

Abstract
The isolated chick ciliary neuron calyx synapse preparation was used to test cysteine string protein (CSP) action on presynaptic N-type Ca2+ channels. Endogenous CSP was localized primarily to secretory vesicle clusters in the presynaptic nerve terminal. Introduction of recombinant CSP into the voltage clamped terminal resulted in a prominent increase in Ca2+ current amplitude. However, this increase could not be attributed to a change in Ca2+ channel kinetics, voltage dependence, prepulse inactivation, or G protein inhibition but was attributed to the recruitment of dormant channels. Secretory vesicle associated endogenous CSP may play an important role in enhancing Ca2+ channel activity at the transmitter release site.
Several studies have indicated that cysteine string protein (CSP) has an important role in the rapid release of transmitters at the presynaptic nerve terminals (Umbach et al. 1994; Zinsmaier et al. 1994). This protein was initially identified in Drosophila melanogaster (Zinsmaier et al. 1990) and independently in the eel electroplax (Gundersen & Umbach, 1992). An association with the Ca2+ channel was suggested early on (Gundersen & Umbach, 1992) but the protein was localized to the secretory vesicle (Umbach et al. 1994). These observations led to the hypothesis that CSP is bound to the secretory vesicle but modulates the presynaptic calcium channel upon docking of the vesicle to the release site (Mastrogiacomo et al. 1994; Ranjan et al. 1998). However, subsequent studies have both contradicted and supported the central question of whether CSP can modulate presynaptic Ca2+ channels. Consistent with this idea, influx of Ca2+ into the nerve terminal was reported to be reduced in Drosophila CSP mutants (Umbach et al. 1998) but no effect on Ca2+ channels was observed in a fly peptidergic neurosecretory terminal (Morales et al. 1999). The finding that a Ca2+ ionophore could overcome transmitter release block in these mutants supports a defect in Ca2+ entry and, in addition, suggested that the effect on Ca2+ channels also results in transmitter release block (Ranjan et al. 1998). However, more recent results were unable to detect changes in Ca2+ influx in mutant flies (Dawson-Scully et al. 2000), in contradiction to the earlier findings.
To establish whether CSP can indeed affect presynaptic Ca2+ channels it is necessary to test its action in a preparation where the nerve terminal Ca2+ current can be recorded directly under voltage clamp. We have explored this question in the calyx-type synapse of the chick ciliary ganglion. This synapse preparation can be fully isolated and used both for imaging native proteins by high resolution immunocytochemistry (Stanley & Mirotznik, 1997; Mirotznik et al. 2000) and for direct recording of presynaptic Ca2+ currents (Stanley & Goping, 1991; Yawo & Momiyama, 1993; Mirotznik et al. 2000). The Ca2+ current in this nerve terminal is almost exclusively N-type (Stanley, 1991; Yawo & Chuhma, 1994). We examined the action of CSP on the calcium channel by direct introduction of the protein into the nerve terminal through a patch pipette. We found that CSP resulted in a marked enhancement of presynaptic Ca2+ current amplitude that could not simply be attributed to a modification in channel biophysical properties.
METHODS
Preparation of recombinant cysteine string protein
pET28c containing the entire CSP open reading frame (Drosophila melanogaster, Genbank no. M63008; Zinsmaier et al. 1990) or the vector alone was transformed into BL-21(DE3) cells (Novagen, Madison, WI, USA) and overnight starter cultures were grown to saturation in Luria-Bertani (LB) medium with Kanamycin (30 μg ml−1). The following day, cultures were split 1:10 in fresh LB medium with antibiotic and grown to an optical density (600 nm) of 0.6. Protein induction was achieved with the addition of 1 mm isopropyl-β-d-thiogalactosidase (IPTG) for 3 h at 30 °C. Protein was purified essentially as described by the manufacturer (Novagen). Briefly, cells were centrifuged and resuspended with binding buffer (5 mm imidazole, 500 mm NaCl, 20 mm Tris, pH 7.9, 10 ml per 250 ml culture). After sonication, insoluble material was removed by centrifugation. The supernatant was filtered through a 0.2 μm filter and loaded onto a Ni-resin column (Novagen). Protein was eluted from the column with a solution containing 120 mm imidazole, 500 mm NaCl, 20 mm Tris pH 8.0. The eluate was concentrated and the buffer exchanged with PBS using Centriprep columns (Amicon, Houston, TX, USA).
Isolation of calyx presynaptic nerve terminals
Chick calyx nerve terminals were isolated as described elsewhere (Stanley & Goping, 1991; Stanley, 1993). Briefly, chick embryos were removed from 15 day chick eggs and immediately decapitated. Ciliary ganglia were enzymatically dissociated in modified Eagle's medium (Calbiochem), plated on glass cover slips and transferred to a cover slip chamber for recording.
Patch-clamp recording
Conventional whole cell recording was used as described (Stanley & Goping, 1991) using pCLAMP (6.03 to 7.0; Axon Instruments). Patch electrodes had tip resistance of 4∼5 MΩ when filled with the internal solution. Data were low-pass filtered at 10 kHz and sampled at 50 μs and leak currents were removed on-line using a P/6 protocol. Data were digitally filtered at 2 kHz for display. All recordings were performed at room temperature.
Ca2+ current analysis
Current pulse amplitudes were measured at the peak value or if there was no clear peak at the end of the standard duration (40 ms) voltage pulse. Voltage-sensitive inhibition was measured as the maximum difference between the current pulse amplitudes with, or without, a prepulse. Slope conductance in ramp voltage protocols was measured between +35 and +45 mV. In this study ‘prepulse inactivation’ refers to a protocol where the amplitude of the Ca2+ current evoked by a test pulse was measured after stepping the holding potential in 10 mV increments for 5 s. The term ‘prepulse inactivation’ was preferred over ‘steady state inactivation’, the usual term for this protocol, as the fragility of this preparation precludes the analysis necessary to justify the latter term. The protocol used here was consistent with a relevant recent study on the modulation of N-type Ca2+ channel inactivation (Jarvis & Zamponi, 2001).
Solutions and chemicals
The composition of the external recording solution was (mm): NaCl 160, CaCl2 5, MgCl2 1, d-glucose 5, 4-aminopyridine 2, tetrodotoxin 0.001, Hepes-Na 10, pH 7.3 (adjusted with NaOH). The internal solution contained (mm): caesium gluconate 120, CsCl 10, EGTA-Cs 10, MgCl2 1, Hepes-Cs 10, tetraethylamonium-Cl 20, MgATP 1, with GTP 0.1 (adjusted to pH 7.3 with CsOH). CSP was included in the internal solution at a concentration of 1 μg ml−1. Control vector solution was diluted by the same ratios.
Immunocytochemistry
Dissociated cells were fixed in 2 % paraformaldehyle for 45 min and permeabilized in 5 % polyoxyethylene-20-cetyl ether with 0.5 % paraformaldehyde for 10 min. The preparation was exposed to polyclonal anti-CSP (Stressgen, Victoria, BC, Canada) and monoclonal anti-SV2 (Developmental Studies Hybridoma Bank, University of Iowa, IA, USA) overnight (Mirotznik et al. 2000). Fluorescein isothiocyanate (FITC) and lissamine rhodamine sulfochloride (LRSC)-conjugated secondary antibodies (Jackson Immunoresearch, West Grove, PA, USA) were applied at 1:50 dilution for 1 h. Cells were visualized under fluorescent illumination on a Zeiss Axiophot with × 63 or × 100, 1.4 NA lenses. Image stacks were acquired at a z-step interval of 200 nm and deblurred by an iterative deconvolution protocol (Axiovision 3.0).
CSP depletion
Immobilized protein-A suspension was washed three times with wash buffer (150 mm NaCl, 20 mm Tris-HCl, 0.1 % Triton X-100, pH 7.4) before use. One-millilitre monoclonal anti-CSP (Zinsmaier et al. 1990; 5 μg ml−1) was mixed with 0.5 ml immobilized protein-A suspension for 3 h at 4 °C with agitation. Beads were collected and washed three times with wash buffer. CSP was depleted by mixing 0.5 ml sample solution (1 μg ml−1) with 0.5 ml immobilized antibody for 3 h at 4 °C, confirming by Western blot.
Statistical analysis
Values are expressed as means ± standard error of the mean. Student's t test was used with either paired data in sequential recordings or unpaired in different cells.
RESULTS
Localization of native CSP protein at the calyx nerve terminal
Dissociated chick ciliary ganglia were fixed and stained using standard immunocytochemical procedures (Mirotznik et al. 2000). Nerve terminals were costained with a polyclonal antibody against SV2, a secretory vesicle marker, and a monoclonal antibody against CSP (and vice versa, which gave a similar pattern of staining). Terminals invariably stained for both SV2 and CSP and at low image resolution the staining patterns were largely colocalized in clusters. However, at high resolution after iterative deconvolution analysis the staining was not totally colocalized with areas of SV2 or CSP staining alone (Fig. 1).
Figure 1. Image slice of a calyx nerve terminal (CX, inset cartoon) attached to a ciliary neuron (CN) and stained for CSP (green) or SV2 (red) with colocalized regions in yellow.
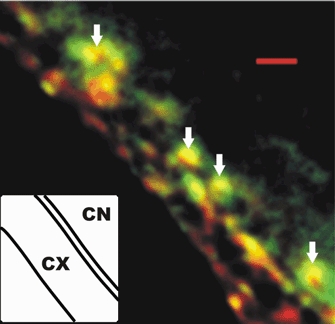
Examples of transmitter release face synaptic vesicle clusters are indicated by arrows. Scale bar, 1 μm.
Action of CSP on the presynaptic calcium current
Effect of intracellular CSP on the presynaptic calcium current amplitude
The calyx nerve terminals have a small internal volume and are readily perfused with substituting ions, allowing the rapid recording of Ca2+ currents. A standard depolarizing voltage pulse to 0 mV from a holding potential of −80 mV was used to evoke Ca2+ currents. In control terminals the amplitude of the evoked current was fairly constant with time after seal rupture (Fig. 2A). However, when recombinant CSP was included in the patch solution (1 μg ml−1) the current exhibited a marked and progressive increase in amplitude (Fig. 2A), beginning almost immediately after seal rupture. Thus, in control terminals the change in the Ca2+ current during 1 min of perfusion was +6 ± 5 % (n = 6) while in the CSP-treated terminals it was +82 ± 24 % (n = 16, P < 0.01). We used a voltage ramp protocol to test whether current enhancement could be observed over the full membrane potential range from current activation to the Ca2+ reversal potential (ER). In control terminals the current did not change whereas with CSP it increased at each potential in approximate proportion to that recorded immediately after seal rupture (Fig. 2B, left panel).
Figure 2. Enhancement of presynaptic calcium current by intracellular CSP.
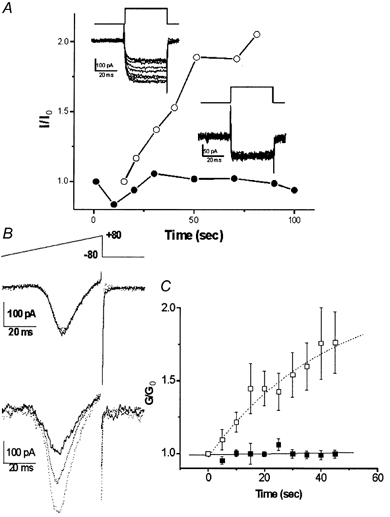
A, current traces from two calyces patched with pipettes containing either plain intracellular solution (inset lower right) or recombinant CSP (inset upper left) and recorded beginning immediately after seal rupture. The voltage protocol was a depolarizing step from −80 to 0 mV. Current amplitudes (I) were measured near the end of the step, normalized to that of the first trial (I0, trace with dark line) and were plotted (with CSP, ○; control, •). B, currents activated by a depolarizing ramp protocol (top panel) recorded in a control calyx (middle panel) or with CSP (lower panel). Traces were recorded immediately after seal rupture (t = 0, bold) and at 20 (dotted) and 45 s (fine-dotted) thereafter. C, a series of ramp depolarizations were given as in Fig. 2B at 5 s intervals to CSP-infused (□) and vector-control-infused (▪) calyx nerve terminals. The slope conductance, G, was determined as the slope of a straight line fitted to the ascending limb of the current peak between +35 and +40 mV and this was normalized to the value of G at t = 0 (G0). n = 4 for each treatment.
Can we attribute the action of the extract to the CSP?
The CSP fusion protein was purified by standard affinity chromatography (see Methods). However, in order to rule out the possibility that a co-purified contaminant, and not the peptide itself, was responsible for the current enhancement we used two controls. The first was to compare CSP with a vector extract. Effects on maximum channel activity were compared by calculating the slope conductance from the steepest part of the voltage-current relation (+35 to +45 mV) using the ramp protocol. The vector-treated terminals had a negligible effect on conductance (vector −1 ± 2 %, n = 4; CSP +52 ± 16 %, n = 4; P < 0.02; Fig. 2C). In the second control CSP was depleted from the extract by immunoprecipitation, which eliminated the effect on current amplitude (0 ± 9 %, n = 5; P > 0.1). Thus, the observed increase in presynaptic Ca2+ current could be attributed to the action of CSP.
The simplest hypothesis for the observed current increase was that CSP alters the biophysical properties of the presynaptic Ca2+ channels. This was tested by examining the time course of the current (channel kinetics), the voltage dependence of activation and the prepulse-current inactivation curve.
CSP and calcium current activation
We used a simple protocol to screen for an effect of CSP on Ca2+ current kinetics by comparing the shapes of a pulse step-induced current before and after current enhancement (Fig. 3A). When the two current pulses were normalized and overlaid they were essentially identical in shape in current growth or, up to the resolution of the patch recording, the tail current decay (Fig. 3A, right panel). Thus, there were no gross changes in channel activation kinetics or deactivation kinetics that could account for the CSP effect.
Figure 3. Effect of CSP on the time course of the Ca2+ current.
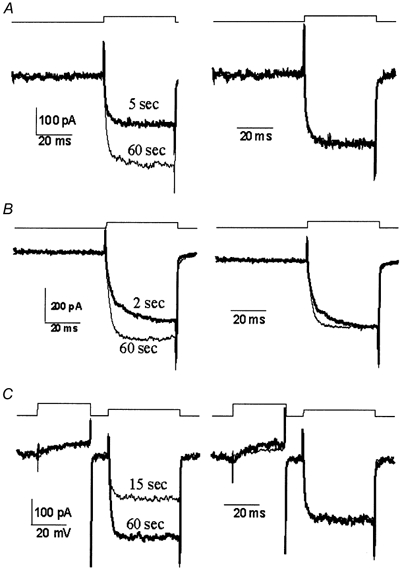
Recordings are from three calyces (A-C) before (thick line) and after (thin line) perfusion through the patch electrode with CSP with voltage protocols above each panel. Raw (leak subtracted) data are shown in the left panel to compare current amplitudes while traces normalized to the current amplitude at the end of the step are on the right to compare current pulse shape. A and B are with a single pulse protocol depolarizing from −80 to 0 mV for 40 ms. In C the test pulse was preceded by a strong (+80 mV, 30 ms) prepulse to remove any voltage-sensitive inhibition.
In a few cases the activation rate was slower prior to CSP action (Fig. 3B). This was attributed to tonic inhibition via the G protein pathway as it could be relieved with the standard protocol of preceding the test pulse by a strong prepulse (Stanley & Mirotznik, 1997; Mirotznik et al. 2000). Since CSP has been linked to this inhibitory pathway (Magga et al. 2000) we tested its involvement in current growth. When the prepulse protocol (Fig. 3C) was used to monitor test pulse current amplitude the increase in Ca2+ current amplitude was still observed (control 1.3 ± 2.4 %, n = 8, CSP 43 ± 14 %, n = 13; P < 0.03). This finding, together with the observation that the CSP-induced growth in current amplitude was much larger than the observed initial tonic inhibition (9.4 ± 3.2 %, n = 21), argues against a CSP effect via the G protein pathway.
CSP and the Ca2+ conductance-voltage relation
Ramp depolarizations (Fig. 3B) were used to obtain single-trial assays of the conductance (G)-voltage (V) relation from −80 to +80 mV. These were recorded immediately after seal rupture and at 45 s later (termed pre- and post-CSP) in nerve terminals in which the Ca2+ current increased +74 ± 24 % (n = 5). The current recorded at each voltage was corrected for driving force with ER = +65 mV (the zero voltage interception of steepest part of curve) and was divided by the maximum conductance for that trial to yield the normalized conductance (G/Gmax, Fig. 4A). Mean G/Gmax-voltage curves for the pre- and post-CSP treatment could be virtually superimposed (Fig. 4A, right panel), with fitted Boltzmann relations with 50 % amplitude (y50 %) and slope factors (dx) of −3 mV and 5.0, and −2 mV and 4.8, respectfully. Thus, CSP did not affect the activation range nor the voltage sensitivity of the presynaptic Ca2+ channels.
Figure 4. Effect of intracellular CSP on calcium current inactivation.
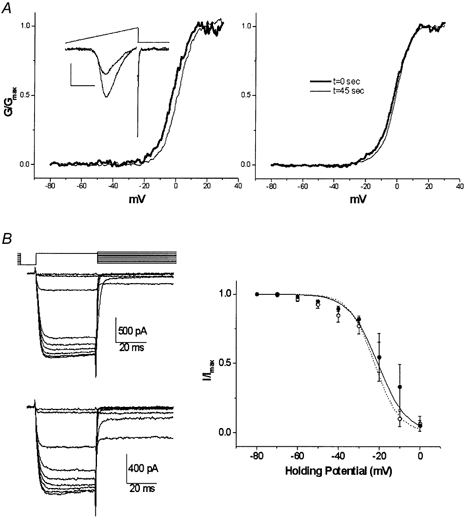
A, conductance-voltage relation. Current traces were recorded during a ramp depolarization protocol (inset, protocol as in Fig. 2B) before (thick line) and after (thin line) CSP-induced growth. Current traces were corrected for driving force using an ECa of +65 mV and were normalized to GMAX (at ∼+30 mV) and were plotted against the membrane potential to yield G/GMAX-voltage curves. Curves before (thick line) and after (thin line) CSP action are shown for a single calyx (left panel) and pooled data (right panel). B, prepulse inactivation (PPI) curve. Ca2+ current PPI was determined using the voltage protocol in the upper left panel with an interval of 5 s between trials. Traces are shown for control (middle left panel) and CSP-facilitated (lower left panel) calyces. Pooled data normalized to current amplitude with a holding potential of −80 mV from four calyces and fitted Boltzmann relations are plotted before (•, continuous line) and after (○, dashed line) the CSP effect in the right panel.
CSP and the Ca2+ current prepulse inactivation
Ca2+ channels typically shift into an inactivated state during prolonged depolarizations, termed prepulse inactivation (PPI). While somatic N-type Ca2+ channels typically exhibit PPI at very hyperpolarized potentials, with I50 % at < −60 mV, calyx presynaptic N-type channels are much less sensitive with I50 % of > −25 mV (Yawo & Momiyama, 1993; Stanley & Mirotznik, 1997). To test if a depolarizing shift in the PPI relation could account for the increase in Ca2+ current amplitude observed with CSP, we measured test current amplitude at holding potentials ranging from −80 to 0 mV (Fig. 4B, left panel). PPI curves in CSP-treated terminals could be superimposed on those from control terminals (Fig. 4B, right panel), ruling out a change in this parameter as the factor responsible for Ca2+ current enhancement. We cannot, however, rule out the involvement of slower inactivating processes that could not be detected by our PPI protocol.
DISCUSSION
The main finding of this study is that the introduction of CSP into the calyx nerve terminal results in an increase in the presynaptic Ca2+ current. This increase does not appear to involve a change in the basic biophysical parameters of the channel but instead suggests the recruitment of previously inactive channels.
Early studies in frog oocytes reported an increased N-type current when the calcium channels were co-expressed with CSP (Gundersen & Umbach, 1992). However, a recent report concluded that the absence of CSP does not affect Ca2+ influx at a mutant Drosophila neuromuscular terminal. In this study Ca2+ levels, detected with an intracellular indicator, were unchanged after a train of impulses (Dawson-Scully et al. 2000), contradicting an earlier study (Umbach et al. 1998). There are several reasons why such an increase in Ca2+ influx might not have been observed. First, there is no evidence that the presynaptic Ca2+ channel at the Drosophila neuromuscular junction is N-type, as in the present study. Second, changes in internal Ca2+ levels after impulse trains reflect not only influx but also buffering, organelle uptake and excretion. Finally, if CSP normally acts only on those channels that are within nanometres of the docked secretory vesicle (Stanley, 1997) its loss should only affect a very small (although critical with respect to transmitter release), and possibly undetectable, fraction of the total Ca2+ channel current (see below).
The present study demonstrates that CSP injected directly into the presynaptic nerve terminal enhances the amplitude of the Ca2+ current. The effect was CSP dependent and could not be accounted for by changes in Ca2+ channel activation or deactivation rates, or the activation voltage dependence. Hence, other than the possibilities that CSP alters the single channel conductance or maximum open probability, which our study does not address, our results indicate that the protein does not increase the presynaptic Ca2+ current by an effect on rapid channel gating properties. It should be noted that injected CSP acts in the presence of the endogenous protein, demonstrating that at least a fraction of the Ca2+ channels remain susceptible to modulation.
It is particularly interesting that we also failed to detect a shift in the voltage range of prepulse-inactivation (PPI). Co-expression of syntaxin with N-type calcium channels in the frog oocyte shifts the steady-state inactivation (PPI) curve of the channels to a more negative voltage range (Bezprozvanny et al. 1995, 2000; Wiser et al. 1996; Jarvis & Zamponi, 2001), effectively inhibiting the channels. Other reports have suggested that the syntaxin effect on PPI is largely eliminated when other release site associated proteins are also expressed, while a reduction in current amplitude is still observed (Wiser et al. 1997; Magga et al. 2000; Jarvis & Zamponi, 2001). This may explain why we and others did not detect the reverse of this effect when syntaxin is cleaved by botulinum toxin C1 (Stanley & Mirotznik, 1997; Marsal et al. 1997). Thus, while interference of syntaxin binding to the Ca2+ channel may be the molecular mechanism of CSP action (Wu et al. 1999) the mechanism of Ca2+ current enhancement does not appear to involve the voltage-dependent inactivation detected with our PPI protocol. Experimental limitations precluded the examination of slower inactivation processes.
The simplest interpretation of our results is that CSP induces the recruitment of a dormant pool of Ca2+ channels. In our case, ‘dormant’ would include disabled, blocked or inactivated (where the process was too slow to resolve with our PPI technique; Degtiar et al. 2000). We essentially ruled out the possibility that the newly recruited channels were of a different, non-N-type on the basis of two observations. First, their biophysical properties would have to be indistinguishable from the pre-existing N-type channel and, second, Ω-conotoxin GVIA, a specific N-type blocker, did not spare the CSP-recruited Ca2+ current (data not shown). Our results indicate that there are three pools of N-type Ca2+ channels at the nerve terminal: A uninhibited, B inhibited and C CSP-inhibition-relieved channels. Based on our previous findings (Stanley, 1991; Haydon et al. 1994), most of the calyx N-type Ca2+ channels are highly clustered and, hence, these three pools are all likely to reflect different states of release site-associated channels. Our interpretation of these pools is that A includes most of the current recorded in resting terminals. This current is relatively resistant to voltage-dependent inactivation (Stanley & Goping, 1991; Yawo & Momiyama, 1993), a property that is unusual for somatic N-type Ca2+ channels, but that is by no means unique (e.g. Cahill et al. 2000). Thus, calyx Ca2+ channels may be intrinsically different or may be modified by interacting proteins. Pool B reflects channels that have been inhibited by a different release site interaction, involving perhaps a shift to a blocked or inactivated state, and C comprises channels where this inhibition has been relieved by CSP.
CSP is associated primarily with the secretory vesicle (Umbach et al. 1994, Fig. 1), and thus, enhancement of presynaptic Ca2+ channel activity by endogenous CSP would be most likely to occur when the vesicle docks to the presynaptic membrane (Mastrogiacomo et al. 1994). Such a mechanism would recruit the Ca2+ channels that are within nanometres of the vesicle docking site (Stanley, 1993, 1997), maximizing Ca2+ efficiency on secretion while minimizing extraneous influx.
Acknowledgments
This project was supported by CIHR award no. 38091 and by a Canada Research Chair to E.F.S.
REFERENCES
- Bezprozvanny I, Scheller RH, Tsien RW. Functional impact of syntaxin on gating of N-type and Q-type calcium channels. Nature. 1995;378:623–626. doi: 10.1038/378623a0. [DOI] [PubMed] [Google Scholar]
- Bezprozvanny I, Zhong P, Scheller RH, Tsien RW. Molecular determinants of the functional interaction between syntaxin and N-type Ca2+ channel gating. Proceedings of the National Academy of Sciences of the USA. 2000;97:13943–13948. doi: 10.1073/pnas.220389697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahill AL, Hurley JH, Fox AP. Coexpression of cloned α1B, β2a, and α2/δ subunits produces non-inactivating calcium currents similar to those found in bovine chromaffin cells. Journal of Neuroscience. 2000;20:1685–1693. doi: 10.1523/JNEUROSCI.20-05-01685.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson-Scully K, Bronk P, Atwood HL, Zinsmaier KE. Cysteine-string protein increases the calcium sensitivity of neurotransmitter exocytosis in Drosophila. Journal of Neuroscience. 2000;20:6039–6047. doi: 10.1523/JNEUROSCI.20-16-06039.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degtiar VE, Scheller RH, Tsien RW. Syntaxin modulation of slow inactivation of N-type calcium channels. Journal of Neuroscience. 2000;20:4355–4367. doi: 10.1523/JNEUROSCI.20-12-04355.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gundersen CB, Umbach JA. Suppression cloning of the cDNA for a candidate subunit of a presynaptic calcium channel. Neuron. 1992;9:527–537. doi: 10.1016/0896-6273(92)90190-o. [DOI] [PubMed] [Google Scholar]
- Haydon PG, Henderson E, Stanley EF. Localization of individual calcium channels at the release face of a presynaptic nerve terminal. Neuron. 1994;13:1275–1280. doi: 10.1016/0896-6273(94)90414-6. [DOI] [PubMed] [Google Scholar]
- Jarvis SE, Zamponi GW. Distinct molecular determinants govern syntaxin 1A-mediated inactivation and G-protein inhibition of N-type calcium channels. Journal of Neuroscience. 2001;21:2939–2948. doi: 10.1523/JNEUROSCI.21-09-02939.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magga JM, Jarvis SE, Arnot MI, Zamponi GW, Braun JE. Cysteine string protein regulates G protein modulation of N-type calcium channels. Neuron. 2000;28:195–204. doi: 10.1016/s0896-6273(00)00096-9. [DOI] [PubMed] [Google Scholar]
- Marsal J, Ruiz-Montasell B, Blasi J, Moreira JE, Contreras D, Sugimori M, Llinas R. Block of transmitter release by botulinum C1 action on syntaxin at the squid giant synapse. Proceedings of the National Academy of Sciences of the USA. 1997;94:14871–14876. doi: 10.1073/pnas.94.26.14871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastrogiacomo A, Parsons SM, Zampighi GA, Jenden DJ, Umbach JA, Gundersen CB. Cysteine string proteins: A potential link between synaptic vesicles and presynaptic Ca2+ channels. Science. 1994;263:981–982. doi: 10.1126/science.7906056. [DOI] [PubMed] [Google Scholar]
- Mirotznik RR, Zheng X, Stanley EF. G-Protein types involved in calcium channel inhibition at a presynaptic nerve terminal. Journal of Neuroscience. 2000;20:7614–7621. doi: 10.1523/JNEUROSCI.20-20-07614.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales M, Ferrus A, Martinez-Padron M. Presynaptic calcium-channel currents in normal and csp mutant Drosophila peptidergic terminals. European Journal of Neuroscience. 1999;11:1818–1826. doi: 10.1046/j.1460-9568.1999.00604.x. [DOI] [PubMed] [Google Scholar]
- Ranjan R, Bronk P, Zinsmaier KE. Cysteine string protein is required for calcium secretion coupling of evoked neurotransmission in Drosophila but not for vesicle recycling. Journal of Neuroscience. 1998;18:956–964. doi: 10.1523/JNEUROSCI.18-03-00956.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley EF. Single calcium channels on a cholinergic presynaptic nerve terminal. Neuron. 1991;7:585–591. doi: 10.1016/0896-6273(91)90371-6. [DOI] [PubMed] [Google Scholar]
- Stanley EF. Single calcium channels and acetylcholine release at a presynaptic nerve terminal. Neuron. 1993;11:1007–1011. doi: 10.1016/0896-6273(93)90214-c. [DOI] [PubMed] [Google Scholar]
- Stanley EF. The calcium channel and the organization of the presynaptic transmitter release face. Trends in Neurosciences. 1997;20:404–409. doi: 10.1016/s0166-2236(97)01091-6. [DOI] [PubMed] [Google Scholar]
- Stanley EF, Goping G. Characterization of a calcium current in a vertebrate cholinergic presynaptic nerve terminal. Journal of Neuroscience. 1991;11:985–993. doi: 10.1523/JNEUROSCI.11-04-00985.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley EF, Mirotznik RR. Cleavage of syntaxin prevents G-protein modulation of presynaptic calcium channels. Nature. 1997;385:340–343. doi: 10.1038/385340a0. [DOI] [PubMed] [Google Scholar]
- Umbach JA, Saitoe M, Kidokoro Y, Gundersen CB. Attenuated influx of calcium ions at nerve endings of csp and shibire mutant Drosophila. Journal of Neuroscience. 1998;18:3233–3240. doi: 10.1523/JNEUROSCI.18-09-03233.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umbach JA, Zinsmaier KE, Eberle KK, Buchner E, Benzer S, Gundersen CB. Presynaptic dysfunction in Drosophila csp mutants. Neuron. 1994;13:899–907. doi: 10.1016/0896-6273(94)90255-0. [DOI] [PubMed] [Google Scholar]
- Wiser O, Bennett MK, Atlas D. Functional interaction of syntaxin and SNAP-25 with voltage sensitive L- and N-type Ca2+ channels. EMBO Journal. 1996;15:4100–4110. [PMC free article] [PubMed] [Google Scholar]
- Wiser O, Tobi D, Trus M, Atlas D. Synaptotagmin restores kinetic properties of a syntaxin-associated N- type voltage sensitive calcium channel. FEBS Letters. 1997;404:203–207. doi: 10.1016/s0014-5793(97)00130-0. [DOI] [PubMed] [Google Scholar]
- Wu MN, Fergestad T, Lloyd TE, He Y, Broadie K, Bellen HJ. Syntaxin 1A interacts with multiple exocytic proteins to regulate neurotransmitter release in vivo. Neuron. 1999;23:593–605. doi: 10.1016/s0896-6273(00)80811-9. [DOI] [PubMed] [Google Scholar]
- Yawo H, Chuhma N. Ω-Conotoxin-sensitive and -resistant transmitter release from the chick ciliary presynaptic terminal. Journal of Physiology. 1994;477:437–448. doi: 10.1113/jphysiol.1994.sp020205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yawo H, Momiyama A. Re-evaluation of calcium currents in pre- and postsynaptic neurones of the chick ciliary ganglion. Journal of Physiology. 1993;460:153–172. doi: 10.1113/jphysiol.1993.sp019464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinsmaier KE, Eberle KK, Buchner E, Walter N, Benzer S. Paralysis and early death in cysteine string protein mutants of Drosophila. Science. 1994;263:977–980. doi: 10.1126/science.8310297. [DOI] [PubMed] [Google Scholar]
- Zinsmaier KE, Hofbauer A, Heimbeck G, Pflugfelder GO, Buchner S, Buchner E. A cysteine-string protein is expressed in retina and brain of Drosophila. Journal of Neurogenetics. 1990;7:15–29. doi: 10.3109/01677069009084150. [DOI] [PubMed] [Google Scholar]