epithelial cells. Cell Calcium. 1999;26:103–110. doi: 10.1054/ceca.1999.0060. [DOI] [PubMed] [Google Scholar]
- Fabiato A, Fabiato F. Effects of pH on the myofilaments and the sarcoplasmic reticulum of skinned cells from cardiac and skeletal muscles. Journal of Physiology. 1978;276:233–255. doi: 10.1113/jphysiol.1978.sp012231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagan KA, Mahey R, Cooper DM. Functional co-localization of transfected Ca2+-stimulable adenylyl cyclases with capacitative Ca2+ entry sites. Journal of Biological Chemistry. 1996;271:12438–12444. doi: 10.1074/jbc.271.21.12438. [DOI] [PubMed] [Google Scholar]
- Fawcett DW, Porter KW. A study of the fine structure of ciliated epithelia. Journal of Morphology. 1954;94:221–281. [Google Scholar]
- Geary CA, Davis CW, Paradiso AM, Boucher RC. Role of CNP in human airways: cGMP-mediated stimulation of ciliary beat frequency. American Journal of Physiology. 1995;268:L1021–1028. doi: 10.1152/ajplung.1995.268.6.L1021. [DOI] [PubMed] [Google Scholar]
- Gheber L, Priel Z, Aflalo C, Shoshan-Barmatz V. Extracellular ATP binding proteins as potential receptors in mucociliary epithelium: characterization using [32P]3′-O-(4-benzoyl)benzoyl ATP, a photoaffinity label. Journal of Membrane Biology. 1995;147:83–93. doi: 10.1007/BF00235399. [DOI] [PubMed] [Google Scholar]
- Gibbons IR. The relationship between the fine structure and direction of beat in gill cilia of a lamellibranch mollusc. Journal of Biophysical and Biochemical Cytology. 1961;11:179–205. doi: 10.1083/jcb.11.1.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grider JR. Interplay of VIP and nitric oxide in regulation of the descending relaxation phase of peristalsis. American Journal of Physiology. 1993;264:G334–340. doi: 10.1152/ajpgi.1993.264.2.G334. [DOI] [PubMed] [Google Scholar]
- Homolya L, Steinberg TH, Boucher RC. Cell to cell communication in response to mechanical stress via bilateral release of ATP and UTP in polarized epithelia. Journal of Cell Biology. 2000;150:1349–1360. doi: 10.1083/jcb.150.6.1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain B, Rubinstein I, Robbins RA, Leise KL, Sisson JH. Modulation of airway epithelial cell ciliary beat frequency by nitric oxide. Biochemical and Biophysical Research Communication. 1993;191:83–88. doi: 10.1006/bbrc.1993.1187. [DOI] [PubMed] [Google Scholar]
- Kase H, Iwahashi K, Nakanishi S, Matsuda Y, Yamada K, Takahashi M, Murakata C, Sato A, Kaneko M. K-252 compounds, novel and potent inhibitors of protein kinase C and cyclic nucleotide-dependent protein kinases. Biochemical and Biophysical Research Communication. 1987;142:436–440. doi: 10.1016/0006-291x(87)90293-2. [DOI] [PubMed] [Google Scholar]
- Kelsen SG, Higgins NC, Zhou S, Mardini IA, Benovic JL. Expression and function of the beta-adrenergic receptor coupled-adenylyl cyclase system on human airway epithelial cells. American Journal of Respiratory and Critical Care Medicine. 1995;152:1774–1783. doi: 10.1164/ajrccm.152.6.8520736. [DOI] [PubMed] [Google Scholar]
- Korngreen A, Ma W, Priel Z, Silberberg SD. Extracellular ATP directly gates a cation-selective channel in rabbit airway ciliated epithelial cells. Journal of Physiology. 1998;508:703–720. doi: 10.1111/j.1469-7793.1998.703bp.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korngreen A, Priel Z. Simultaneous measurement of ciliary beating and intracellular calcium. Biophysical Journal. 1994;67:377–380. doi: 10.1016/S0006-3495(94)80492-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korngreen A, Priel Z. Purinergic stimulation of rabbit ciliated airway epithelia: control by multiple calcium sources. Journal of Physiology. 1996;497:53–66. doi: 10.1113/jphysiol.1996.sp021749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lansley AB, Sanderson MJ. Regulation of airway ciliary activity by Ca2+: simultaneous measurement of beat frequency and intracellular Ca2+ Biophysical Journal. 1999;77:629–638. doi: 10.1016/S0006-3495(99)76919-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lansley AB, Sanderson MJ, Dirksen ER. Control of the beat cycle of respiratory tract cilia by Ca2+ and cAMP. American Journal of Physiology. 1992;263:L232–242. doi: 10.1152/ajplung.1992.263.2.L232. [DOI] [PubMed] [Google Scholar]
- Lazarowski ER, Boucher RC, Harden TK. Constitutive release of ATP and evidence for major contribution of ecto-nucleotide pyrophosphatase and nucleoside diphosphokinase to extracellular nucleotide concentrations. Journal of Biological Chemistry. 2000;275:31061–31068. doi: 10.1074/jbc.M003255200. [DOI] [PubMed] [Google Scholar]
- Matsui H, Grubb BR, Tarran R, Randell SH, Gatzy JT, Davis CW, Boucher RC. Evidence for periciliary liquid layer depletion, not abnormal ion composition, in the pathogenesis of cystic fibrosis airways disease. Cell. 1998a;95:1005–1015. doi: 10.1016/s0092-8674(00)81724-9. [DOI] [PubMed] [Google Scholar]
- Matsui H, Randell SH, Peretti SW, Davis CW, Boucher RC. Coordinated clearance of periciliary liquid and mucus from airway surfaces. Journal of Clinical Investigation. 1998b;102:1125–1131. doi: 10.1172/JCI2687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morse DM, Smullen JL, Davis CW. Differential effects of UTP, ATP, and adenosine on ciliary activity of human nasal epithelial cells. American Journal of Physiology. 2001;280:C1485–1497. doi: 10.1152/ajpcell.2001.280.6.C1485. [DOI] [PubMed] [Google Scholar]
- Muniz M, Martin ME, Hidalgo J, Velasco A. Protein kinase A activity is required for the budding of constitutive transport vesicles from the trans-Golgi network. Proceedings of the National Academy of Sciences of the USA. 1997;94:14461–14466. doi: 10.1073/pnas.94.26.14461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodin JAG. Ultrastructure and function of the human tracheal mucosa. American Review of Respiratory Disease. 1966;93:1–15. doi: 10.1164/arrd.1966.93.3P2.1. [DOI] [PubMed] [Google Scholar]
- Rhodin JAG, Dalhamn T. Electron microscopy of the tracheal ciliated mucosa in rat. Zeitschrift fuer Zellforschung und Mikroskopische Anatomie. 1956;44:345–412. doi: 10.1007/BF00345847. [DOI] [PubMed] [Google Scholar]
- Rimon G, Rubin M. Regulation of a common, low-affinity binding site for primary prostanoids on bovine aortic endothelial cells. Biochimica et Biophysica Acta. 1998;1380:289–296. doi: 10.1016/s0304-4165(97)00153-0. [DOI] [PubMed] [Google Scholar]
- Sackner MA, Rosen MJ, Wanner A. Estimation of tracheal mucus velocity by bronchofiberoscopy. Journal of Applied Physiology. 1973;34:495–499. doi: 10.1152/jappl.1973.34.4.495. [DOI] [PubMed] [Google Scholar]
- Salathe M, Bookman RJ. Coupling of [Ca2+]i and ciliary beating in cultured tracheal epithelial cells. Journal of Cell Science. 1995;108:431–440. doi: 10.1242/jcs.108.2.431. [DOI] [PubMed] [Google Scholar]
- Salathe M, Bookman RJ. Mode of Ca2+ action on ciliary beat frequency in single ovine airway epithelial cells. Journal of Physiology. 1999;520:851–865. doi: 10.1111/j.1469-7793.1999.00851.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salathe M, Ivonnet PI, Lieb T, Bookman RJ. Agonist-stimulated calcium decreases in ovine ciliated airway epithelial cells: role of mitochondria. Journal of Physiology. 2001;531:13–26. doi: 10.1111/j.1469-7793.2001.0013j.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salathe M, Lieb T, Bookman RJ. Lack of nitric oxide involvement in cholinergic modulation of ovine ciliary beat frequency. Journal of Aerosol Medicine. 2000;13:219–229. doi: 10.1089/jam.2000.13.219. [DOI] [PubMed] [Google Scholar]
- Salathe M, Lipson E, Ivonnet PI, Bookman RJ. Muscarinic signal transduction in tracheal epithelial cells: effects of acetylcholine on intracellular Ca2+ and ciliary beating. American Journal of Physiology. 1997;272:L301–310. doi: 10.1152/ajplung.1997.272.2.L301. [DOI] [PubMed] [Google Scholar]
- Salathe M, Pratt MM, Wanner A. Cyclic AMP-dependent phosphorylation of a 26 kDa axonemal protein in ovine cilia isolated from small tissue pieces. American Journal of Respiratory Cell and Molecular Biology. 1993;9:306–314. doi: 10.1165/ajrcmb/9.3.306. [DOI] [PubMed] [Google Scholar]
- Sanderson MJ, Dirksen ER. Mechanosensitive and beta-adrenergic control of the ciliary beat frequency of mammalian respiratory tract cells in culture. American Review of Respiratory Disease. 1989;139:432–440. doi: 10.1164/ajrccm/139.2.432. [DOI] [PubMed] [Google Scholar]
- Serafini SM, Wanner A, Michaelson ED. Mucociliary transport in central and intermediate size airways: effect of aminophylline. Bulletin European Physiopathologie Respiratoire. 1976;12:415–422. [PubMed] [Google Scholar]
- Tamaoki J, Kobayashi K, Sakai N, Kanemura T, Horii S, Isono K, Takeuchi S, Chiyotani A, Yamawaki I, Takizawa T. Atrial natriuretic factor inhibits ciliary motility in cultured rabbit tracheal epithelium. American Journal of Physiology. 1991;260:C201–205. doi: 10.1152/ajpcell.1991.260.2.C201. [DOI] [PubMed] [Google Scholar]
- Tamaoki J, Kondo M, Takizawa T. Adenosine-mediated cyclic AMP-dependent inhibition of ciliary activity in rabbit tracheal epithelium. American Review of Respiratory Disease. 1989a;139:441–445. doi: 10.1164/ajrccm/139.2.441. [DOI] [PubMed] [Google Scholar]
- Tamaoki J, Kondo M, Takizawa T. Effect of cyclic AMP on ciliary function in rabbit tracheal epithelial cells. Journal of Applied Physiology. 1989b;66:1035–1039. doi: 10.1152/jappl.1989.66.3.1035. [DOI] [PubMed] [Google Scholar]
- Uzlaner N, Priel Z. Interplay between the NO pathway and elevated [Ca2+]i enhances ciliary activity in rabbit trachea. Journal of Physiology. 1999;516:179–190. doi: 10.1111/j.1469-7793.1999.179aa.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watt WC, Lazarowski ER, Boucher RC. Cystic fibrosis transmembrane regulator-independent release of ATP. Its implications for the regulation of P2Y2 receptors in airway epithelia. Journal of Biological Chemistry. 1998;273:14053–14058. doi: 10.1074/jbc.273.22.14053. [DOI] [PubMed] [Google Scholar]
- Wong LB, Yeates DB. Luminal purinergic regulatory mechanisms of tracheal ciliary beat frequency. American Journal of Respiratory Cell and Molecular Biology. 1992;7:447–454. doi: 10.1165/ajrcmb/7.4.447. [DOI] [PubMed] [Google Scholar]
- Wyatt TA, Spurzem JR, May K, Sisson JH. Regulation of ciliary beat frequency by both PKA and PKG in bovine airway epithelial cells. American Journal of Physiology. 1998;275:L827–835. doi: 10.1152/ajplung.1998.275.4.L827. [DOI] [PubMed] [Google Scholar]
- Yang B, Schlosser RJ, McCaffrey TV. Dual signal transduction mechanisms modulate ciliary beat frequency in upper airway epithelium. American Journal of Physiology. 1996;14:L745–751. doi: 10.1152/ajplung.1996.270.5.L745. [DOI] [PubMed] [Google Scholar]
- Yeates DB, Aspin N, Levison H, Jones MT, Bryan AC. Mucociliary tracheal transport rates in man. Journal of Applied Physiology. 1975;30:487–495. doi: 10.1152/jappl.1975.39.3.487. [DOI] [PubMed] [Google Scholar]