

Abstract
The Calu-3 human cell line exhibits features of submucosal gland serous cells and secretes HCO3−. The aim of this study was to identify the HCO3− transporters present in these cells by studying their role in the regulation of intracellular pH (pHi). Calu-3 cells were grown on coverslips, loaded with the pH-sensitive fluorescent dye BCECF, and their fluorescence intensity monitored as an indication of pHi. Cells were acidified with NH4Cl (25 mm, 1 min) and pHi recovery recorded. In the absence of HCO3−, initial recovery was 0.208 ± 0.016 pH units min−1 (n = 37). This was almost abolished by removal of extracellular Na+ and by amiloride (1 mm), consistent with the activity of a Na+-H+ exchanger (NHE). In the presence of HCO3− and CO2, recovery (0.156 ± 0.018 pH units min−1) was abolished (reduced by 91.8 ± 6.7 %, n = 7) by removal of Na+ but only attenuated (by 63.3 ± 5.8 %, n = 9) by amiloride. 4,4-Dinitrostilbene-2,2-disulfonic acid (DNDS) inhibited recovery by 45.8 ± 5.0 % (n = 7). The amiloride-insensitive recovery was insensitive to changes in membrane potential, as confirmed by direct microelectrode measurements, brought about by changing extracellular [K+] in the presence of either valinomycin or the K+ channel opener 1-EBIO. In addition, forskolin (10 μm), which activates the cystic fibrosis transmembrane conductance regulator Cl− conductance in these cells and depolarises the cell membrane, had no effect on recovery. Removal of extracellular Cl− trebled pHi recovery rates, suggesting that an electroneutral, DNDS-sensitive, Cl−-HCO3− exchanger together with a NHE may be involved in pHi regulation and HCO3− secretion in these cells. RT-PCR detected the expression of the electrogenic Na+-HCO3− cotransporter NBC1 and the Cl−-HCO3− exchanger (AE2) but not the electroneutral Na+-HCO3− cotransporter NBCn1.
HCO3− secretion is a feature of airway epithelial ion transport (Smith & Welsh, 1992; Van Scott et al. 1995; Inglis et al. 1996), and in particular of secretion by submucosal glands (Inglis et al. 1997, 1998; Ballard et al. 1999). The normal physiological role of this secretion is not well understood, but there is evidence that it is reduced in cystic fibrosis (CF) (Smith & Welsh, 1992) as a result of mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) anion channel. The mechanisms by which these mutations cause the lung disease that characterises CF are not yet fully understood, but it is possible that impaired HCO3− secretion may play a role by reducing gland liquid secretion (Inglis et al. 1997, 1998; Trout et al. 1998) and thus compromising the mucociliary clearance mechanisms (King, 1980; Clary-Meinesz et al. 1998).
The Calu-3 human cell line models a number of features of submucosal gland serous cells, expressing serous cell markers such as lysozyme and lactoferrin (Finkbeiner et al. 1993) and high levels of CFTR (Shen et al. 1994). When grown to confluence, these cells form resistive monolayers that secrete HCO3−, both under basal conditions (Lee et al. 1998) and after stimulation with forskolin (Devor et al. 1999). The mechanisms of this HCO3− secretion have been actively debated and a number of models proposed, all of which are based on the results of Ussing chamber studies (Singh et al. 1997; Lee et al. 1998; Devor et al. 1999). However, no clear consensus has emerged and so the aim of the present study was to take a different approach to the study of this problem. Since the movement of HCO3− and H+ across the cell membrane will affect intracellular pH (pHi), we have monitored the activity of the transporters involved in this transport by measuring changes in pHi in Calu-3 cells.
METHODS
Cell culture
Calu-3 cells were grown in 80 cm2 tissue culture flasks containing Dulbecco's modified Eagle's medium (DMEM)-NUT mix F-12 with fetal bovine serum (10 %), penicillin (10 000 U (500 ml)−1), streptomycin (10 mg (500 ml)−1) and amphotericin (25 μg (500 ml)−1). The cells were incubated in a humidified air containing 5 % CO2 at 37 °C. Confluent monolayers were subcultured by trypsinisation and, for experiments, cells were seeded onto coverslips and grown for 2–3 days.
Measurement of pHi
pHi recovery from acidification
Cells on coverslips were loaded with the pH-sensitive fluorescent dye 2′7′-bis(2-carboxyethyl)-5(6)-carboxyfluorescein (BCECF, 10 μm) in Hepes-buffered physiological salt solution (composition given below) for 10 min at room temperature. Coverslips were then mounted onto the heated stage of an inverted microscope (Nikon) and the cells initially perfused with Hepes-buffered saline (37 °C) for at least 10 min. During each experiment the cells were excited alternately at 490 and 440 nm and the BCECF fluorescence emitted from single cells was detected at 540 nm using a photomultiplier tube (Thorn, UK). The BCECF fluorescence ratio (440 nm/490 nm), which is an indicator of pHi, was continually displayed and recorded (1 Hz) to computer disk using a Cairn Research interface and associated software (Faversham, Kent, UK). To study the regulation of pHi, cells were exposed to pulses of NH4Cl (25 mm for 1 min). It is well documented that the application of NH4Cl causes a rapid intracellular alkalinisation, due to the diffusion of NH3 into the cell, and that, once NH4Cl is removed, pHi falls to a value more acidic than the initial resting pHi (Roos & Boron, 1981). The application of uniform pulses of NH4Cl thus allows a series of standard acid loads to be imposed upon the cell. By quantifying the rate at which pHi is restored to its resting level under different experimental conditions, it is possible to characterise the ion transport systems that allow acid equivalents to be extruded from the cytoplasm.
In situ calibration
At the end of each experiment, BCECF fluorescence ratios were calibrated in situ by permeabilising the cells with nigericin (20 μm) and perfusing with a high K+ solution of various pH values within the range pH 6–8.2 (KCl 130 mm, NaCl 10 mm, CaCl2 1.5 mm, MgCl2 1 mm, Hepes 10 mm; pH was adjusted with KOH at room temp). Under these conditions pHi is determined by extracellular pH and it is therefore possible to determine the relationship between BCECF fluorescence ratio and pHi, which was invariably linear over the tested pH range.
Intrinsic buffer capacity (βi)
In each experiment we measured the initial rate at which pHi recovered from the peak of the acid overshoot evoked by 1 min pulses of 25 mm NH4Cl. However, the rates of pHi recovery measured in such experiments will be determined not only by the rate at which acid equivalents are extruded from the cytoplasm (H+ extrusion rate), but also by the total H+-buffering capacity of the intracellular fluid (βt). The H+ ion extrusion rate cannot, therefore, be calculated unless βt is known. It is possible to resolve βt into two physiologically distinct components; βi, which is a measure of the cytoplasm's intrinsic buffering capacity, and βCO2, which is the buffering attributable to HCO3−-CO2. To determine βi, cells were initially superfused with a HCO3−, CO2 and Na+-free, Hepes-buffered solution that contained 1 mm BaCl2. Na+ was removed as preliminary experiments had shown that all H+-extruding systems present in these cells were dependent upon external Na+, whilst Ba2+ was included to prevent the movement of NH4+ through K+ channels. We therefore assumed that all pHi-regulating mechanisms were blocked under these conditions. The cells were first exposed to 10 mm extracellular NH4Cl, which, as anticipated (Roos & Boron, 1981), evoked a sustained alkalisation (Fig. 1A). The decreases in pHi evoked by successively lowering the external NH4Cl concentration ([NH4Cl]o) to 5, 2.5, 1, 0.5 and 0 mm were then measured (Fig. 1A). Each time [NH4Cl]o is lowered, there is a corresponding fall in the external concentration of NH3 which establishes a driving force for the efflux of this highly mobile gas molecule. Although NH4+ cannot leave the cell directly under these conditions, it will rapidly dissociate into NH3, which can leave the cell by diffusion. This will continue until a new equilibrium is reached. The released H+, however, remains trapped inside the cytoplasm as acid extrusion is essentially impossible under Na+-free conditions. Under these conditions each reduction in [NH4Cl]o deposits a defined quantity of H+ into the cytoplasm and it is therefore possible to calculate βi from the expression βi = Δ[NH4+]i/ΔpHi. Moreover, if the pK for NH4+ (8.9 at 37 °C) is assumed to be the same in intracellular and extracellular compartments, then [NH4+]i can be determined from the expression [NH4+]i = [NH4+]o × 10(pHo - pHi). It is therefore possible to construct a plot relating βi to pHi (Fig. 1B). Under HCO3−- and CO2-free conditions, βt was assumed to be equal to βi, whilst in cells exposed to HCO3−-CO2-buffered solutions it was assumed to be determined by the expression βi + βCO2. Under such conditions βCO2 will be equal to 2.303[HCO3−]i, where [HCO3−]i is the intracellular HCO3− concentration, calculated from the Henderson-Hasselbalch equation assuming that the cell behaves as a system open to CO2 (Roos & Boron, 1981).
Figure 1. Measurement of buffering capacity.
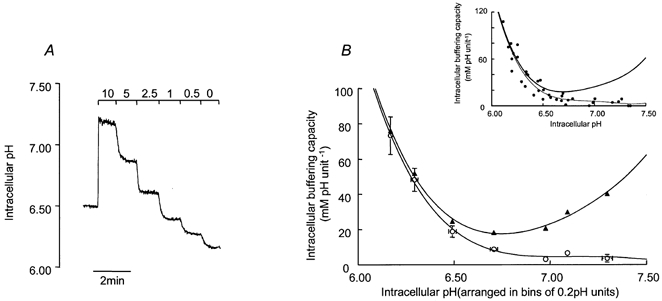
A, cells were acidified with solutions containing various concentrations of NH4Cl (10, 5, 2.5, 1, 0.5 and 0 mm). Solutions were Na+ free and contained BaCl2 (1 mm) to prevent entry of NH4+ into the cells through K+ channels. The stepwise falls in pHi were used to calculate intrinsic buffering capacity (see Methods). B, open circles represent intrinsic buffering capacity (βi). Values were arranged into bins of 0.2 pH units beginning 6.0–6.2, and the means ± s.e.m. plotted against the mid-point of each step fall in pHi. Beginning with the bin representing pHi 6.0–6.2, 6.2–6.4 and so on, n = 5, 8, 8, 7, 3, 2 and 4. The filled triangles represent estimates for the total H+-buffering capacity (βt), calculated as described in Methods. Both βi and βt were fitted with a fourth-order polynomial. The inset graph shows all the data used to produce the graph in B. These data are fitted with a fourth-order polynomial. The thick line indicates βt.
Data analysis
Fluorescence intensities resulting from excitation at 440 and 490 nm were saved to spreadsheet software (Microsoft Excel) and the ratio 490 nm/440 nm calculated. Initial rates of recovery from NH4-induced acidification were determined by fitting a line to the data collected during the first minute of the recovery period using least squares regression (Slidewrite Plus, Advanced Graphics Software, Encinitas, CA, USA).
Control experiments showed that the rates of recovery from acidification after consecutive NH4 pulses were not significantly different (initial rates of pHi recovery from an acid load were: first NH4+ pulse, 0.307 ± 0.034 pH units min−1; second NH4+ pulse, 0.256 ± 0.037 pH units min−1). Experiments thus followed a strictly paired regime, with recoveries under ‘test’ conditions compared directly with recoveries under control conditions measured in the same cells. Maximal rates of H+ extrusion were calculated by multiplying initial rates of pHi recovery by βt (calculated at the initial pHi of the pH recovery period).
Measurement of cell membrane potential
Cells grown on coverslips were placed in a chamber on the stage of an inverted microscope (Nikon) and perfused with HCO3−-buffered Ringer solution (37 °C, 1 ml min−1). Techniques for measuring cell membrane potential were similar to those described previously (Acevedo et al. 1990). Microelectrodes were pulled from 1.5 mm (o.d.) borosilicate glass capillaries with filaments (Clark Electromedical, Reading, UK) and filled with 500 mm KCl so that they had resistances of 60–180 MΩ when placed in control Ringer solution. They were connected through a Ag-AgCl wire to a high impedance electrometer (Model S-7100A, World Precision Instruments, New Haven, CT, USA). The reference Ag-AgCl electrode was connected to the tissue chamber with an agar-150 mm KCl bridge. Output from the electrometer was filtered at 5 Hz (low pass filter model 900, Frequency Devices, Inc., Haverhill, MA, USA).
RNA extraction and reverse transcriptase-polymerase chain reaction (RT-PCR)
Total RNA was extracted from cells using Trizol reagent (Gibco BRL, Paisley, UK) according to the manufacturer's instructions. mRNA was reverse transcribed into cDNA using a specific anti-sense primer and M-MLV reverse transcriptase (Promega, Southampton, UK). The resultant cDNA was then amplified, using specific primers, by PCR.
The primers used were designed to amplify cDNA sequences specific to electrogenic pNBC1 (Gross et al. 2001), electroneutral NBCn1(Choi et al. 2000), Na+-dependent Cl−-HCO3− exchanger (NDAE1) (Romero et al. 2000) and Cl−-HCO3− exchanger (AE2) (Loffing et al. 2000; Table 1).
Table 1.
Oligonucleotide sequences of RT-PCR primers
| Name | Sequence (5′→ 3′) | Position (base pairs) | Size (base pairs) | Accession number |
|---|---|---|---|---|
| pNBC1 sense | ATGTGTGTGATGAAGAAGAAGTAGAAG | 47–73 | 621 | AF107265 |
| pNBC1 antisense | GACCGAAGGTTGGATTTCTTG | 648–668 | ||
| AE2 sense | AAGATTCAGGAAGTCAAGGAGC | 3418–3439 | 482 | U62531 |
| AE2 antisense | CTACACAGGCATGGGCATCTC | 3870–3900 | ||
| NBCn sense | AGTTCCTCGGAATTCGTGA | 3171–3189 | 500 | AF069511 |
| NBCn antisense | TTCGAATGGGAGGTGCACA | 3653–3671 | ||
| NDAE1 sense | TCAATGGCAGCGTGATGC | 415–432 | 564 | AF047468 |
| NDAE1 antisense | AAGTTGCTCTGACTGCCAGG | 960–979 |
The reverse transcription step was carried out at 48 °C for 45 min followed by a 2 min denaturation step at 94 °C. The cDNA was then cycled 30 times as follows: 94 °C for 30 s (denaturation); 52 °C for 1 min (annealing); 72 °C for 2 min (elongation) followed by a 7 min extension at 72 °C. To confirm that the products generated originated from RNA, and not contaminating DNA, control reactions where performed where the reverse transcription step was omitted. Negative controls were also undertaken by running RT-PCR reactions in which neither RNA nor cDNA had been added to the reaction tubes in order to verify that there had been no contamination of the reagents used. The reaction products were separated on 1 % (w/v) agarose-TAE-ethidium bromide gels and visualised under ultraviolet light. The sizes of the amplified fragments were determined by comparing their migration distance through the gel with that of DNA standards. The RT-PCR products were extracted from the gels (QIAquick Gel Extraction Kit, Qiagen, Crawley, West Sussex, UK), ligated into the cloning vector pGEM-T (Promega) and transformed into competent JM109 cells (Promega). Plasmids were extracted from cultures of transformed JM109 cells and the inserts were sequenced to confirm their origin (ABI Prism automated DNA sequencer).
Cells and solutions
Calu-3 cells were a generous gift from Dr J. A. Plumb (University of Glasgow, Glasgow, Scotland, UK). Hepes-buffered Ringer solution contained (mm): NaCl, 112; KCl, 4.7; CaCl2, 2.5; MgSO4, 2.4; KH2PO4, 1.2; Hepes, 25; and d-glucose, 11.6; pH adjusted to 7.4 with HCl). In HCO3−-buffered solution, Hepes was replaced with equimolar NaHCO3− and the solution was continuously bubbled with 5 % CO2 in O2 to maintain pH 7.4. Na+ was isosmotically replaced with N-methyl-d-glucamonium (NMDG+) in all Na+-free solutions. In Na+-free solutions containing HCO3−, NaCl was replaced with equimolar NMDG+ and pH was titrated to 7.0 with HCl. A further 25 mm NMDG+ was then added to replace NaHCO3− and the solution was equilibrated with 5 % CO2 in O2 to lower the pH to the value (7.4) predicted by the Henderson-Hasselbalch equation. In solutions containing NH4Cl, 25 mm NaCl was replaced with equimolar NH4Cl. Low and high K+ solutions were similar to HCO3−-buffered solution (above) but contained 60 mm NaCl and either 1.8 mm KCl and 60 mm NMDGCl (low K+) or 62 mm KCl (high K+). 4,4-Dinitrostilbene-2,2-disulfonic acid (DNDS) was obtained from Pfaltz and Bauer (CT, USA), 1-ethyl-2-benzimidazolinone (1-EBIO) from Tocris Cookson (Bristol, UK) and BCECF from Molecular Probes (OR, USA). All other chemicals were obtained from Sigma-Aldrich (Dorset, UK).
RESULTS
Steady-state pHi and measurement of βi in Hepes-buffered solutions
The mean ± s.e.m., steady-state pHi of Calu-3 cells bathed in Hepes-buffered solution was 7.14 ± 0.04 (n = 88). Figure 1B shows that βi was very low at this average resting pHi and increased steeply at acidic pH. βt, calculated as described in Methods, is depicted by the filled triangles in Fig. 1B. Removal of extracellular Na+ significantly acidified the cells by 0.62 ± 0.09 pH units in 33 ± 4 min (Student's paired t test, P < 0.05, n = 10), suggesting that a Na+-dependent process was maintaining steady-state levels of pHi.
Recovery from an acid load in Hepes-buffered solutions
To study the acid-extruding systems active in Calu-3 cells, intracellular acidification was achieved by brief exposure to NH4+ (25 mm, 1 min) (see Roos & Boron, 1981). Figure 2 shows that extracellular NH4+ caused pHi to rise to a value of 8.22 ± 0.04 (n = 37). This alkalinisation is thought to result from the rapid influx of NH3 and the subsequent combination of these molecules with intracellular H+ (Roos & Boron, 1981). This was followed by a slow fall in pHi (to pHi = 8.00 ± 0.04 after 1 min), caused by the entry of NH4+ and its dissociation into NH3 and H+ (Roos & Boron, 1981). Subsequent removal of extracellular NH4+ caused a rapid fall in pHi due to dissociation of NH4+ into H+, which remains in the cells, and NH3, which can rapidly leave the cytoplasm (Roos & Boron, 1981). This caused a large undershoot of the pHi to 6.78 ± 0.04, significantly below the starting value (P < 0.05). pHi subsequently recovered (average rate, 0.208 ± 0.016 pH units min−1), as a result of the activity of acid-extruding systems present in the cells. The rest of this study was designed to characterise these systems.
Figure 2. Recovery from NH4Cl-induced intracellular acidification in Hepes-buffered solution.
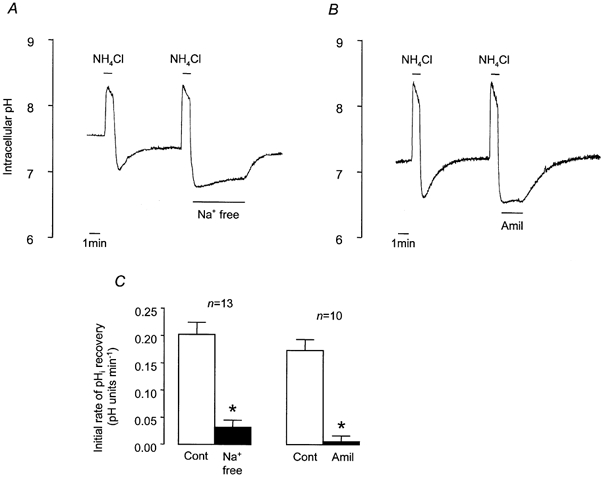
A and B, representative experiments showing changes in pHi in response to 1 min pulses of NH4Cl (25 mm) and the effects of Na+-free solution (A) and 1 mm amiloride (Amil; B) on pHi recovery. C, effect of Na+-free solution and amiloride on mean initial rates of pHi recovery. * Significantly different from paired control (Cont) recovery rate (Student's paired t test, P < 0.05). Error bars depict s.e.m.
Na+ dependence of pHi recovery and sensitivity to amiloride in Hepes-buffered solution
To test for the involvement of NHE activity in recovery from acidosis, we explored the effect of removing extracellular Na+. Figure 2A and C shows that removal of extracellular Na+ almost abolished recovery from an acid load and that recovery was restored when extracellular Na+ was reintroduced. Recovery was also extremely sensitive to amiloride (1 mm), an inhibitor of NHE (Fig. 2B and C). The recovery rate in the absence of extracellular Na+ (0.032 ± 0.013 pH units min−1, n = 13) was not significantly different from the rate in the presence of amiloride (0.005 ± 0.011 pH units min−1, n = 10). This inhibitory effect of amiloride was reversible and mimicked by 5-(N-ethyl-N-isopropyl)-amiloride (EIPA; 100 μm), an amiloride analogue that inhibits NHE, whilst having a minimal effect upon Na+ channels (control recovery rate, 0.162 ± 0.067 pH units min−1; EIPA, 0.009 ± 0.010 pH units min−1, n = 4). These results suggest that an amiloride-sensitive, Na+-dependent acid extruder, NHE, is almost entirely responsible for recovery from an induced acid load under nominally HCO3−- and CO2-free conditions.
Recovery in HCO3−-CO2-buffered solution
Replacing the Hepes-buffered solution with HCO3−-CO2-buffered solution induced an acidification of pHi, presumably resulting from the rapid entry of CO2 into the cells, the formation of H2CO3 and its subsequent dissociation into H+ and HCO3−. This acidification was followed by rapid recovery (Fig. 3A). Subsequent exposure to NH4Cl (25 mm, 1 min) evoked a pattern of pHi changes similar to those seen during superfusion with Hepes-buffered solutions. The rate of recovery (0.181 ± 0.013 pH units min−1) was not significantly different from the recovery rate in Hepes-buffered solution (0.190 ± 0.015 pH units min−1, n = 23; Fig. 3A and D). However, the H+ extrusion rate required to achieve this rate of pHi recovery was significantly higher in the presence of HCO3− (3.57 ± 0.45 versus 1.53 ± 0.27 mm min−1 in the absence of HCO3−, P < 0.05; Fig. 4), because βt is greater during exposure to HCO3−-CO2. Removal of extracellular Na+ almost abolished recovery from NH4-induced intracellular acidification (91.8 ± 6.7 % inhibition, n = 7; Fig. 3B and D), indicating that recovery was dependent on extracellular Na+. The increased H+ extrusion rate seen in the presence of HCO3−-CO2 may therefore result from either an increase in the activity of the NHE or from the activity of an additional, HCO3−-dependent acid extrusion mechanism.
Figure 3. Recovery from NH4Cl-induced intracellular acidification in HCO3−-CO2-buffered solution.
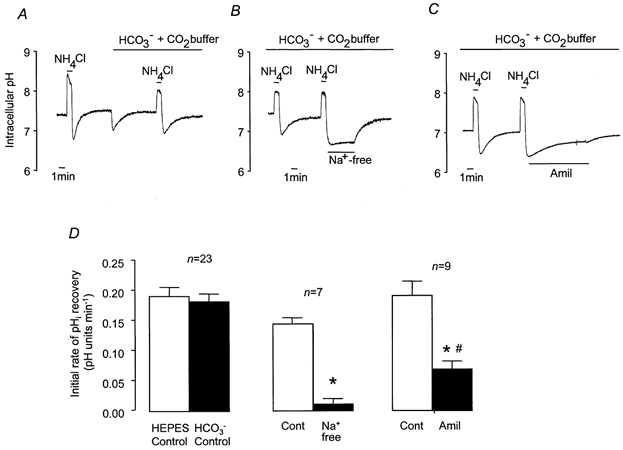
A-C, representative experiments showing changes in pHi in response to 1 min pulses of NH4Cl (25 mm). A, control NH4Cl pulse in Hepes-buffered solution, switch to HCO3−-CO2-buffered solution, and control NH4Cl pulse in HCO3−-CO2-buffered solution. B, effect of Na+-free solution on pHi recovery in HCO3−-CO2-buffered solution. C, effect of amiloride (1 mm) on pHi recovery in HCO3−-CO2-buffered solution. D, effect of HCO3−-CO2-buffered solution, Na+-free solution and amiloride on mean initial rates of pHi recovery. * Significantly different from paired control recovery rate (Student's paired t test, P < 0.05). # Significantly different from initial rate of recovery in Na+-free solution (Student's unpaired t test, P < 0.05). Error bars depict s.e.m.
Figure 4. Calculated maximum rates of H+ extrusion in Hepes- and HCO3−-CO2- buffered solutions.
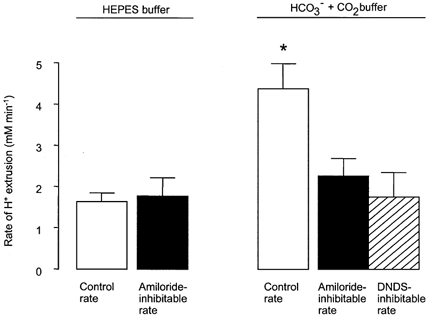
H+ extrusion rate = maximal (i.e. initial) rate of pHi recovery × βt (see Methods). □, mean control rate; ▪, rate inhibited by amiloride (1 mm); and  , rate inhibited by DNDS (3 mm). * Significant difference from control extrusion rate in Hepes buffer (Student's t test, P < 0.05). Error bars depict s.e.m.
, rate inhibited by DNDS (3 mm). * Significant difference from control extrusion rate in Hepes buffer (Student's t test, P < 0.05). Error bars depict s.e.m.
Amiloride (1 mm) significantly slowed the recovery from acidification (Fig. 3C and D) but, in contrast to the complete abolition of recovery seen in Hepes-buffered solutions, in the presence of HCO3− amiloride inhibited recovery by only 63.3 ± 5.8 % (n = 9). It is unlikely, however, that this reduced effect on pHi recovery is due to a reduced activity of the NHE, because the amiloride-sensitive component of H+ extrusion was similar in the presence and absence of HCO3− (Fig. 4). It thus seemed likely that the increased control rate of H+ extrusion from cells containing HCO3− resulted from the activity of an additional, HCO3−-dependent process.
Sensitivity of pHi recovery to DNDS in HCO3−-CO2-buffered solution
Since it has been proposed that a Na+-HCO3− cotransporter (NBC) is involved in Calu-3 cell HCO3− secretion (Lee et al. 1998; Devor et al. 1999), we tested the possibility that this transporter was involved in pHi recovery by investigating the effect of DNDS, an inhibitor of NBC. Figure 5 shows that pHi recovery from an acid load was significantly reduced (by 45.8 ± 5.0 %, n = 7, P < 0.05) in the presence of 3 mm DNDS, and DNDS inhibited the H+ extrusion rate by 1.75 ± 0.59 mm min−1 (Fig. 4), consistent with the notion that pHi recovery in the presence of HCO3− results from the activity of both NHE and NBC.
Figure 5. Effect of DNDS on recovery from NH4Cl-induced intracellular acidification in HCO3−-CO2-buffered solution.
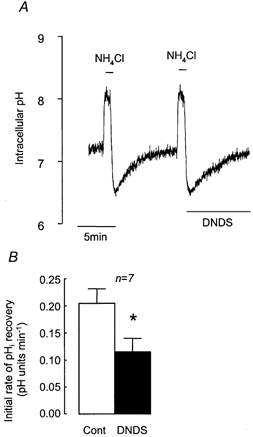
A, representative experiment showing the effect of 3 mm DNDS on recovery from acidification induced by 1 min pulses of NH4Cl (25 mm). B, effect of DNDS on mean initial rates of pHi recovery. * Significant difference from paired control recovery rate (Student's paired t test, P < 0.05). Error bars depict s.e.m.
Effect of changing cell membrane potential in HCO3−-CO2-buffered solution
Since the NBC transporter is reported to be electrogenic in Calu-3 cells (Devor et al. 1999) with a probable stoichiometry for Na+:HCO3− of 1:2, we would expect any pHi recovery mediated by NBC to be dependent on the cell membrane potential (Vm). We therefore used a number of methods to alter Vm and investigated their effects on amiloride-insensitive pHi recovery. Devor et al. (1999) showed that 1-EBIO opened Ca2+-activated K+ channels in these cells and proposed that this hyperpolarised Vm, thus inhibiting the entry of HCO3− via the NBC. Figure 6 shows that 1-EBIO (1 mm) significantly slowed the rate of amiloride-insensitive recovery from acidosis in a reversible manner. This effect of 1-EBIO is consistent with the involvement of an electrogenic NBC transporter in the recovery from acidosis. To check that the effect of 1-EBIO was indeed brought about by the activation of K+ channels and consequent changes in Vm, we investigated the effects of 1-EBIO in cells exposed to different extracellular K+ concentrations ([K+]o), which would be expected to change Vm. pHi recovery of cells exposed to 1-EBIO in solutions containing low [K+]o (1.8 mm; 0.085 ± 0.016 pH units min−1) was not significantly different from that in solutions containing high [K+]o (62 mm; 0.088 ± 0.023 pH units min−1, n = 9, P > 0.05). The order of addition of low or high [K+]o solutions did not affect recovery rates.
Figure 6. Effect of 1-EBIO on recovery from NH4Cl-induced intracellular acidification in HCO3−-CO2-buffered solution containing amiloride (1 mm).
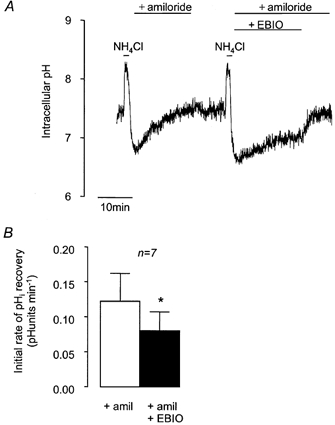
A, representative experiment showing the effect of 1 mm 1-EBIO on the recovery from acidification induced by 1 min pulses of NH4Cl (25 mm). B, effect of 1-EBIO on mean initial rates of pHi recovery. * Significant difference from paired control recovery rate (Student's paired t test, P < 0.05). Error bars depict s.e.m.
To confirm that exposure to solutions containing different [K+]o did alter Vm, we used microelectrodes to measure Vm at different [K+]o (Fig. 7). The Vm of cells in control HCO3−-buffered solution was −61.1 ± 2.1 mV (n = 15). Exposure to high [K+]o solution significantly depolarised Vm, whilst low [K+]o hyperpolarised Vm (Fig. 7A). Addition of 1-EBIO had no significant effect on membrane potential (−57.3 ± 1.7 mV, n = 11), and exposing 1-EBIO-treated cells to high and low [K+]o had effects on Vm that could not be distinguished from control (Fig. 7B and C). It is thus clear that 1-EBIO has no effect on Vm. When taken together with the measurements of pHi, these results show that pHi recovery does not depend on membrane potential and is therefore unlikely to be electrogenic. To further explore the relationship between Vm and the rate of pHi recovery, we investigated the effect of valinomycin, a K+ ionophore. After exposure to NH4Cl (25 mm, 1 min), cells were superfused with Na+-free buffer containing valinomycin (5 μm, 3 min), and then either high [K+]o or low [K+]o buffer containing valinomycin (5 μm) and amiloride (1 mm). Figure 8 shows that there was no difference in the recovery rates in the two different buffers. Microelectrode studies, however, showed that Vm was significantly hyperpolarised in the presence of valinomycin (control, −61.1 ± 2.1 mV; valinomycin, −69.2 ± 2.6 mV, n = 11), as would be expected following an increase in the K+ permeability of the cell membrane, and established that this parameter depended upon [K+]o (Fig. 7B and C).
Figure 7. Effect of valinomycin, 1-EBIO and changes in [K+]o on membrane potential (Vm).
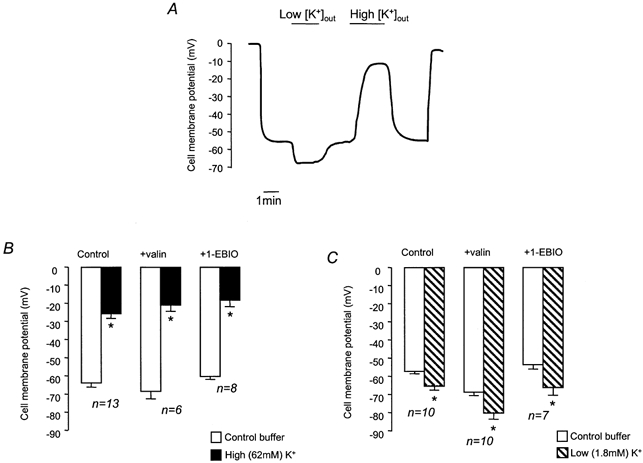
A, representative trace showing the effect of high and low [K+]o on Vm. B, effect of high [K+]o on mean Vm (control), in the presence of valinomycin (+valin, 5 μm), and in the presence of 1-EBIO (+1-EBIO, 1 mm). C, effect of low [K+]o on mean Vm (Control), in the presence of valinomycin (+valin), and in the presence of 1-EBIO (+1-EBIO). * Significant difference from Vm in control buffer (Student's paired t test, P < 0.05).
Figure 8. Effect of high and low [K+]o on recovery from NH4Cl-induced intracellular acidification in HCO3−-CO2-buffered solution containing valinomycin and amiloride.
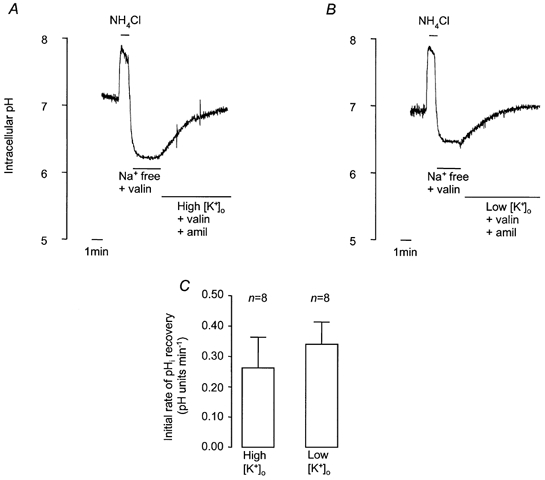
A and B, representative experiments showing effect of high [K+]o (62 mm; A) and low [K+]o (1.8 mm; B) on recovery. After NH4Cl treatment, cells were exposed to Na+-free solution containing 5 μm valinomycin (3 min) before solutions of either high or low [K+] containing valinomycin and 1 mm amiloride. C, effect of high and low [K+]o containing valinomycin and amiloride on mean initial rates of pHi recovery. Error bars depict s.e.m.
Effect of forskolin on pHi recovery and Vm in HCO3−-CO2-buffered solution
Forskolin stimulates ion transport in Calu-3 cells (Shen et al. 1994; Devor et al. 1999), activating apical CFTR channels (Haws et al. 1994). Microelectrode studies demonstrated that forskolin (10 μm) significantly depolarised Vm from −55.8 ± 3.3 mV to −41.2 ± 2.8 mV (n = 5, P < 0.05). However, forskolin (10 μm) had no significant effect on pHi recovery rate (control recovery rate, 0.135 ± 0.015 pH units min−1; rate in the presence of forskolin, 0.165 ± 0.038 pH units min−1, n = 9, P > 0.05).
Involvement of Cl−-HCO3− exchange in pHi recovery in HCO3−-CO2-buffered solution
To investigate the possible role of Cl−-HCO3− exchange in pHi recovery from acidosis, the effect of extracellular Cl− removal was investigated. Cells were treated with valinomycin to permeabilise the cell membranes to K+ and hence prevent changes in membrane potential resulting from Cl− efflux through anion channels. Figure 9 shows that removal of extracellular Cl− from cells pretreated with valinomycin significantly increased the rate of pHi recovery. These results are consistent with the activity of a Cl−-HCO3− exchanger (AE).
Figure 9. Effect of extracellular Cl− removal on recovery from NH4Cl-induced intracellular acidification in HCO3−-CO2-buffered solution containing valinomycin and amiloride.
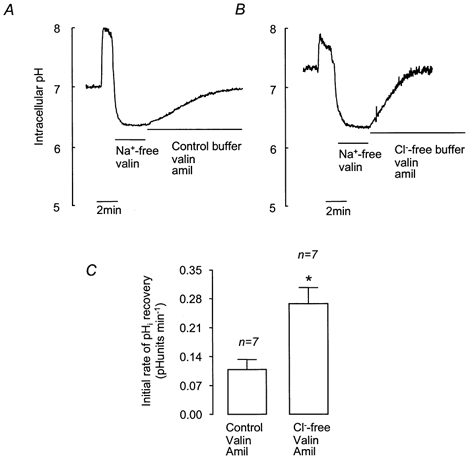
A and B, representative experiments showing the effect of Cl− -free solution on recovery. After NH4Cl treatment, cells were exposed to Na+-free solution containing 5 μm valinomycin (3 min) before either control buffer containing amiloride (1 mm) and valinomycin (5 μm; A) or Cl−-free buffer with valinomycin and amiloride (B). C, effect of Cl−-free solution containing valinomycin and amiloride on mean initial rates of pHi recovery. Error bars depict s.e.m. * Significant difference from recovery in control buffer (Student's unpaired t test, P < 0.05).
RT-PCR
RT-PCR detected the presence of mRNA for both pNBC1 and AE2, a Cl−-HCO3− exchanger, in Calu-3 cells (Fig. 10A and B). mRNA for the electroneutral NBC (NBCn) was not detected in Calu-3 cells, but was found in mRNA isolated from rat kidney (Fig. 10C). The Na+-dependent Cl−-HCO3− exchanger (NDAE) was not detected in Calu-3 cells, rat heart or rat kidney (data not shown).
Figure 10. Ethidium bromide-stained agarose gels of PCR products.
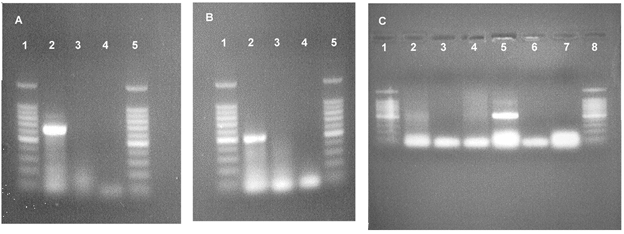
A, electrogenic NBC (pNBC1), 621 bp; B, Cl−-HCO3− exchanger (AE2), 482 bp; C, electroneutral NBC (NBCn1), 500 bp. Lanes: 1 and 5 (A and B), 1 and 8 (C), 100 bp DNA ladder; 2, Calu-3 cells; 3 (and 6, C), RNA template omitted; 4 (and 7, C), reverse transcriptase omitted; 5 (C), rat kidney.
DISCUSSION
The data in the present study show that Calu-3 cells have at least two major means of recovering from acidification, both of which require the presence of extracellular Na+. At present, the nature of the slow pHi recovery seen in the absence of extracellular Na+ is unknown. In preliminary experiments, bafilomycin (0.1 μm), an inhibitor of H+-ATPase, had no effect on the rate of pHi recovery in the absence of extracellular Na+ (S. K. Inglis, unpublished data), suggesting that this transporter is not involved in pHi regulation in these cells. It is possible that the K+-H+-ATPase, reported to be expressed in human bronchial epithelium, may be involved (Coakley et al. 2000). However, the majority of pHi recovery (over 90 %) in these cells is dependent on the presence of extracellular Na+.
The first major means of pHi recovery is Na+ dependent, sensitive to amiloride and EIPA, and likely to reflect the activity of a NHE. NHE activity has also been described in nasal and tracheal surface epithelial cells (Acevedo & Steele, 1993; Paradiso, 1997) and mRNA encoding NHE has been isolated from human bronchi (Dudeja et al. 1999). Lee et al. (1998) proposed that NHE is involved in HCO3− secretion by Calu-3 cells, since a component of HCO3− secretion is sensitive to EIPA. It is also sensitive to acetazolamide, an inhibitor of carbonic anhydrase, which catalyses the conversion of CO2 and H2O into carbonic acid. Lee et al. (1998) proposed that the H+ subsequently formed from the dissociation of carbonic acid is extruded from the cells by the NHE, preventing a large fall in pHi developing during secretory activity. Similar mechanisms have been proposed for the secretion of HCO3− in pancreatic (Schulz, 1987) and airway epithelia (Smith & Welsh, 1992; Inglis et al. 1998).
The second major means of pHi recovery is dependent upon HCO3−, insensitive to amiloride but sensitive to DNDS. These features are consistent with the activity of a NBC, and RT-PCR studies showed that mRNA for the pNBC transporter is present in these cells. The results of Ussing chamber studies have suggested that an electrogenic NBC contributes to HCO3− secretion by Calu-3 cells, cotransporting two HCO3− with each Na+ (Lee et al. 1998; Devor et al. 1999). This suggestion was largely based on the results of studies using EBIO, which increases the basolateral K+ conductance of Calu-3 cells (Devor et al. 1999). Although Vm was not measured, it was predicted that this effect of EBIO on K+ conductance would hyperpolarise Vm and thus reduce the electrical driving force for HCO3− entry on an electrogenic NBC (Devor et al. 1999). Such a prediction has clear consequences for pHi recovery; if an electrogenic NBC is indeed responsible for amiloride-insensitive pHi recovery from acidosis, we would expect EBIO to slow this recovery.
Consistent with this prediction, we found that EBIO significantly reduced pHi recovery from acidosis. To confirm that this effect of EBIO was mediated through the activation of K+ channels and consequent hyperpolarisation of the cell membrane potential, we compared the pHi recovery of cells exposed to EBIO in either high or low [K+]o. Under these conditions we would predict that cells exposed to high [K+]o would be depolarised relative to those exposed to low [K+]o and that any pHi recovery mediated by an electrogenic NBC would therefore be faster in high [K+]o. Precisely this finding was made in similar studies of pancreatic duct cells, which are known to express electrogenic NBC activity (Shumaker et al. 1999). However, somewhat unexpectedly, changing [K+]o in our study had no effect on the rate of pHi recovery in the presence of EBIO. Moreover, direct measurements of Vm confirmed that altering [K+]o had the expected effects on Vm, both in the presence and absence of EBIO. These experiments thus indicate that amiloride-resistant pHi recovery is unaffected by changes in Vm, suggesting that the HCO3− transport system underlying this recovery process is not electrogenic. Further evidence of this came from experiments using valinomycin. Electrophysiological studies confirmed that this K+ ionophore hyperpolarised the cells, and showed that changing [K+]o caused substantial excursions in Vm. However, these changes in Vm had no effect upon the rate of pHi. These data are thus entirely inconsistent with the involvement of an electrogenic NBC transport process in pHi recovery.
In contrast, a similar study of pancreatic cells showed that after treatment with valinomycin, changing [K+]o had profound effects on the rate of pHi recovery from ammonium-induced acidosis (Shumaker et al. 1999), indicating that NBC activity can be detected by measurements of pHi recovery. These pancreatic cells express both CFTR and electrogenic NBC and further experiments showed that the rate of pHi recovery was significantly increased by membrane-permeable cAMP. This appeared to be mediated by cAMP-induced activation of CFTR, membrane depolarisation, and subsequent increased activity of the electrogenic NBC, since the effect was not observed in cells expressing mutated CFTR (Shumaker et al. 1999). In a further attempt to change Vm in Calu-3 cells in the present study, we therefore explored the effects of forskolin, which increases intracellular cAMP levels, activating CFTR and stimulating HCO3− secretion (Haws et al. 1994; Shen et al. 1994; Devor & Pilewski, 1999). In contrast to the effects in pancreatic cells, forskolin had no effect on pHi recovery in Calu-3 cells, though it significantly depolarised Vm. This is consistent with our other findings that indicate the lack of involvement of an electrogenic NBC transport process in pHi recovery.
The reason for this apparent lack of involvement of electrogenic NBC in pHi recovery is not clear. It is possible that the non-polarised nature of the Calu-3 cells used in this study prohibits the activity of the electrogenic NBC. However, it is clear that cell polarity is not an essential requirement for electrogenic NBC activity, since the above-described study of pancreatic cells used non-polarised cells grown on coverslips. In addition, numerous studies have detected electrogenic Na+-HCO3− cotransport in oocytes transfected with electrogenic NBC subtypes (see (Romero & Boron, 1999). Interestingly, a recent study has suggested that the molecular structure of NBC transporters, which share very close homology, is not the sole determinant of their stoichiometries (Gross et al. 2001). Pancreatic and kidney NBC1, which are 93 % identical but have different N-termini (Abuladze et al. 1998) and are thought to have stoichiometries of 2 HCO3−:1 Na+ and 3 HCO3−:1 Na+, respectively, in their native cell types (for review see Romero & Boron, 1999), were expressed in different cell types. When expressed in proximal convoluted tubule cells both NBC1 subtypes had stoichiometries of 2 HCO3−:1 Na+, whilst their stoichiometries were both 3 HCO3−:1 Na+ when expressed in collecting duct cells. This suggests that as yet unidentified cellular regulators may modify the stoichiometry of these transporters. Thus it is possible that the stoichiometries of these transporters may be subject to change under different cellular conditions. Our results indicate that despite the presence of mRNA for pNBC1 in Calu-3 cells, the processes involved in pHi recovery from acidosis are not dependent on membrane potential and therefore do not include electrogenic NBC activity.
Since forskolin is reported to stimulate HCO3− secretion across these cells, we may have expected to detect some change in pHi recovery rate. It is possible that the increased efflux of HCO3− through forskolin-activated CFTR is matched by an increased entry of HCO3− into the cell, or increased metabolic generation of HCO3− in the presence of forskolin. Thus the overall rate of change of pHi is unaffected. Recent preliminary experiments have shown, however, that the rate of pHi recovery can be affected by certain other agonists that stimulate HCO3− secretion. ACh, which acts primarily by increasing [Ca2+]i rather than cAMP to stimulate HCO3− secretion across Calu-3 cell monolayers, increases the rate of pHi recovery from acidosis (S. K. Inglis, M. Constable & L. Finlay, unpublished data). These data confirm that pHi recovery rates from acidosis can be altered by some agonists that induce HCO3− secretion.
The inhibitory effect of EBIO on H+ extrusion cannot be due to membrane hyperpolarisation following activation of K+ channels as predicted since EBIO had no significant effect on Vm. It is possible that the effect of EBIO is brought about by the activation of CFTR (Devor et al. 1999; Cuthbert, 2001), which would allow an increased efflux of HCO3−, thereby slowing the pHi recovery from acidification. Alternatively, EBIO may have a direct inhibitory effect on the amiloride-insensitive transporter that contributes to pHi recovery in these cells.
At the present time we cannot be certain of the identity of the transporter mediating amiloride-insensitive, DNDS-sensitive, electroneutral pHi recovery. However, since the imposition of an outwardly directed Cl− gradient almost trebled the rate of amiloride-insensitive pHi recovery, we believe that a Cl−-HCO3− exchanger is involved. Consistent with this prediction, mRNA for the Cl−-HCO3− exchanger AE2 was detected by RT-PCR in both this and a previously published study (Loffing et al. 2000). We failed to detect mRNA for either Na+-dependent Cl−-HCO3− exchanger (NDAE) or electroneutral NBC, suggesting that these transporters are not expressed by Calu-3 cells. However, since the RT-PCR primers were based on the NDAE sequence cloned from Drosophila (Romero et al. 2000), it is possible that our primers reacted with a Drosophila-specific region of mRNA. This may explain why we also failed to detect NDAE in rat kidney, in which Na+-dependent Cl−-HCO3− exchanger functional activity has been detected (Boyarsky et al. 1988). We therefore cannot rule out the possible involvement of Na+-dependent Cl−-HCO3− exchange in pHi recovery in Calu-3 cells.
In summary, pHi recovery from acidosis in Calu-3 cells is independent of membrane potential and dependent for the most part on extracellular Na+. It is mediated by NHE and by a DNDS-sensitive, HCO3−-dependent transporter that may be AE2, NDAE or an electroneutral form of pNBC1 activity.
Acknowledgments
The authors would like to thank Elaine A. Waller and Maree Constable for their excellent technical help and the Wellcome (Wellcome Research Career Development Fellowship, S.K.I.), Tenovus Tayside and Anonymous Trusts for financial support.
REFERENCES
- Abuladze N, Lee I, Newman D, Hwang J, Boorer K, Pushkin A, Kurtz I. Molecular cloning, chromosomal localization, tissue distribution, and functional expression of the human pancreatic sodium bicarbonate cotransporter. Journal of Biological Chemistry. 1998;273:17689–17695. doi: 10.1074/jbc.273.28.17689. [DOI] [PubMed] [Google Scholar]
- Acevedo M, Olver RE, Ward MR. Ionic permeabilities of the cell membranes of sheep tracheal epithelium. Journal of Physiology. 1990;422:67–81. doi: 10.1113/jphysiol.1990.sp017973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acevedo M, Steele LW. Na+-H+ exchanger in isolated epithelial tracheal cells from sheep. Involvement in tracheal proton secretion. Experimental Physiology. 1993;78:383–394. doi: 10.1113/expphysiol.1993.sp003692. [DOI] [PubMed] [Google Scholar]
- Ballard ST, Trout L, Bebok Z, Sorscher EJ, Crews A. CFTR involvement in chloride, bicarconate and liquid secretion by airway submucosal glands. American Journal of Physiology. 1999;277:L694–699. doi: 10.1152/ajplung.1999.277.4.L694. [DOI] [PubMed] [Google Scholar]
- Boyarsky G, Ganz MB, Sterzel RB, Boron WF. pH regulation in single glomerular mesangial cells II. Na+-dependent and -independent Cl−-HCO3− exchangers. American Journal of Physiology. 1988;255:C857–869. doi: 10.1152/ajpcell.1988.255.6.C857. [DOI] [PubMed] [Google Scholar]
- Choi I, Aalkjaer C, Boulpaep EL, Boron W. An electroneutral sodium/bicarbonate cotransporter NBCn1and associated sodium channel. Nature. 2000;405:571–575. doi: 10.1038/35014615. [DOI] [PubMed] [Google Scholar]
- Clary-Meinesz C, Mouroux J, Cosson J, Huitorel P, Blaive B. Influence of pH on ciliary beat frequency in human bronchi and bronchioles. European Respiratory Journal. 1998;11:330–333. doi: 10.1183/09031936.98.11020330. [DOI] [PubMed] [Google Scholar]
- Coakley RD, Grubb BR, Gatzy JT, Chadburn JL, Boucher RC. Differential airway surface liquid (asl) pH, HCO3− and K+ homeostasis in cultured human and bronchial epithelium. Pediatric Pulmonology. 2000;(suppl.)(20):194. [Google Scholar]
- Cuthbert AW. Assessment of CFTR chloride channel openers in intact normal and cystic fibrosis murine epithelia. British Journal of Pharmacology. 2001;132:659–668. doi: 10.1038/sj.bjp.0703859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devor DC, Pilewski JM. UTP inhibits Na absorption in wild-type and ΔF508 CFTR-expressing human bronchial epithelia. American Journal of Physiology. 1999;276:C827–837. doi: 10.1152/ajpcell.1999.276.4.C827. [DOI] [PubMed] [Google Scholar]
- Devor DC, Singh AK, Lambert LC, Deluca A, Frizzell RA, Bridges RJ. Bicarbonate and chloride secretion in Calu-3 human airway epithelial cells. Journal of General Physiology. 1999;113:743–760. doi: 10.1085/jgp.113.5.743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudeja PK, Hafez N, Tyagi S, Gailey CA, Toofanfard M, Alrefai WA, Nazir TM, Ramaswamy K, Al-Bazzaz F J. Expression of the Na+/H+ and Cl−/HCO3− exchanger isoforms in proximal and distal human airways. American Journal of Physiology. 1999;276:L971–978. doi: 10.1152/ajplung.1999.276.6.L971. [DOI] [PubMed] [Google Scholar]
- Finkbeiner WE, Carrier SD, Teresi CE. Reverse transcription-polymerase chain reaction (RT-PCR) phenotypic analysis of cell cultures of human tracheal epithelium, tracheobronchial glands and lung carcinomas. American Journal of Respiratory Cell Molecular Biology. 1993;9:547–556. doi: 10.1165/ajrcmb/9.5.547. [DOI] [PubMed] [Google Scholar]
- Gross E, Hawkins K, Abuladze N, Pushkin A, Cotton CU, Hopfer U, Kurtz I. The stoichiometry of the electrogenic sodium bicarbonate cotransporter NBC1 is cell-type dependent. Journal of Physiology. 2001;531:597–603. doi: 10.1111/j.1469-7793.2001.0597h.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haws C, Finkbeiner WE, Widdicombe JH, Wine JJ. CFTR in Calu-3 human airway cells: channel properties and role in cAMP-activated Cl− conductance. American Journal of Physiology. 1994;266:L502–512. doi: 10.1152/ajplung.1994.266.5.L502. [DOI] [PubMed] [Google Scholar]
- Inglis SK, Corboz MR, Ballard ST. Effect of anion secretion inhibitors on mucin content of airway submucosal glands. American Journal of Physiology. 1998;274:L762–766. doi: 10.1152/ajplung.1998.274.5.L762. [DOI] [PubMed] [Google Scholar]
- Inglis SK, Corboz MR, Taylor AE, Ballard ST. Regulation of ion transport across porcine distal bronchi. American Journal of Physiology. 1996;270:L289–297. doi: 10.1152/ajplung.1996.270.2.L289. [DOI] [PubMed] [Google Scholar]
- Inglis SK, Corboz MR, Taylor AE, Ballard ST. Effect of anion transport inhibition on mucus secretion by airway submucosal glands. American Journal of Physiology. 1997;272:L372–377. doi: 10.1152/ajplung.1997.272.2.L372. [DOI] [PubMed] [Google Scholar]
- King M. Viscoelastic properties of airway mucus. Federation Proceedings. 1980;39:3080–3085. [PubMed] [Google Scholar]
- Lee MC, Penland CM, Widdicombe JH, Wine JJ. Evidence that Calu-3 human airway cells secrete bicarbonate. American Journal of Physiology. 1998;274:L450–453. doi: 10.1152/ajplung.1998.274.3.L450. [DOI] [PubMed] [Google Scholar]
- Loffing J, Moyer BD, Reynolds D, Shmukler BE, Alper SL, Stanton BA. Functional and molecular characterization of an anion exchanger in airway serous epithelial cells. American Journal of Physiology. 2000;279:C1016–1023. doi: 10.1152/ajpcell.2000.279.4.C1016. [DOI] [PubMed] [Google Scholar]
- Paradiso AM. ATP-activated basolateral Na+/H+ exchange in human normal and cystic fibrosis airway epithelium. American Journal of Physiology. 1997;273:L148–158. doi: 10.1152/ajplung.1997.273.1.L148. [DOI] [PubMed] [Google Scholar]
- Romero MF, Boron WF. Electrogenic Na+/HCO3− cotransporters: cloning and physiology. Annual Review of Physiology. 1999;61:699–723. doi: 10.1146/annurev.physiol.61.1.699. [DOI] [PubMed] [Google Scholar]
- Romero MF, Henry D, Nelson S, Harte PJ, Dillon AK, Sciortino CM. Cloning and characterisation of a Na+-driven anion exchanger (NDAE1) Journal of Biological Chemistry. 2000;275:24552–24559. doi: 10.1074/jbc.M003476200. [DOI] [PubMed] [Google Scholar]
- Roos A, Boron WF. Intracellular pH. Physiological Reviews. 1981;61:296–434. doi: 10.1152/physrev.1981.61.2.296. [DOI] [PubMed] [Google Scholar]
- Schulz I. Electrolyte and Fluid Secretion in the Exocrine Pancreas. New York: Raven Press; 1987. [Google Scholar]
- Shen B-Q, Finkbeiner WE, Wine JJ, Mrsny RJ, Widdicombe JH. Calu-3: a human airway epithelial cell line that shows cAMP-dependent Cl− secretion. American Journal of Physiology. 1994;266:L493–501. doi: 10.1152/ajplung.1994.266.5.L493. [DOI] [PubMed] [Google Scholar]
- Shumaker H, Amlal H, Frizzell R, Ulrich CD, Soleimani M. CFTR drives Na+-nHCO3− cotransport in pancreatic duct cells: a basis for defective HCO3− secretion in CF. American Journal of Physiology. 1999;276:C16–25. doi: 10.1152/ajpcell.1999.276.1.C16. [DOI] [PubMed] [Google Scholar]
- Singh M, Krouse M, Moon S, Wine JJ. Most basal Isc in Calu-3 human airway cells is bicarbonate-dependent Cl− secretion. American Journal of Physiology. 1997;272:L690–698. doi: 10.1152/ajplung.1997.272.4.L690. [DOI] [PubMed] [Google Scholar]
- Smith JJ, Welsh MJ. cAMP stimulates bicarbonate secretion across normal, but not cystic fibrosis airway epithelia. Journal of Clinical Investigation. 1992;89:1148–1153. doi: 10.1172/JCI115696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trout L, Gatzy JT, Ballard ST. Acetylcholine-induced liquid secretion by bronchial epithelium: role of Cl− and HCO3− transport. American Journal of Physiology. 1998;275:L1095–1099. doi: 10.1152/ajplung.1998.275.6.L1095. [DOI] [PubMed] [Google Scholar]
- Van Scott MR, Chinet TC, Burnette AD, Paradiso AM. Purinergic regulation of ion transport across nonciliated bronchiolar epithelial (Clara) cells. American Journal of Physiology. 1995;269:L30–37. doi: 10.1152/ajplung.1995.269.1.L30. [DOI] [PubMed] [Google Scholar]