

Abstract
Uniquantal endplate currents (EPCs) were recorded simultaneously at the proximal, central and distal parts of the frog neuromuscular synapse, and their minimal synaptic latencies, latency dispersions and sensitivity to noradrenaline, cAMP and protein kinase A inhibition were measured. The latency dispersion was highest in the proximal part (P90 = 1.25 ms); it decreased to P90 = 0.95 ms in the central part and to P90 = 0.75 ms (60 % of the proximal part) in the distal part. In the proximal parts of the long neuromuscular synapse, stimulation-evoked EPCs with long release latencies were eliminated when the intracellular cAMP was increased by β1 activation by noradrenaline, by the permeable analogue db-cAMP, by activation of adenylyl cyclase or by inhibition of cAMP hydrolysis. This makes the evoked release more compact, and the amplitude of the reconstructed multiquantal currents increases. Protein kinase A is a target of this regulation, since a specific inhibitor, Rp-cAMP, prevents the action of cAMP in the proximal parts and increases the occurrence of long-latency events in the distal parts of the synapse. Our results show that protein kinase A is involved in the timing of quantal release and can be regulated by presynaptic adrenergic receptors.
The molecular mechanisms that underlie transmitter release are still in the realm of hypotheses rather than being a firmly established theory. To understand these mechanisms, it is useful to study two variables: the number of released vesicles (quantal content) and the release parameters as reflected by the synaptic latency distribution (Katz & Miledi, 1965; Minenko & Magazanik, 1986). In our previous study (Bukharaeva et al. 1999) we measured both parameters simultaneously and found that after application of noradrenaline (NA), the quantal content remained unaltered, whereas the duration of release became shorter. This suggests that different mechanisms are involved in the control of quantal content and the time course of release (Zucker, 1999). The molecular mechanisms underlying this finding were followed in two parallel lines of research.
One line was pharmacological. Inhibitor and agonist experiments showed that NA acts on the β1 receptor (Bukharaeva et al. 1999), which might be coupled with the cAMP cascade (Carlson et al. 1994; Majewski & Barrington, 1995; Sulakhe & Vo, 1995). We report here on changes in the latencies of focally recorded uniquantal endplate currents (EPCs) after the application of several agents that regulate intracellular cAMP levels, and demonstrate that the synchronizing action of NA is mediated by cAMP and protein kinase A (PKA). Interestingly, an increased fluctuation and asynchronicity of evoked release were described recently for several Drosophila mutants in which the synthesis and degradation of cAMP were affected (Renger et al. 2000).
Another aspect of our research was physiological. It is known that catecholamines facilitate neuromuscular transmission (Orbeli, 1923; Burn, 1945) by increasing EPC amplitude. This has been attributed to an increase in quantal content (Jenkinson et al. 1968; Hidaka & Kuriyama, 1969; Kuba, 1970; Kuba & Tomita, 1971; Wessler & Anschuetz, 1988; Vizi, 1991), an increase in quantum size (Van der Kloot & Van der Kloot, 1986) and a greater sensitivity of the subsynaptic membrane to ACh (Chen et al. 1991). For example, when a decrease in quantal content and longer latencies are observed, NA removes activity-induced fatigue (Ruzzier & Scuka, 1986). We showed that the larger synaptic potential may result from a more synchronized release, even though the quantal content is the same (Bukharaeva et al. 1999). These data were obtained by focal recording of EPCs in the proximal part of the 150- to 200-μm-long nerve terminal at the frog endplate. However, it is known that there are different patterns of spike amplitudes and time courses along the frog nerve terminal (Benoit & Mambrini, 1970; Mallart, 1984; Shakiryanova et al. 1994), non-uniform spontaneous and evoked release (Zefirov, 1983; Mallart, 1984; D'Alonzo & Grinnell, 1989) as well as different structural characteristics of the proximal and distal endplate regions (Davey & Bennett, 1982; Robitaille & Tremblay, 1987). We therefore estimated the latencies of EPCs simultaneously at the proximal, central and distal parts of the synapse and found substantial differences between these parts in minimal synaptic latencies, latency dispersions and sensitivity to NA, cAMP manipulations and PKA inhibition. Preliminary results with cAMP in the proximal part of the synapse have already been presented as a short report (Bukharaeva et al. 2000).
METHODS
Animals and drugs
The experimental preparation, bathing solutions and focal extracellular recording technique have been described in a previous paper (Bukharaeva et al. 1999). Briefly, experiments were carried out on the cutaneous pectoris muscle from the frog Rana ridibunda from November to April. Animals were anaesthetized with ether before being stunned and double-pithed. The preparations were pinned to a translucent chamber and superfused with (mm): NaCl 113.0, KCl 2.5, CaCl2 0.2, NaHCO3 3.0, and MgCl2 4.0. The pH was 7.3 at 20.0 ± 0.3 °C. The Animal Care and Use Committee of the Institute of Physiology, Czech Academy of Sciences, approved the protocol.
The following drugs were used (Sigma, St Louis, MO, USA): NA, dibutyryl cAMP, adenosine 3,5-monophosphothioate (Rp-cAMP), forskolin, and 3-isobutyl-1-methylxanthine (IBMX). The drugs were added to the superfusing solution, and the measurements were started 20 min after drug application, unless stated otherwise. In most cases, the drugs were washed out for a further 30 min before new recordings were made.
Electrophysiology
Suprathreshold stimuli of 0.1 ms duration were applied to the nerve at 2 s intervals. Nerve action potentials and extracellular EPCs were recorded using three focal Ringer-filled extracellular pipettes with a tip diameter of 2–3 μm and 1–3 MΩ resistance. Extracellular pipettes were positioned under visual control in the proximal, central and distal endplate regions of a long nerve terminal, 3–5 μm, 40–50 μm and 80–100 μm, respectively from the end of the myelinated segments of the axon. In these regions, three-component nerve spikes were recorded simultaneously with extracellular EPCs (Mallart, 1984; Shakiryanova et al. 1994). The recorded signals were filtered between 0.03 Hz and 10 kHz, digitized at 3 μs intervals by an analog-to-digital 9 bit converter, fed into a computer and processed by our application program package.
Uniquantal EPCs (Katz & Miledi, 1965; Barrett & Stevens, 1972) were recorded in the presence of 0.2 mm Ca2+ and 4.0 mm Mg2+. The quantal content (mo) of the low-quanta EPCs were determined from 5 or 6 stimulation periods (256 stimuli each) in the absence and in the presence of particular drugs. The number of failures in a series of 250–400 uniquantal responses was measured and mo calculated as equal to ln N/no, where N is the total number of stimuli and no is the number of failures (Del Castillo & Katz, 1954; Martin, 1955). The number of stimuli was therefore between 1280 and 1536 in each series of stimulation periods. Unless mo is quite low, at least some of the responses in a series of 250–400 responses will be larger than uniquantal.
Latency was measured as the time interval between the peak of the inward presynaptic Na+ currents and the time at which the rising phase of the concomitantly recorded quantal event reached 20 % of maximum (Fig. 1A). Because the amplitude of the reference Na+ current decreases along the nerve terminal (Mallart, 1984, Shakyrianova et al. 1994), the distal recording site was selected so that the Na+ peak, from which the latency was measured, was still clearly seen on averaged records from this part of the nerve terminal (e.g. Fig. 1A, DIST). The limit of the latency measurement was 6 ms for experiments performed at 20 °C.
Figure 1. Latencies of quantal releases in the proximal (PROX), central (CENTR) and distal (DIST) parts of the synapse.
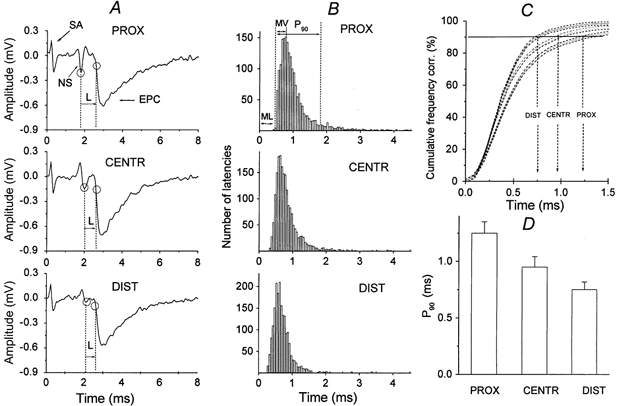
A, extracellularly recorded presynaptic nerve spikes (NS) and individual endplate currents (EPCs). The stimulus artefact (SA) is followed by a three-phase action potential, the shape of which changes along the nerve terminal (Shakyrianova et al. 1994). The time interval between the peak of the inward presynaptic Na+ current and the time at which the rising phase of the EPC reached 20 % of maximum (ovals) is defined as the release latency (L). B, non-corrected latency histograms. Values of minimal synaptic latencies (ML, were not subtracted) of 2268 uniquantal EPCs from 9 experiments. The bin size is 0.05 ms. The modal value (MV) of the histogram and the time at which 90 % of all measured uniquantal EPCs had occurred (P90) are indicated. C, the cumulative plots of latencies for the same data, corrected for minimal synaptic latencies, are shown. The vertical arrows indicate P90 in the distal (DIST), central (CENTR) and proximal (PROX) parts. The dashed lines parallel to the curves are confidence limits at P = 0.05. D, corrected P90 (values of minimal synaptic latencies subtracted) at PROX, CENTR and DIST parts of the nerve terminal (means ± s.e.m. from 10 experiments).
The stability of the recording electrode position next to the membrane region studied was crucial during long-lasting extracellular recordings. Therefore, we monitored the amplitude of the nerve terminal action potentials throughout each data set. Only data sets in which the terminal spike changed by less than 10 % during the drug application and washout were analysed further (Bukharaeva et al. 1999). Statistical analyses of pre- and postsynaptic events were performed using Student's t test for paired data.
The quantitative characteristics of the change in the time course of evoked secretion can be obtained by cumulative curve analysis (Van der Kloot, 1991) and by the release probability function (Barrett & Stevens, 1972; Baldo et al. 1986). From our experience (Bukharaeva et al. 1999), both methods provide reliable information about the time course of evoked release; therefore in the present paper only cumulative curves were used (Fig. 1C) together with an estimation of the modal values of latency histograms (MV in Fig. 1B) and the minimal synaptic latencies (ML in Fig. 1B). Cumulative curves were constructed from latency histograms of the uniquantal EPCs. The histograms of the amplitudes of the total population of EPCs were analysed using a standard computer procedure. Uniquantal EPCs were characterized by amplitudes that fell in the first peak in the amplitude histograms (not shown).
The mean value of the shortest 5 % of the latencies in each series was taken as the minimal synaptic latency. For the comparison of dispersion histograms in different parts of the terminal, minimal synaptic latencies, which differ along the nerve terminal, were subtracted. The interval between the minimal synaptic latency and the time at which 90 % of all measured uniquantal EPCs had occurred was designated as P90 (Fig. 1B, C). The statistical significance of the difference between two cumulative curves was assessed by the Kolmogorov-Smirnov statistic; P ≤ 0.05 was taken as significant (Bronstein & Semendjaev, 1986; Van der Kloot, 1991). Other statistical tests were performed with SigmaStat 0.1 (Jandel Corporation 1992–1994). Analyses of variance (ANOVA) of the experimental groups versus the control group were performed by multiple comparisons using the ANOVA Bonferroni t test. The amplitudes, rise times of EPCs (20–80 % of the maximal amplitude) and exponential decay constants (τ) of the responses are presented as means ± s.e.m. Differences between two groups were considered statistically significant at the probability level P = 0.05. The letter n indicates the number of endplates measured in each group.
RESULTS
Proximal to distal gradient of release dispersion
Evoked presynaptic nerve spikes recorded in three parts of the synapse were different (‘NS’ in Fig. 1A), as were the mean quantal contents of EPCs. In a low-calcium, high-magnesium solution, mo was 0.33 ± 0.09 (n = 12) in the proximal part, 0.25 ± 0.07 (n = 12) in the central part and 0.18 ± 0.09 (n = 12) in the distal part. In the proximal region, the nerve spikes had three components (Fig. 1A, PROX): an outward (positive) passive capacitance current, a large inward Na+ current and an outward K+ current (Mallart, 1984; Shakiryanova et al. 1994). The spikes recorded in the central part and the most remote distal part were characterized by two phases (Fig. 1A). The EPCs appeared after a synaptic latency (‘L’ in Fig. 1A; Katz & Miledi, 1965). The time courses of quantal release latencies were also different in these regions. This is documented by native records (Fig. 4), synaptic latency histograms (Fig. 1B) and the cumulative plots of latencies (Fig. 1C). The vertical arrows in Fig. 1C indicate the times when 90 % of the quanta have been released (P90) in the distal, central and proximal parts. The dashed lines parallel to the curves are confidence limits at P = 0.05. According to the Kolmogorov-Smirnov criterion, the difference between the curves is statistically significant if the confidence limits of the two curves do not overlap. For example, significant differences between the distal and proximal parts can already be seen when about 30 % of EPCs are released.
Figure 4. Effect of db-cAMP on the latencies in three parts of the synapse.
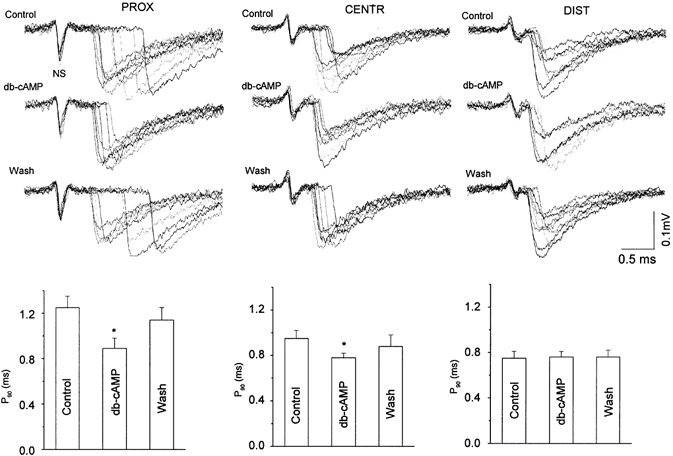
Upper records are 8–10 superimposed extracellular recordings of presynaptic NSs and individual EPCs before (Control), in the presence of and after washout of 1 × 10−5m db-cAMP (Wash). All recordings were obtained simultaneously in one experiment, by three extracellular microelectrodes located in three zones of the same endplate. Columns: P90 parameter (ordinates) in controls, in the presence of 1 × 10−5m db-cAMP and after 60 min washout of the drug (n = 6).
The minimal synaptic latencies differed significantly (P ≤ 0.05) and were 0.50 ± 0.01 ms in the proximal part, 0.41 ± 0.01 ms in the central part and 0.31 ± 0.01 ms in the distal part (n = 12 each). For this reason, the latency distribution histograms were shifted to the left in the central and distal parts. They were asymmetrical due to EPCs with long latencies (late release according to Barrett & Stevens, 1972). However, the latency dispersion expressed as P90, corrected for minimal synaptic latency (Fig. 1C, D), was highest in the proximal part (1.25 ± 0.10 ms), decreasing to 0.95 ± 0.09 ms in the central part and 0.75 ± 0.07 ms (60 % of the proximal part) in the distal part; all differences were significant at P ≤ 0.05 (n = 12). The modal values of the histograms, when measured from the minimal synaptic latencies in the proximal, central and distal parts (Fig. 1B), were similar: 0.25 ± 0.02 ms, 0.21 ± 0.02 ms and 0.28 ± 0.01 ms, respectively (P ≤ 0.05, n = 12 each). These observations demonstrate that the highest degree of synchronization in the remote parts of the long nerve terminal were due to a small number of long-latency EPCs.
The effect of pressure on release dispersion
In five experiments, the extracellular electrode was located near the nerve ending under different pressures to check whether the differences observed along the endplate were caused mechanically while searching for the optimal recording site along the progressively narrowing nerve ending (Davey & Bennett, 1982). Figure 2 shows one of five experiments in which the effect of mechanical pressure was tested. The data on this figure demonstrate that the time course of the release (latency histograms and P90, Fig. 2B, C) was not altered when greater pressure was applied and when presynaptic spikes, quantal content and EPC amplitudes increased (Fig. 2Ab). In particular, the increase in mo from 0.29 ± 0.04 to 0.41 ± 0.07 without any changes in the latency dispersion pattern, provides further evidence (Parnas & Parnas, 1999) that under particular conditions, quantum content and quantal latencies are controlled independently, and that probably no presynaptic quantal interaction exists in the frog, as was suggested recently for the lobster neuromuscular junction (Bykhovska et al. 1999).
Figure 2. Effect of slight and strong microelectrode pressure on the latencies of quantal releases in the proximal part of the synapse.
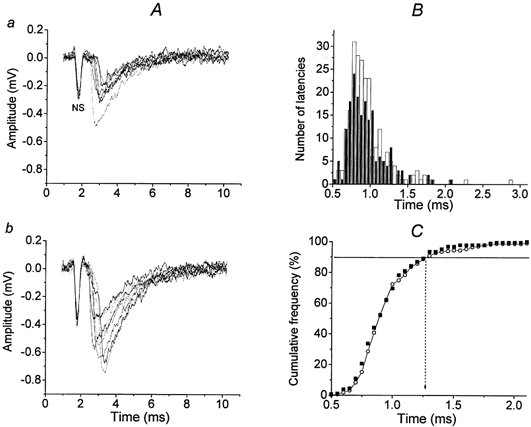
A, 10 superimposed randomly selected recordings from a total of 220 presynaptic NSs and extracellular EPCs before (a) and after the application of stronger pressure (b) applied by moving the recording microelectrode 20 μm vertically towards the muscle fibre. Recordings with failures were omitted. Quantum content increased from 0.32 in panel a to 0.60 in this case, apparently due to mechanical stretching in the release area (Turkanis, 1973; Chen & Grinnell, 1997). B, corrected latency histograms of 220 uniquantal EPCs from this experiment. The bin size is 0.05 ms. Filled columns, slight pressure; open columns, strong pressure. C, the cumulative plots of latencies for the same data before (○) and after pressure (▪). The vertical dotted arrow indicates P90, which is the same (1.25 ms) for slight and strong pressure of the microelectrode.
NA decreases the latency dispersion in the proximal part
Control quantal content mo was 0.33 ± 0.09 (n = 6) in the proximal part. After application of 1 × 10−5m NA, mo remained unchanged at 0.39 ± 0.08 (n = 6, P ≥ 0.05). The mean frequency of miniature EPCs (mEPCs) in the presence of NA (1.73 ± 0.45 s−1, n = 6, P ≤ 0.05) was also similar to that recorded in the controls before the drug was administered (1.4 ± 0.50 s−1). NA did not change the presynaptic action potential, the EPC amplitude or the minimal synaptic latency, whereas the modal values of the latency histograms shifted slightly from 0.27 ± 0.02 ms to 0.18 ± 0.03 ms (Bukharaeva et al. 1999). NA did, however, change the latency distribution by removing long synaptic latencies in the proximal part where the latency dispersion was originally high (Fig. 3). The control value of the corrected P90, 1.25 ± 0.10 ms, was significantly and reversibly reduced by 35 % (as assessed using the Kolmogorov-Smirnov criteria) to 0.81 ± 0.10 ms (n = 6, P ≤ 0.05). On the other hand, the changes in the central parts were marginal (minimal latency was 0.41 ± 0.01 ms before and 0.40 ± 0.02 ms after NA; modal values were 0.21 ms and 0.20 ms, respectively, and P90 was 0.95 ms and 0.80 ms, respectively), and were not significant in the distal parts, where they were characterized by the most compact release (minimal latency was 0.31 ± 0.02 ms before and 0.31 ± 0.01 ms after NA; modal values were 0.25 ± 0.05 ms and 0.27 ± 0.04 ms, respectively, and P90 0.78 ms and 0.75 ms, respectively).
Figure 3. Effects of drugs changing cAMP levels and the protein kinase A (PKA) inhibitor Rp-cAMP on P90 values at PROX, CENTR and DIST parts of the endplate.
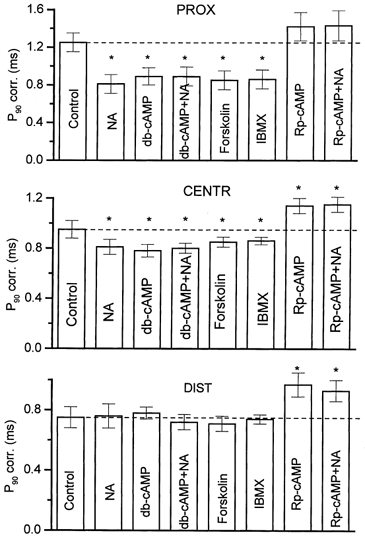
Left ordinates represent the corrected (corr) P90 (i.e. minimal synaptic latencies were subtracted). Dashed lines indicate the control level in each group. The drugs applied were: noradrenaline (NA), 1 × 10−5m; db-cAMP, 1 × 10−6m; forskolin, 1 × 10−6m; 3-isobutyl-1-methylxanthine (IBMX) 1 × 10−4m; Rp-cAMP, 2 × 10−5m; Rp-cAMP + NA-Rp-cAMP together with 1 × 10−5m NA. Asterisks indicate values that were significantly different from controls at the probability level P = 0.05.
db-cAMP imitates NA action
The permeable derivative of cAMP, db-cAMP (1 × 10−6m), when present for 1 h, markedly shortened the latency distribution in a way resembling the action of NA (Fig. 4). The most pronounced effect on long-latency EPCs was found in the proximal part: the number of EPCs with longer synaptic latencies was decreased, but the minimal synaptic latency was unchanged (0.50 ± 0.02 ms in nine control experiments versus 0.47 ± 0.04 ms in the presence of db-cAMP for minimal latencies), whereas the modal values of the latency histograms shifted slightly (from 0.28 ± 0.04 ms to 0.20 ± 0.03 ms, respectively, P ≤ 0.05). Corrected cumulative curves of the synaptic latencies resulted in a control P90 value of of 1.25 ± 0.10 ms, which was significantly reduced to 0.89 ± 0.10 ms (n = 6, P ≤ 0.05, Fig. 4) in the presence of db-cAMP. The ratio between the P90 value in db-cAMP versus that in the controls was 0.71. This means that this drug shortened the release by 29 %.
The corrected P90 in the central part was decreased to 0.78 ± 0.05 ms in db-cAMP, versus 0.95 ± 0.07 ms (n = 6, P ≤ 0.05) in controls. In the distal part, P90 was not significantly changed: 0.75 ± 0.07 ms in controls, and 0.78 ± 0.09 ms in db-cAMP (Fig. 4).
No other effects of 1 × 10−6m db-cAMP were found: the quantal content in the central part was 0.34 ± 0.15 (n = 6) before, 0.35 ± 0.25 (n = 6) 1 h after db-cAMP application, and 0.36 ± 0.17 (n = 5) after washing out of the drug for 2 h. The mean frequency of mEPCs (0.83 ± 0.45 s−1) was very similar to that recorded in the controls before application of the drug (0.74 ± 0.50 s−1, n = 5, P ≤ 0.05). It is worth mentioning that a higher concentration of the drug, in particular 1 × 10−4m, shortened the release phase but increased the frequency of occurrence of miniature endplate potentials (mEPPs; Miyamoto & Breckenbridge, 1974) to an extent that made the latency estimation incorrect due to the unpredictable appearance of mEPPs in the latency window. Like NA, db-cAMP did not change the amplitude and decay time of mEPCs, suggesting that at a concentration of 1 × 10−6m, it had no effect on postsynaptic receptor sensitivity.
The effects of db-cAMP and NA were not cumulative. When applied 1 h after db-cAMP, NA did not further shorten the early release phase in the proximal zone. The P90 was 0.89 ± 0.10 ms in the presence of 1 × 10−6m db-cAMP and 0.85 ± 0.05 ms (n = 5, P ≤ 0.05) when NA was added for another 60 min. The absence of an effect could be expected if the NA action were completely mediated by endogenous cAMP.
Forskolin imitates NA and db-cAMP actions
The adenylyl cyclase activator forskolin (Laurenza et al. 1987) was tested at a concentration of 1 × 10−6m, which is not high enough to influence the frequency of mEPCs or postsynaptic ACh sensitivity (Khirough et al. 1998). The time course of evoked quantal secretion in the presence of forskolin was affected almost the same as in the presence of NA and db-cAMP. This effect is demonstrated by the reduction of P90 from 1.25 ± 0.10 ms in the controls to 0.85 ± 0.09 ms (n = 6, P ≤ 0.05), which means that forskolin shortened the release by 32 %. A much smaller effect was observed in the central part, where P90 decreased by 11 % from 0.95 ± 0.07 ms in the controls to 0.85 ± 0.06 ms (n = 6, P ≤ 0.05). Forskolin was almost ineffective in the distal part (Fig. 3). Other parameters, such as minimal synaptic latencies and modal values of latency histograms, were affected similarly, as in the case of NA and db-cAMP in all parts of the synapse (data not given).
The minimal latency in controls in the proximal part was 0.49 ± 0.02 ms, and in forskolin it was 0.50 ± 0.03 ms; the modal value in controls was 0.30 ± 0.01 ms, which slightly but significantly decreased in forskolin (0.25 ± 0.03 ms). As with db-cAMP, the effects of forskolin and NA in the proximal part were not cumulative, and P90 was 0.85 ± 0.11 ms (n = 5, P ≥ 0.05) in the presence of both drugs (Fig. 3).
IBMX imitates the actions of NA, db-cAMP and forskolin
The level of cAMP and of other cyclic nucleotides in the cell is regulated not only by their production, but also by the rate of their degradation to 5′-AMP (Nestler & Greengard, 1989; Majewski & Barrington, 1995). We therefore studied the effect of 1 × 10−4m of the phosphodiesterase inhibitor IBMX (Beavo et al. 1970) on latency distribution; a similar pattern of action was found as when db-cAMP and forskolin were used (Fig. 3). P90 was decreased by 31 % in the proximal part, a lesser shortening by 10 % was observed in the central part and no effect was seen in the distal part. NA was ineffective when applied together with IBMX in the proximal part (not shown). The above concentration of the drug slightly increased the quantal content, from 0.33 ± 0.01 before to 0.45 ± 0.03 after 1 × 10−4m IBMX. Once again, other parameters (i.e. minimal synaptic latencies and the modal values of latency histograms) were affected similarly in all parts of the synapse (data not given), as in the case of NA and db-cAMP. The minimal latency in the proximal part in controls was 0.48 ± 0.04 ms and was unchanged in IBMX (0.49 ± 0.05 ms). The modal values in controls (0.24 ± 0.03 ms) and in IBMX (0.19 ± 0.02 ms) were not significantly different.
PKA inhibition increases latency dispersion
All hitherto presented data indicate that an increase of intraterminal cAMP can slightly shorten the early release period and substantially decrease the late phase of the release. It is known that cAMP activates the phosphorylation of many functional and regulatory proteins (Majewski & Barrington, 1995). The inhibition of a particular class of protein kinases should thus increase the latency dispersion. In release timing, the activation of PKA might be involved, since in a preliminary screening of several protein kinase inhibitors, we found that Rp-cAMP, a specific inhibitor of PKA (Dostmann et al. 1990), was the most effective. When applied at a concentration of 2 × 10−5m, Rp-cAMP induced the appearance of long-latency EPCs mostly in remote release sites, originally the most synchronized, but to a lesser extent in proximal parts characterized by high dispersion (Fig. 3). Thus, P90 increased by 29 ± 5 % in the distal part, by 20 ± 5 % in the central part, and insignificantly by 14 ± 5 % (n = 6) in the proximal part. At this concentration, the drug slightly decreased the quantal content, from 0.33 ± 0.09 before to 0.24 ± 0.03 after application. No effect of NA or db-cAMP was observed in any part of the junction when they were applied together with Rp-cAMP to the endplates already treated with this PKA inhibitor (Fig. 3, the far right columns). Rp-cAMP prolonged slightly but significantly the minimal synaptic latencies in the proximal and central parts by 10 % (from 0.50 ± 0.01 ms to 0.55 ± 0.03 ms and from 0.41 ± 0.01 ms to 0.46 ± 0.02 ms, respectively) and by 12 % (from 0.31 ± 0.01 ms to 0.35 ± 0.03 ms) in the distal part (n = 10 each, P ≤ 0.05). In contrast, cAMP manipulation, had no effect on this parameter. Nevertheless, the modal values of the histograms, when measured from the minimal synaptic latencies in each part, were similar before and after Rp-cAMP application: 0.31 ± 0.03 ms and 0.28 ± 0.04 ms, respectively, in the proximal part, 0.25 ± 0.04 ms and 0.22 ± 0.05 ms, respectively, in the central part and 0.22 ± 0.02 ms and 0.21 ± 0.03 ms, respectively, in the distal part (P ≥ 0.05, n = 12 each). Thus, the main group of short-latency quanta (early release) remained unchanged, and the PKA inhibition increased the number of EPCs with longer latencies. The increase in P90 was reversible because after a 30–60 min washout of Rp-cAMP, P90 returned to 96 % of its control value. Such good recovery probably indicates that phosphorylation and dephosphorylation take place at sites already prepared for quantal release and not during priming and docking of the vesicles, because once docked, vesicles are very stable at the release sites (Betz et al. 1993).
A similar conclusion can be drawn from results with tetanic stimulation. In five experiments, neuromuscular preparations were stimulated for 60 min with a frequency of 10 Hz in a solution containing 1.8 mm Ca2+ and in the presence of 2 × 10−5m Rp-cAMP. The stimulation was aimed to reach exhaustion of the immediately available quanta from the sites of release when multiquantal full-size EPCs are evoked (Ceccareli et al. 1972). After stimulation, the solution was again changed to one containing 0.35 mm Ca2+, +4.0 mm Mg2+ and +2 × 10−5m Rp-cAMP. Following 30 min of solution exchange, recordings of uniquantal EPCs were made every 2 s in the distal part in the presence of Rp-cAMP, for a period of 60 s (second post-tetanic recording period). P90, corrected for minimal synaptic latency, was prolonged during the first post-stimulation period to 1.06 ± 0.09 ms (n = 5) as compared with mean values obtained in the distal zone without stimulation (Fig. 3, lower panel). After washout of the drug, P90 decreased to normal and was 0.75 ± 0.07 ms (n = 5, P ≤ 0.05) during the second recording period. This represents a shift of 41 % if the washout values are taken as 100 %. In control experiments without the drug, P90 values were 0.76 ± 0.04 ms and 0.71 ± 0.09 ms during the first and second post-tetanic recording periods, respectively, without any significant difference. Minimal synaptic latencies were slightly shortened during the second recording period after the washout of Rp-cAMP (0.31 ± 0.02 ms) as compared with the first stimulation period in the presence of Rp-cAMP (0.38 ± 0.05 ms). Without Rp-cAMP, the latencies were 0.33 ± 0.04 ms and 0.34 ± 0.03 ms, respectively, in controls without the drug. The modal values of latency histograms were not influenced by stimulation and were similar to controls during both recording periods (not shown).
Construction of multiquantal EPCs
Our data have shown that an increase in cAMP makes the evoked release of single quanta more synchronous by eliminating long-latency events and that PKA inhibition leads to a more dispersed release. To assess the impact of these changes on the EPC peak amplitude, multiquantal EPCs were constructed by summation of the experimentally obtained uniquantal EPCs and their real latencies, before and after treatment with db-cAMP or Rp-cAMP (Fig. 5). In the proximal part, the amplitudes of the multiquantal EPCs constructed from uniquantal EPCs in the presence of db-cAMP, which decreased P90 by 29 %, were larger than those of the controls by as much as 27 % (Fig. 5A). The rise time to 20–80 % of the maximal amplitude decreased from 0.4 ms to 0.3 ms and the τ of the exponential decay was also slightly shortened from 2.1 ms to 2.0 ms. This is in accord with previous model studies (Sou'cek, 1971; Giniatullin et al. 1995, Zefirov & Gafurov, 1997) predicting that changes in the latency dispersion of standard-size single quanta affect mostly the amplitude and rise time and, to a lesser extent, the decay time of reconstructed EPCs, provided that standard quanta with τ = 1.5 are summated. On the other hand, the summed EPCs in the distal parts were not affected by db-cAMP, as the drug had no effect on the dispersion pattern (Fig. 5B).
Figure 5. Multiquantal EPCs summated from uniquantal EPCs recorded in the absence (Control) and presence of 1 × 10−6m db-cAMP (A, B) or 2 × 10−5m Rp-cAMP (C, D).
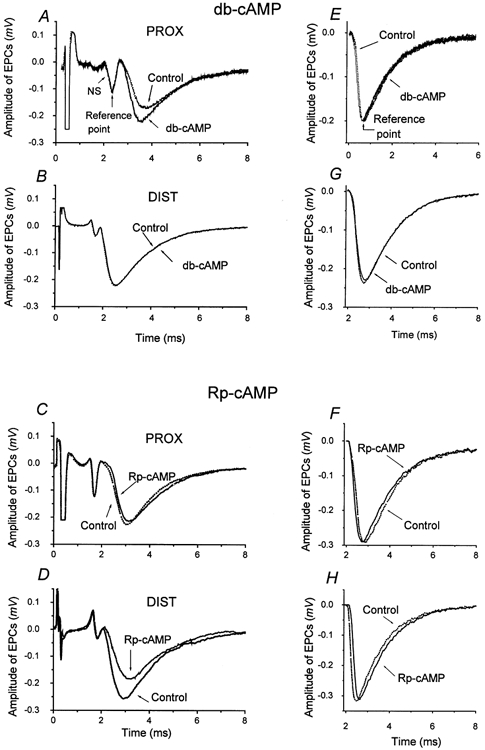
Uniquantal EPCs used for construction were recorded in the PROX and DIST parts of the endplate. Abscissae represent time in ms, ordinates represent amplitude in mV. Summations of 50–70 uniquantal EPCs measured before (Controls, dotted lines) and 30 min after the application of either drug (continuous lines) were performed. The summations were performed according to two different reference points (arrows): either according to the downward deflection of the NS (i.e. each EPC was considered with its own latency before and during the application of the drugs; A, B, C, D), or the same uniquantal EPCs were summated and normalized according to their amplitude peaks (E, F, G, H). In these latter reconstructions, controls and drug-treated EPCs are either indistinguishable or very close.
The opposite was found for the PKA inhibitor Rp-cAMP. EPCs from the proximal parts were the same in controls and after treatment with Rp-cAMP (Fig. 5C), apparently because there was no change in release pattern in these areas. Reconstruction based on recordings in the distal parts of the terminal was affected significantly by Rp-cAMP (Fig. 5D). The amplitude of the EPC reconstructed from uniquantal EPCs recorded in the presence of the PKA inhibitor Rp-cAMP (which increased P90 by 24 %) was lower by 28 % than in the controls, evidently due to the participation of events with longer latencies. The rise time increased from 0.35 ms to 0.50 ms, and the τ of the exponential decay was slightly increased from 1.8 ms to 2.0 ms. Parts E, F, G, and H in Fig. 5 show the amplitudes and time courses of the EPCs reconstructed from the same experimental uniquantal EPCs, normalized according to their amplitude maxima. In these reconstructions, multiquantal EPCs were virtually identical before and during the application of both drugs; this also indicates that no changes occurred in the quantum size and in postsynaptic sensitivity in the presence of either drug, and once again demonstrates the stability of the recordings.
DISCUSSION
We have demonstrated that long-latency EPCs are removed to a great extent when intracellular cAMP is increased following β1 activation by NA, by the permeable analogue db-cAMP, by forskolin activation of adenylyl cyclase or by inhibition of cAMP hydrolysis. On the other hand, the number of long-latency EPCs increased when PKA was inhibited. NA was ineffective in the presence of the PKA inhibitor Rp-cAMP.
The effects of PKA activation and inhibition depend upon the initial level of the late release along the nerve terminal. The late release is characterized by the presence of long-latency EPCs, and can be revealed by the P90 parameter, provided that the modal value of the early release latency histograms is not substantially changed. In the proximal part of the synapse, many EPCs are released with latencies longer than 2 ms, and they are eliminated to a great extent when the cAMP level is increased. In contrast, the inhibition of PKA had no effect. In the distal parts, EPCs were released mostly during the early release period of about 1 ms, and upon cAMP application, the P90 did not significantly change. The inhibition of PKA leads to the appearance of the longest latencies, which can be found in the frog muscle endplate at room temperature. In the distal parts, the EPCs have shorter minimal latencies and a more compact release than in the proximal parts. Saturating concentrations of cAMP may occur in the distal parts of the terminal, probably due to smaller axonal volumes (Robitaille & Tremblay, 1987, Pawson et al. 1998a,b). Other morphological differences between the proximal and distal sites do not seem to be a necessary prerequisite for the release pattern. This follows from the appearance of long-latency EPCs after PKA inhibition in the distal parts, which would not be expected if only differences in micro-morphological properties (e.g. the amount, size and shape of the active zones of release) were responsible. Even though this difference was not found in a recent freeze-fracture analysis (Pawson et al. 1998b), it may exist as indicated by the proximo-distal gradient of the minimal latencies, which are 0.5 ms in the proximal parts and only 0.3 ms in the distal parts; minimal latencies are not influenced by any changes in cAMP levels. The value of 0.3 ms is close to previous estimates (Almers, 1990), indicating that the opening of voltage-dependent Ca2+ channels, the entry of Ca2+, the activation of the exocytotic machinery, and the release and diffusion of the neurotransmitter to the postsynaptic membrane can occur within 0.2 ms. Synaptic vesicles that undergo exocytosis must therefore be docked very close to or in physical contact with Ca2+ channels (Stanley, 1997; Wiser et al. 1999). This distance might, however, be larger in the proximal part than in the distal part, and this might be reflected in the longer minimal latencies.
What really determines the synaptic latencies is unknown. Minimal latencies are probably the most constant parts of the release time course and represent the quanta (probably vesicles) that are optimally located with respect to Ca2+ entry sites and that have their soluble N-ethyl-maleimide-sensitive factor (NSF)-attachment protein receptors (SNARE proteins), such as the calcium-binding protein synaptotagmin I (Fernández-Chacon et al. 2001), optimally assembled for release. After quanta with minimal latencies, a main pool of quanta is released and characterized by the modal value of latency histograms. This early release (Barrett & Stevens, 1972) is followed by a late release with long latencies. Are these quanta released with latencies as long as 5 ms, or even longer, and therefore forced to move to the release site from more remote docking sites? Are they not docked properly, or do they belong to another population of vesicles? A comparison of the amplitudes and time parameters (Table 1) of fast-released EPCs with latencies between 0.5 and 0.7 ms with the rest of the EPCs showed no difference in quantum size or rise and decay times. This means that they most probably belong to the same release pool. However, other parameters of docking and release are difficult to measure in real time, at least at vertebrate endplates. The molecular organization of Ca2+ channels and synaptic proteins are generally studied using immunobinding studies, channel expression and protein-to-protein interactions revealed by separation techniques (Wiser et al. 1999), but no direct method exists to reproduce experimentally the fusion process that occurs in small synapses in real time. Even such important findings as those concerning SNARE pins as essential machinery for vesicle fusion (Weber et al. 1998) have been obtained on a time scale of minutes.
Table 1.
A comparison of the amplitude and time parameters of short- and long-latency uniquantal endplate currents (EPCs)
| Aepc (mV) | RT (μs) | τdec (ms) | A (mVms) | |
|---|---|---|---|---|
| Short-latency EPCs | 0.26 ± 0.01 | 165 ± 70 | 1.30 ± 0.18 | 0.91 ± 0.11 |
| Long-latency EPCs | 0.27 ± 0.01 | 163 ± 80 | 1.00 ± 0.22 | 1.00 ± 0.18 |
150 EPCs were recorded in the proximal part of 3 synapses (50 in each). The EPCs released with latencies in the interval between 0.5 ms (minimal synaptic latencies) and 0.7 ms were considered as short-latency EPCs. Long-latency EPCs were those that exhibited latencies of 1.2 ms and longer. Aepcs, EPC amplitude in mV; RT, rise time of EPC between 20 and 80 % of maximum; τdec, decay time constant; A, mean area of the EPC.
In the present study we demonstrate the regulatory cascade NA-β1 receptors-cAMP-PKA. Is this cascade operating only after a nerve stimulus, or is it acting permanently by aligning vesicles in an optimal release position? Regulation of the time lag between stimulus and vesicle fusion would imply phosphorylation and dephosphorylation of v- and t-SNARE proteins. Many other proteins proposed for fusion regulation may also undergo phosphorylation (Pfeffer, 1996; Fergestad et al. 1999). However, the speed of exocytosis in fast synapses such as the motor endplate would not allow enough time for a series of sequential protein interactions and PKA-dependent hydrolysis of ATP during nerve stimulation, but would provide time only for a rapid conformational change in fusion proteins during quantum release. The present experiments in which the early releasable pool of vesicles was exhausted during tetanic stimulation were aimed at distinguishing where phosphorylation via PKA takes place. The number of long-latency EPCs increased after tetanic stimulation, when new vesicles are apparently primed and docked, in the presence of a PKA inhibitor. However, this effect was readily reversible, and we can therefore speculate that, similarly to the already described phosphorylation of α-soluble NSF attachment protein (α-SNAP; Hirling & Scheller, 1996), PKA can phosphorylate release proteins when vesicles are already docked at the release zone. Some observations indicate that ATP-dependent priming of secretion occurs before calcium-triggered exocytosis in chromaffin and PC12 cells (Hay & Martin, 1992; Holz et al. 1992). As shown, the inhibition of PKA, even without any tetanic stimulation, increases the number of long-latency EPCs. This means that previously phosporylated release protein(s) can be slowly dephosphorylated by phosphatases in the absence of functional PKA, and this disqualifies these quanta for short-latency release. Yet another remote possibility is that without PKA-mediated phosphorylation, the turnover of otherwise fixed vesicles at docking sites increases and ‘fast’ vesicles are then replaced by ‘slow’ ones. This can be tested in frog terminals (e.g. using dye-loaded vesicles; Betz et al. 1993). Thus, exocytotic fusion may occur and be differentially regulated by phosphorylation at several stages in the process of transmitter release (Fesce, 1999).
Acknowledgments
We thank Dr James Dutt for reading the manuscript and valuable suggestions. Dr Robert Gaynulov is acknowledged for his participation in the creation of evaluation programmes. This work was supported by Grants EU ‘Nesting’, Grant Agency of the Czech Academy of Sciences A7101902, MSMT 113100003 and RFBR 99-04-48286.
REFERENCES
- Almers W. Exocytosis. Annual Reviews of Physiology. 1990;52:607–624. doi: 10.1146/annurev.ph.52.030190.003135. [DOI] [PubMed] [Google Scholar]
- Baldo G, Cohen I, Vander Kloot W. Estimating the time course of evoked quantal release at the frog neuromuscular junction using end-plate current latencies. Journal of Physiology. 1986;374:503–513. doi: 10.1113/jphysiol.1986.sp016094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett EE, Stevens CF. The kinetics of transmitter release at the frog neuromuscular junction. Journal of Physiology. 1972;227:691–708. doi: 10.1113/jphysiol.1972.sp010054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beavo JA, Rogers NL, Crofford OB, Hardman JG, Sutherland EW, Newman EV. Effects of xanthine derivatives on lipolysis and on adenosine 3′,5′-monophosphate phosphodiesterase activity. Molecular Pharmacology. 1970;6:597–603. [PubMed] [Google Scholar]
- Benoit P, Mambrini J. Modification of transmitter release by ions which prolong the presynaptic action potential. Journal of Physiology. 1970;210:681–695. doi: 10.1113/jphysiol.1970.sp009235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betz WJ, Ridge RMAP, Bewick GS. Comparison of FM 1–43 staining patterns and electrophysiological measures of transmitter release at the frog neuromuscular junction. Journal of Physiology, Paris. 1993;87:193–202. doi: 10.1016/0928-4257(93)90030-w. [DOI] [PubMed] [Google Scholar]
- Bronstein IN, Semendjaev KA. Handbook of Mathematics. Moscow: Science Publishing House; 1986. [Google Scholar]
- Bukcharaeva E, Kim K, Moravec J, Nikolsky E, Vyskocil F. Noradrenaline synchronizes evoked quantal release at frog neuromuscular junctions. Journal of Physiology. 1999;517:879–888. doi: 10.1111/j.1469-7793.1999.0879s.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bukcharaeva E, Samigullin D, Nikolsky E, Vyskočil F. Cyclic AMP synchronizes evoked quantal release at frog neuromuscular junctions. Physiological Research. 2000;49:475–479. [PubMed] [Google Scholar]
- Burn JH. The relation of adrenaline to acetylcholine in the nervous system. Physiological Reviews. 1945;25:377–394. [Google Scholar]
- Bykhovska M, Hackett JT, Worden K. Asynchrony of quantal events in evoked multiquantal responses indicates presynaptic quantal interaction. Journal of Neurophysiology. 1999;81:2234–2242. doi: 10.1152/jn.1999.81.5.2234. [DOI] [PubMed] [Google Scholar]
- Carlson S, Trauth K, Brooks W, Roszman T. Enhancement of beta-adrenergic-induced cAMP accumulation in activated T-cells. Journal of Cell Physiology. 1994;161:39–48. doi: 10.1002/jcp.1041610106. [DOI] [PubMed] [Google Scholar]
- Ceccarelli B, Hurlbut WL, Mauro A. Depletion of vesicles from frog neuromuscular junction by prolonged tetanic stimulation. Journal of Cell Biology. 1972;54:30–38. doi: 10.1083/jcb.54.1.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Dryden WF, Singh YN. Transduction of the modulatory effect of catecholamines at the mammalian motor neuron terminal. Synapse. 1991;7:93–98. doi: 10.1002/syn.890070202. [DOI] [PubMed] [Google Scholar]
- Chen BM, Grinnell AD. Kinetics, Ca2+dependence and biophysical properties of integrin-mediated mechanical modulation of transmitter release from frog motor nerve. Journal of Neurosciences. 1997;17:904–916. doi: 10.1523/JNEUROSCI.17-03-00904.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Alonzo A, Grinnell AD. Profiles of evoked release along the length of frog motor nerve terminals. Journal of Physiology. 1989;359:235–258. doi: 10.1113/jphysiol.1985.sp015583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davey DF, Bennett MR. Variation in the size of synaptic contacts along developing and mature motor terminal branches. Brain Research. 1982;281:11–22. doi: 10.1016/0165-3806(82)90108-0. [DOI] [PubMed] [Google Scholar]
- Del Castillo J, Katz B. Statistical factors involved in neuromuscular facilitation and depression. Journal of Physiology. 1954;124:574–585. doi: 10.1113/jphysiol.1954.sp005130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dostmann WR, Taylor SS, Genieser HG, Jastorff B, Doskeland SO, Ogreid D. Probing the cyclic nucleotide binding sites of cAMP-dependent protein kinases I and II with analogs of adenosine 3′,5′-cyclic phosphorothioates. Journal of Biological Chemistry. 1990;265:10484–10491. [PubMed] [Google Scholar]
- Fergestad T, Davis WS, Broadie K. The stoned proteins regulate synaptic vesicle recycling in the presynaptic terminal. Journal of Neuroscience. 1999;19:5847–5860. doi: 10.1523/JNEUROSCI.19-14-05847.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FernÁndez-ChacÓn R, Königstorfer A, Gerber SH, Garcia J, Matos M, Stevens CF, Rizo J, Rosemund C, Südhof TC. Synaprotagmin I functions as calcium regulator of release probability. Nature. 2001;410:41–49. doi: 10.1038/35065004. [DOI] [PubMed] [Google Scholar]
- Fesce R. The kinetics of nerve-evoked quantal secretion. Philosophical Translations of the Royal Society B. 1999;354:319–329. doi: 10.1098/rstb.1999.0383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giniatullin RA, Kheeroug LS, Vyskocil F. Modeling endplate current: dependence on quantum secretion probability and postsynaptic miniature current parameters. European Biophysics Journal. 1995;23:443–446. doi: 10.1007/BF00196832. [DOI] [PubMed] [Google Scholar]
- Hay JC, Martin TF. Resolution of regulated secretion into sequential MgATP-dependent and calcium-dependent stages mediated by distinct cytosolic proteins. Journal of Cell Biology. 1992;119:139–151. doi: 10.1083/jcb.119.1.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hidaka T, Kuriyama H. Effects of catecholamines on the cholinergic neuromuscular transmission in fish red muscle. Journal of Physiology. 1969;201:61–71. doi: 10.1113/jphysiol.1969.sp008742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirling H, Scheller RH. Phosphorylation of synaptic vesicle proteins: modulation of the alpha SNAP interaction with the core complex. Proceedings of the National Academy of Sciences of the USA. 1996;93:11945–11949. doi: 10.1073/pnas.93.21.11945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holz RW, Bittner MA, Senter RA. Regulated exocytotic fusion I: chromaffin cells and PC12 cells. Methods in Enzymology. 1992;219:165–178. doi: 10.1016/0076-6879(92)19019-3. [DOI] [PubMed] [Google Scholar]
- Jenkinson D, Stamenovic B, Whitaker B. The effect of noradrenaline on the end-plate potential in twitch fibres of the frog. Journal of Physiology. 1968;195:743–754. doi: 10.1113/jphysiol.1968.sp008486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz B, Miledi R. The measurement of synaptic delay, and the time course of acetylcholine release at the neuromuscular junction. Proceedings of the Royal Society B. 1965;161:483–495. doi: 10.1098/rspb.1965.0016. [DOI] [PubMed] [Google Scholar]
- Khirough L, Sokolova E, Giniatullin R, Afzalov R, Nistri A. Recovery from desensitization of neuronal nicotinic acetylcholine receptors of rat chromaffin cells is modulated by intracellular calcium through distinct second messengers. Journal of Neuroscience. 1998;18:2458–2466. doi: 10.1523/JNEUROSCI.18-07-02458.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuba K. Effects of catecholamines on the neuromuscular junction in the rat diaphragm. Journal of Physiology. 1970;211:551–570. doi: 10.1113/jphysiol.1970.sp009293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuba K, Tomita T. Noradrenaline action on nerve terminal in the rat diaphragm. Journal of Physiology. 1971;217:19–31. doi: 10.1113/jphysiol.1971.sp009557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurenza A, Khadelwal Y, De-Souza N, Rupp R, Metzger H, Seamon K. Stimulation of adenylate cyclase by water-soluble analogues of forskolin. Molecular Pharmacology. 1987;32:133–139. [PubMed] [Google Scholar]
- Majewski H, Barrington M. Second messenger pathways in the modulation of neurotransmitter release. In: Powis D, Bunn S, editors. Neurotransmitter Release and its Modulation. Cambridge, UK: Cambridge University Press; 1995. pp. 163–178. [Google Scholar]
- Mallart A. Presynaptic currents in frog motor endings. Pflügers Archiv. 1984;400:8–20. doi: 10.1007/BF00670529. [DOI] [PubMed] [Google Scholar]
- Martin AR. A further study of the statistical composition of the endplate potential. Journal of Physiology. 1955;130:114–112. doi: 10.1113/jphysiol.1955.sp005397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minenko ML, Magazanik LG. Phenomena of asynchronous evoked transmitter release at the neuro-muscular junction of the frog. Russian Journal of Physiology. 1986;18:346–354. [Google Scholar]
- Miyamoto M, Breckenbridge B. A cyclic adenosine monophosphate link in the catecholamine enhancement of transmitter release at the neuromuscular junction. Journal of General Physiology. 1974;63:609–624. doi: 10.1085/jgp.63.5.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nestler EJ, Greengard P. Protein phosphorylation and regulation of neuronal function. In: Siegler GJ, Agranoff BW, Albers RW, Molinoff PB, editors. Basic Neurochemistry: Molecular, Cellular and Medical Aspects. New York: Raven; 1989. pp. 378–398. [Google Scholar]
- Orbeli LA. Die sympatetische Innervation der Skelettmuskeln. Bulletin of the Institute of Science. 1923;6:194–197. [Google Scholar]
- Parnas I, Parnas H. Different mechanisms control the amount and time course of neurotransmitter release. Journal of Physiology. 1999;517:629. doi: 10.1111/j.1469-7793.1999.0629s.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawson PA, Grinnell AD, Wolowske B. Quantitative freeze-fracture analysis of the frog neuromuscular junction synapse-I. Naturally occurring variability in active zone structure. Journal of Neurocytology. 1998a;27:361–377. doi: 10.1023/a:1006942909544. [DOI] [PubMed] [Google Scholar]
- Pawson PA, Grinnell AD, Wolowske B. Quantitative freeze-fracture analysis of the frog neuromuscular junction synapse-II. Proximal-distal measurements. Journal of Neurocytology. 1998b;27:379–391. doi: 10.1023/a:1006995010453. [DOI] [PubMed] [Google Scholar]
- Pfefer SR. Transport vesicle docking: SNAREs and associates. Annual Reviews of Cell Development Biology. 1996;12:441–461. doi: 10.1146/annurev.cellbio.12.1.441. [DOI] [PubMed] [Google Scholar]
- Renger JJ, Ueda A, Atwood HL, Govind CK, Wu CF. Role of cAMP cascade in synaptic stability and plasticity: ultrastructural and physiological analyses of individual synaptic boutons in Drosophila memory mutants. Neuroscience. 2000;20:3980–3992. doi: 10.1523/JNEUROSCI.20-11-03980.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robitaille R, Tremblay J. Non-uniform release at the frog neuromuscular junction, evidence of morphological and physiological plasticity. Brain Research Reviews. 1987;12:95–116. doi: 10.1016/0165-0173(87)90019-1. [DOI] [PubMed] [Google Scholar]
- Ruzzier F, Scuka M. The effect of repetitive neuromuscular activity on the sensitivity of acetylcholine receptors. Pflügers Archiv. 1986;406:99–103. doi: 10.1007/BF00586669. [DOI] [PubMed] [Google Scholar]
- Shakiryanova DM, Zefirov AL, Nikolsky EE, Vyskocil F. The effect of acetylcholine and related drugs on currents at the frog motor nerve terminal. European Journal of Pharmacology. 1994;263:107–114. doi: 10.1016/0014-2999(94)90530-4. [DOI] [PubMed] [Google Scholar]
- Sou’cek B. Influence of latency fluctuations and the quantal process of transmitter release on the end-plate potential's amplitude distribution. Biophysical Journal. 1971;11:127–139. doi: 10.1016/S0006-3495(71)86202-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley EF. The calcium channel and the organization of the presynaptic transmitter release face. Trends in Neurosciences. 1997;20:404–409. doi: 10.1016/s0166-2236(97)01091-6. [DOI] [PubMed] [Google Scholar]
- Sulakhe P, Vo X. Regulation of phospholamban and troponin-I phoshphorylation in the intact rat cardiomyocytes by adrenergic and cholinergic stimuli: roles of cyclic nucleotides, calcium, protein kinases and phosphatases and depolarization. Molecular Cell Biochemistry. 1995;150:149, 103–126. doi: 10.1007/BF01076569. [DOI] [PubMed] [Google Scholar]
- Turkanis SA. Effects of muscle stretch on transmitter release at endplate of rat diaphragm and frog sartorius muscle. Journal of Physiology. 1973;230:391–403. doi: 10.1113/jphysiol.1973.sp010194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Kloot W. The regulation of quantal size. Progress in Neurobiology. 1991;36:93–130. doi: 10.1016/0301-0082(91)90019-w. [DOI] [PubMed] [Google Scholar]
- Vander Kloot W, Vander Kloot T. Catecholamines, insulin and ACTH increase quantal size at the frog neuromuscular junction. Brain Research. 1986;376:378–381. doi: 10.1016/0006-8993(86)90203-9. [DOI] [PubMed] [Google Scholar]
- Vizi S. Evidence that catecholamines increase acetylcholine release from neuromuscular junction through stimulation of alpha-1 adrenoreceptors. Naunyn-Schmiedeberg's Archives of Pharmacology. 1991;343:435–438. doi: 10.1007/BF00169543. [DOI] [PubMed] [Google Scholar]
- Weber T, Zemelman BV, McNew JA, Westermann B, Gmachl M, Parlati F, Sollner TH, Rothman JE. SNAREpins: minimal machinery for membrane fusion. Cell. 1998;92:759–772. doi: 10.1016/s0092-8674(00)81404-x. [DOI] [PubMed] [Google Scholar]
- Wiser O, Trus M, HernÁndez A, RenstrÖm E, Barg S, Rorsman P, Atlas D. The voltage sensitive Lc-type Ca2+ channel is functionally coupled to the exocytic machinery. Proceedings of the National Academy of Sciences of the USA. 1999;96:248–253. doi: 10.1073/pnas.96.1.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wessler J, Anschuetz S. Beta-adrenoreceptor stimulation enhances transmitter output from the rat phrenic nerve. British Journal of Pharmacology. 1988;94:669–674. doi: 10.1111/j.1476-5381.1988.tb11574.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zefirov AL. Transmitter release in proximal and distal parts of a nerve terminal of frog sartorius muscle. Neirofiziologya. 1983;15:36–370. (in Russian) [Google Scholar]
- Zefirov AL, Gafurov MS. The influence of the asynchroneity on the amplitude-time parameters of the evoked postsynaptic currents and potentials at the neuromuscular synapse. Fiziologiceski Zhournal. 1997;83:22–31. (in Russian) [PubMed] [Google Scholar]
- Zucker RS. Calcium- and activity-dependent synaptic plasticity. Current Opinion in Neurobiology. 1999;9:305–313. doi: 10.1016/s0959-4388(99)80045-2. [DOI] [PubMed] [Google Scholar]