

Abstract
Neurotoxins such as aconitine (AC) bind to receptor site 2 on voltage-gated sodium channels and modify channel kinetics. Although AC modification typically induces hyperpolarizing shifts in sodium channel activation, the effects on channel inactivation seem to vary depending on the tissue origin of the channel. In the present study, the α subunits of human heart (hH1) and rat skeletal muscle (μ1) sodium channels were transiently expressed in human embryonic kidney (HEK293t) cells. Whole-cell currents were examined before and after AC modification of the channels to determine whether the toxin had isoform-specific effects on channel kinetics. The magnitudes of the hyperpolarizing shifts in steady-state current activation and inactivation were similar for AC-modified hH1 and μ1 channels, and AC modification did not alter the voltage dependence of macroscopic current decay of either channel subtype. There were two notable differences between hH1 and μ1 channels after AC modification. First, the steady-state availability of AC-modified μ1 channels decreased by 5–10 % after very negative conditioning pulses. Second, AC-modified μ1 channels inactivated completely at all voltages, whereas AC-modified hH1 channels exhibited sustained inward currents at voltages near the threshold of current activation. Interestingly, AC-modified hH1 channels inactivated completely if the external solution did not contain sodium ions. The data demonstrate that AC modification affects the activation of hH1 and μ1 channels similarly but affects inactivation of the two channels distinctly. The results also imply that the reduced inactivation of AC-modified hH1 channels at least partially depends on the presence of extracellular sodium.
Voltage-gated sodium channels are primarily responsible for action potential propagation in excitable tissues. As a crucial component in the physiology of excitable tissues, sodium channels are primary targets for several naturally occurring neurotoxins. To date, sodium channels have been shown to possess at least six receptor sites for neurotoxins that inhibit or modify channel function. Batrachotoxin (BTX), aconitine (AC) and veratridine are lipophilic compounds that markedly alter sodium channel kinetics by binding to receptor site 2 (Strichartz et al. 1987; Catterall et al. 1992; Ulbricht, 1998). In general, site 2 toxins elicit hyperpolarizing shifts in the voltage dependence of channel activation, thereby inducing repetitive action potentials in excitable tissues. Under voltage clamp conditions, site 2 toxins shift sodium current activation towards more hyperpolarized potentials and increase sodium channel permeability to ion species that are nominally permeable through unmodified channels. In addition to the effects on channel activation and permeability, the toxins often reduce or eliminate fast inactivation of open channels.
One of the interesting differences among site 2 toxins is the degree to which they alter channel inactivation. For example BTX virtually eliminates fast inactivation of neuronal (Mozhaeva et al. 1986; Wang & Wang, 1994) or skeletal muscle sodium channels (Wang & Wang, 1998) because sodium currents under voltage clamp may be sustained for several seconds with little decrement. In contrast, AC (Schmidt & Schmitt, 1974) or veratridine (Barnes & Hille, 1988; Ulbricht, 1998) modification elicits a less dramatic reduction of fast inactivation. Although site 2 neurotoxins share an overlapping binding region (Catterall, 1980), the fact that poison dart frogs (Phyllobates terribilis), which secrete BTX, are insensitive to BTX but remain sensitive to other site 2 toxins (Daly et al. 1980) suggests that the binding sites for the toxins are not identical. In addition, the different effects on fast inactivation could suggest that there are subtle differences between BTX and other site 2 toxins such as AC in the mechanism of altering channel behaviour. An interesting characteristic of AC modification is that the extent of reduced inactivation differs depending on the tissue origin of the channel. For example AC more markedly reduces the inactivation of frog node of Ranvier sodium channels (Schmidt & Schmitt, 1974; Mozhaeva et al. 1980) compared with the reduced inactivation of sodium channels in frog skeletal muscle (Campbell, 1982), mouse heart (Nilius et al. 1986), or sodium channels cloned from rat brain (Rao & Sikdar, 2000). Furthermore, AC-modified sodium channels in neuroblastoma cells (Grishchenko et al. 1983) inactivate nearly completely at all voltages.
The fact that AC modification has different effects on sodium channels from different tissues raises the possibility that the neurotoxin has distinct effects on different sodium channel isoforms. Alternatively, the observed differences in channel modification could be explained by subtle differences in experimental protocol. The focus of the present study was to compare the effects of AC modification on cloned sodium channels from human heart (hH1, Gellens et al. 1992) and rat skeletal muscle (μ1, Trimmer et al. 1989) under identical conditions. AC modification of hH1 and μ1 channels had similar effects on several properties of the whole cell currents. First, intracellular or extracellular application of AC were both effective in modifying channel kinetics. Second, as described for sodium channels from native tissues, AC modification greatly reduced the current amplitude and shifted the voltage dependence of current activation towards more negative voltages. Third, AC modification of hH1 and μ1 channels elicited similar negative shifts in the midpoint voltages of steady-state inactivation. Fourth, the voltage dependence of macroscopic current decay did not change after AC modification. Despite these similarities, there were two obvious differences between AC-modified hH1 and μ1 channels. First, the steady-state availability (h∞) of AC-modified μ1 channels decreased by 5 to 10 % after conditioning pulses ranging from −140 to −120 mV (normalized to the availability at −160 mV). Second, AC-modified hH1 currents exhibited incomplete inactivation during voltage clamp steps between −90 to −70 mV but inactivated completely at voltages more positive than −60 mV. In contrast, AC-modified μ1 currents inactivated nearly completely at all voltages. Unexpectedly, AC-modified hH1 currents inactivated completely at all voltages when the external solution did not contain sodium ions. The other changes in channel kinetics associated with AC modification, such as the hyperpolarizing shifts in channel activation and steady-state inactivation, still occurred in the absence of external sodium. Together, these data suggested that AC modification of hH1 or μ1 sodium channels elicited similar changes in channel activation but distinct changes in channel inactivation. Finally, the data demonstrated that external sodium ions influence the completeness of open channel inactivation of AC-modified hH1 channels.
METHODS
Transient transfection of HEK293t cells
Human embryonic kidney (HEK293t) cells were transfected with hH1 (Gellens et al. 1992) or μ1 (Trimmer et al. 1989) channel plasmid (2–4 μg) and reporter plasmid CD8-pih3m (1 μg) by the calcium phosphate precipitation method (Cannon & Strittmatter, 1993) as described previously (Wright et al. 1997, 1999). The transfected cells were replated onto 35 mm culture dishes and used for experiments up to 3 days after plating. Individual transfected cells that expressed the CD8 antigen, as determined by binding CD8 Dynabeads (Dynal, Lake Success, NY, USA) were selected for whole-cell patch clamp experiments. The hH1 clone was a gift from Dr Roland Kallen, and the μ1 clone, HEK293t cell line and CD8-pih3m plasmid were gifts from Drs Sho-Ya Wang and Ging Kuo Wang.
Solutions and chemicals
Three extracellular solutions, containing 0, 65, or 130 mm sodium, were used to perfuse the HEK cells. Respectively, these solutions contained (mm): (1) 0 NaCl, 150 choline chloride, 2 CaCl2 and 10 Hepes; (2) 65 NaCl, 85 choline chloride, 2 CaCl2 and 10 Hepes; or (3) 130 NaCl, 20 choline chloride, 2 CaCl2 and 10 Hepes. All external solutions were titrated with tetramethyl ammonium hydroxide to pH 7.4. In most of the experiments the pipette solution contained (mm): 100 NaF, 30 NaCl, 10 EGTA and 10 Hepes. In a few experiments a low sodium pipette solution was used that contained (mm): 10 NaCl, 120 CsF, 10 EGTA and 10 Hepes. The pipette solutions were titrated with CsOH to pH 7.2. BTX was added to the pipette solution to obtain a final concentration of 5 μm. Aconitine (Sigma, St Louis, MO, USA) was dissolved in 100 % ethanol and was stored as 10 mm aliquots at −80 °C. The 10 mm aconitine stock was diluted at the appropriate concentration in extracellular or intracellular solution on the day of the experiment.
Whole-cell voltage clamp and data analysis
Glass electrodes with resistances ranging from 0.5 to 1.0 MΩ were used to study macroscopic hH1 or μ1 sodium currents under whole-cell voltage clamp conditions (Hamill et al. 1981). Command voltages were programmed by pCLAMP 7.0 software (Axon Instruments, Burlingame, CA, USA) and were delivered by a Warner PC501A voltage clamp (Warner Instrument Corporation, Hamden, CT, USA). The data were sampled at 50 kHz and filtered at 5 kHz. The holding potential for all experiments was −140 mV and all experiments were conducted at room temperature (21 ± 2 °C). As described previously (Wright et al. 1997, 1999), data acquisition did not begin until 25–30 min after establishing whole-cell conditions such that the steady-state inactivation curve would have shifted by about 5–7 mV during the time course (20–30 min) of cell dialysis (Wang et al. 1996). Most of the capacitance current was cancelled by the voltage clamp circuitry, and the remaining capacitance artefact and the leakage current were subtracted by the P/-4 method. Voltage errors were ≤ 4 mV after series resistance compensation.
To determine the midpoint voltage of steady-state activation, peak currents were measured during voltage clamp steps ranging from −120 to +50 mV, and the data were plotted as the normalized conductance:
where INa was the peak sodium current, V was the amplitude of the voltage step, and Vrev was the reversal potential determined for each cell. These data were fitted with an empirical Boltzmann function:
where V0.5 was the midpoint voltage of the function and k was the slope factor in mV/e-fold change in current. As a precaution for spatial voltage control, the only experiments presented in the study were those in which the slope factor of the Boltzmann function was ≥ 6 mV.
Steady-state fast inactivation of the current was determined by delivering 100 ms conditioning pulses to potentials ranging from −160 to −30 mV and the available current was measured during a test pulse to +30 mV. The peak current measured during the test pulse was normalized to the peak current recorded following the conditioning pulse to −160 mV. These data were fitted with a Boltzmann function (1/(1 + exp(V - V0.5)/k)) to determine V0.5 and k values as described above for normalized conductance. Least squares curve fitting was performed with Microcal Origin software (Northampton, MA, USA). Statistical comparisons (Student's t test, one-way ANOVA) were performed using SigmaStat (Jandel Scientific Software, San Rafael, CA, USA), and P values of < 0.05 were considered statistically significant. Plotted data are presented as means ± s.e.m.
RESULTS
Modification of hH1 and μ1 currents by internal application of BTX or AC
In whole-cell studies of sodium channel modification by site 2 neurotoxins, the toxins are often applied internally via the patch pipette because the toxins gain access to the receptor of open channels from the cytoplasm or through lipid regions of the channel. Figure 1A shows BTX-modified outward hH1 and μ1 currents in a reversed sodium gradient (0 mm [Na+]/130 mm [Na+]i) after channel modification by intracellular application of 5 μm BTX. After delivery of 1000 depolarizing pulses to modify the channels (not shown) the currents did not inactivate completely during a 2 s voltage clamp step to +30 mV. The hH1 current decayed slightly and reached a steady state, whereas the μ1 current decayed very little during the pulse. The small decrease (∼20–25 %) in the AC-modified hH1 current during a long pulse suggested that BTX modification did not completely eliminate slow inactivation of hH1 channels. This was an interesting observation because slow inactivation of wild-type hH1 channels under similar conditions has a much slower onset, is much less voltage dependent, and is far less complete compared with slow inactivation of wild-type μ1 channels (O'Reilly et al. 1999). These data implied that BTX modification elicits identifiable alterations in the inactivation kinetics of hH1 and μ1 channels. Nevertheless, BTX modification of either isoform under these conditions virtually eliminated fast inactivation from the open state.
Figure 1. Modification of hH1 and μ1 channels by internal application of 5 μm BTX (A) or 30 μm AC (B, C).
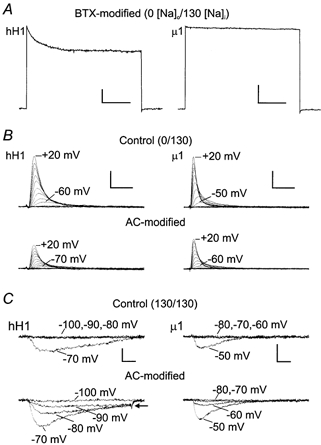
A, BTX-modified hH1 (left) and μ1 (right) currents after delivery of 1000 pulses to +30 mV. Modification of the channels by BTX virtually eliminated fast inactivation during a 2 s pulse to +30 mV. External and internal sodium concentrations were 0 and 130 mm, respectively. Scale, 500 pA, 500 ms. B, outward currents through hH1 (left) and μ1 (right) channels during voltage clamp steps ranging from −100 to +20 mV. The upper traces in B are currents recorded before channel modification, and the lower traces are currents from the same cell after delivery of 1000 pulses to +20 mV at 2 Hz. AC modification of the channels shifted the voltage dependence of current activation by about −10 mV. External and internal sodium concentrations were 0 and 130 mm, respectively. Scale, 1 nA, 2 ms. C, inward currents through hH1 and μ1 channels near the threshold of current activation. External and internal sodium concentrations were both 130 mm, and the channels were modified as described in B. AC modification shifted the voltage dependence of current activation of both isoforms and hH1 channels exhibited a sustained inward current during voltage steps ranging from −90 to −70 mV (arrow). In contrast, AC modified μ1 channels inactivated completely near the threshold of current activation. Scale, 400 pA, 2 ms.
Figure 1B shows that AC modification of hH1 and μ1 channels in 0 mm external sodium solution elicited shifts in current activation but failed to modify the inactivation of hH1 or μ1 currents. In these experiments, the pipette solution contained 30 μm AC. The upper traces are the control records during 15 ms voltage steps from the holding potential to voltages ranging from −120 to +20 mV. The hH1 current activated near −60 mV whereas the μ1 current activated near −50 mV. The lower traces are currents from the same cells after delivery of 1000 pulses (2 Hz) to +20 mV to modify the channels. AC modification of the channels decreased the amplitude of the current and shifted current activation in the hyperpolarizing direction by ∼10 mV. Although AC modification induced a notable shift in the activation of hH1 and μ1 currents, inactivation of the currents was complete at all voltages.
Because AC reduces the inactivation of inward sodium currents in frog heart cells (Nilius et al. 1986) and frog skeletal muscle (Campbell, 1982), a few experiments were conducted using a symmetrical sodium gradient (130 mm [Na+]o/130 mm [Na+]i). The channels were again modified by including 30 μm AC in the patch pipette and by delivering 1000 depolarizing pulses at 2 Hz. Before AC modification, hH1 and μ1 currents activated near −70 mV and −50 mV, respectively (Fig. 1C, upper traces). After AC modification, activation of the hH1 current shifted in the hyperpolarizing direction to −90 mV and inactivation of the current was incomplete during 15 ms voltage steps near the threshold of current activation (Fig. 1C, arrow). Activation of μ1 currents shifted from −50 mV in control solution to −60 mV after AC modification, but the AC-modified μ1 current inactivated completely. Although AC modification again induced hyperpolarizing shifts in hH1 and μ1 current activation, these data suggested that the inactivation of AC-modified hH1 channels became reduced only if the external solution contained sodium ions. In contrast, the presence of extracellular sodium had no obvious effect on the inactivation of AC-modified μ1 channels.
Modification of the channels by external application of AC
Although internally applied AC and delivery of up to 1000 depolarizing pulses did modify the channels, external perfusion of the cells with saline containing 100 μm AC and delivery of ≥ 50 depolarizing pulses allowed for more stable recordings and more consistent modification of the current. As shown in the insets of Fig. 2, modification of hH1 and μ1 channels by delivering 100 depolarizing pulses in external saline containing 0 mm sodium and 100 μm AC resembled the modification of the channels when AC was included in the pipette solution (Fig. 1B). That is, AC modification reduced the current amplitude and the currents inactivated completely at all voltages. AC modification shifted the activation thresholds of hH1 (Fig. 2A) and μ1 (Fig. 2B) currents towards more hyperpolarized potentials by 15–20 mV.
Figure 2. Normalized current-voltage relationships of hH1 (A) and μ1 (B) channels in 0 mm external sodium.
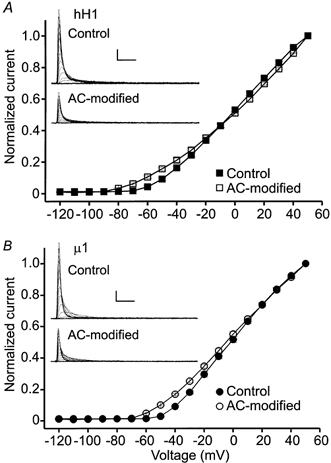
The channels were modified by perfusing the cells with external solution containing 100 μm AC and delivering ≥ 50 depolarizing pulses to +20 mV. The currents in the insets are from representative experiments. Scale, 1 nA, 2 ms. A, current-voltage relationship of hH1 channels before (▪, n = 13) and after (□, n = 11) AC modification. B, current-voltage relationship of μ1 channels before (•, n = 6) and after (○, n = 6) AC modification. AC modification shifted the voltage dependence of current activation of both channel isoforms by −15 to −20 mV.
The data shown in Fig. 1C provided the initial suggestion that there were differences between hH1 and μ1 currents after AC modification if the extracellular solution contained sodium ions. To further investigate the role of external sodium, AC modification of hH1 and μ1 currents were compared using external solutions containing 65 mm or 130 mm sodium (Fig. 3). The upper traces are control hH1 (left) and μ1 (right) currents recorded in 65 mm (Fig. 3A) or 130 mm sodium (Fig. 3B). The 15 ms step commands from the holding potential were to potentials ranging from −120 to +50 mV. The lower traces in Fig. 3A and B are the currents recorded from the same cells after external perfusion of saline containing 100 μm AC and delivery of 100 depolarizing pulses to modify the channels. AC modification of the channels in either external sodium concentration again decreased the amplitude of the current. Inactivation of the hH1 current was incomplete during voltage steps ranging from −90 to −70 mV (Fig. 3A and B, arrows), whereas the current evoked during voltage steps more positive than −60 mV inactivated completely. After AC modification, the μ1 current inactivated nearly completely at all voltages in either external solution. These data were consistent with the results shown in Fig. 1 and indicated that in the presence of external sodium ions AC-modified hH1 channels did not inactivate completely from the open state at voltages near the activation threshold. In contrast, external sodium seemed to have little influence on the inactivation of AC-modified μ1 channels. The plots in Fig. 3C and D show the normalized current-voltage relationships in external solutions containing 65 and 130 mm sodium, respectively. The data were normalized according to the outward current evoked at +50 mV in order to compare the shifts in current activation. In either external solution, AC modification shifted current activation of the two channel subtypes by −25 to −30 mV, and the reversal potential for sodium changed very little.
Figure 3. Current-voltage relationships of hH1 and μ1 channels in solutions containing external sodium.
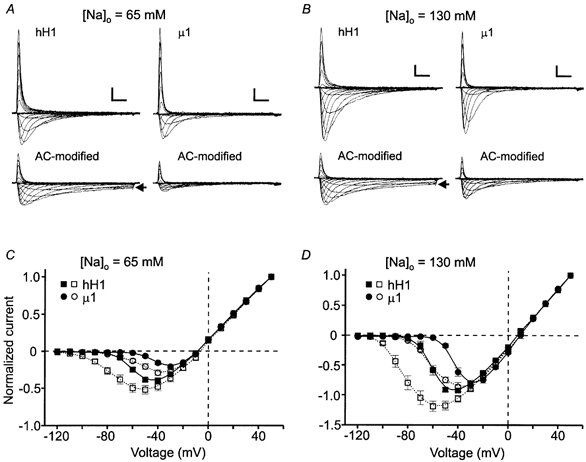
The channels were modified as described in Fig. 2. A and B, representative hH1 (left) and μ1 (right) currents recorded from cells bathed in 65 (A) or 130 mm (B) external sodium solution. Note the sustained inward hH1 currents after AC modification (arrows in A and B). Scale in A and B, 1 nA, 2 ms. C, normalized current-voltage relationships of hH1 (▪, □) and μ1 (•, ○) channels recorded in external solution containing 65 mm sodium before (filled symbols; hH1, n = 8; μ1, n = 6) and after (open symbols; hH1, n = 7; μ1, n = 5) AC modification. D, normalized current-voltage relationships of hH1 (▪, □) and μ1 (•, ○) channels recorded in external solution containing 130 mm sodium before (filled symbols; hH1, n = 8; μ1, n = 6) and after (open symbols; hH1, n = 8; μ1, n = 6) AC modification. The data for each cell were normalized according to the peak current amplitude at +50 mV. Compared with the control data, AC modification shifted the voltage dependence of current activation by −25 to −30 mV.
The plots in Fig. 4A (65 mm [Na+]o) and B (130 mm [Na+]o) show the normalized conductance-voltage relationships before and after AC modification. A standard Boltzmann function was used to determine the midpoint voltage (V0.5) of current activation and the slope factor (k) for each of the data sets. In 65 mm external sodium, the control V0.5 values were 4–5 mV more positive and the k values were slightly larger compared with the control V0.5 and k values measured in 130 mm external sodium, but these changes were not significant (P > 0.05, Student's t test) for either hH1 or μ1. AC modification of the channels induced hyperpolarizing shifts in V0.5 and decreased the voltage dependence of current activation (Fig. 4A and B). Compared with the control data in 65 mm and 130 mm external sodium, AC modification shifted the V0.5 values of hH1 conductance by 11 mV and 16 mV, respectively, and shifted the V0.5 values of μ1 conductance by 9 mV and 12 mV, respectively (Table 1). The negative shifts in the V0.5 values after AC modification in either external sodium solution were significant (P < 0.05) for both channel subtypes. Note, however, that the overall effect of increasing the external sodium concentration on the V0.5 values of normalized conductance after AC modification was relatively small for both hH1 (6 mV) and μ1 (3 mV) given that the control V0.5 values were 4–5 mV more negative in the higher external sodium concentration. AC modification also decreased the voltage dependence of channel activation as indicated by the increased k values. In 65 mm external sodium, AC modification increased the k values of hH1 and μ1 normalized conductance by 2 and 3 mV per e-fold change in current, respectively, and in 130 mm external sodium the increases were 4 and 5 mV per e-fold change in current, respectively. Together, these data suggested that AC modification of hH1 and μ1 channels elicited similar changes in the voltage dependence of current activation (i.e. normalized conductance) that were relatively independent of external sodium concentration. The major difference between the channels was that AC modification reduced the inactivation of the hH1 current but had little influence on the inactivation of the μ1 current.
Figure 4. Normalized conductance-voltage relationships in external solutions containing 65 (A) or 130 mm (B) sodium before (filled symbols) and after (open symbols) channel modification by 100 μm AC.
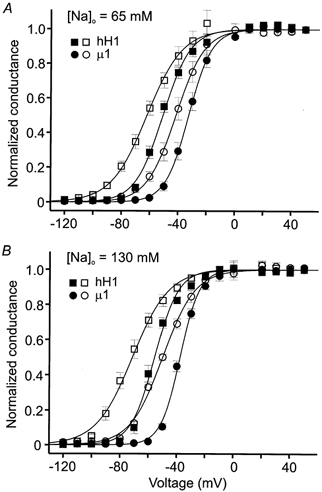
Conductance (gm) measurement and subsequent fit with an empirical Boltzmann function are described in Methods. A, normalized membrane conductance of hH1 (▪, □) and μ1 (•, ○) channels in 65 mm external sodium solution. B, normalized membrane conductance in 130 mm external sodium solution. Data in A and B are from the same cells used to plot the current-voltage relationships in Fig. 3. The midpoint voltages (V0.5), slope factors (k) of the fitted Boltzmann function, and number of cells (n) for each data set are listed in Table 1.
Table 1.
V0.5 and k values of steady-state current activation before (control) and after ACmodification of the channels in external solution containing 65 or 130 mM sodium
| Control | AC-modified | ||||
|---|---|---|---|---|---|
| V0.5 (mV) | k (mV) | V0.5 (mV) | k (mV) | ||
| 65 mNa+ | hH1 | −50.3 ± 1.6 | 9.7 ± 0.9 (8) | −61.2 ± 2.6* | 12.9 ± 1.3 (7) |
| μ1 | −32.4 ± 2.0 | 7.7 ± 0.3 (6) | −41.5 ± 2.9* | 9.7 ± 0.3* (5) | |
| 130 mNa+ | hH1 | −54.6 ± 1.5 | 8.3 ± 0.6 (8) | −71.1 ± 2.5* | 12.3 ± 1.1* (8) |
| μ1 | −37.6 ± 1.5 | 6.7 ± 0.4 (6) | −49.6 ± 1.4* | 11.6 ± 0.8* (6) | |
P < 0.05 (Student's t test) compared with control data in the same external sodium concentration.
External sodium has little influence on steady-state inactivation of AC-modified channels
Figure 5 shows the effect of AC modification on the normalized steady-state inactivation curves of hH1 (Fig. 5A) and μ1 (Fig. 5B) channels in 0, 65 and 130 mm external sodium solutions. To determine steady-state availability of the current, the cells were given 100 ms conditioning pulses to the voltages indicated on the abscissa and the available current was measured during a test pulse to +30 mV. The filled symbols in Fig. 5A and B are the control steady-state inactivation data in each external sodium concentration, whereas the open symbols are the steady-state inactivation data after AC modification. The channels were modified by perfusing the cells with saline containing 100 μm AC and then delivering 75–100 pulses to ≥ +10 mV. External sodium concentration did not significantly affect the control V0.5 and k values of steady-state inactivation of either channel (P > 0.05, one way ANOVA, Table 2). In all three external solutions, AC modification elicited significant (P < 0.05) hyperpolarizing shifts in steady-state inactivation compared with the control data. The only significant change in k value was in the comparison between control and AC-modified μ1 currents in 0 mm external sodium. One interesting alteration in μ1 channel behaviour after AC modification was a 5–10 % decrease in available current between −140 and −120 mV (Fig. 5B, arrow). At conditioning voltages more positive than −120 mV, steady-state inactivation of modified channels progressed to completion near −70 mV.
Figure 5. Normalized steady-state inactivation of hH1 (A) and μ1 (B) channels before (filled symbols) and after (open symbols) channel modification by 100 μm aconitine.
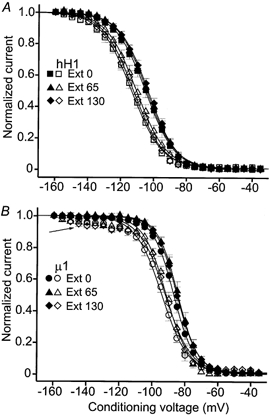
The pulse protocol for determination of steady-state availability and the Boltzmann equation used to fit the data are described in Methods. A, steady-state inactivation of hH1 channels in 0 mm (▪, □), 65 mm (▴, ▵) and 130 mm (♦, ⋄) external sodium solution. B, steady-state inactivation of μ1 channels in 0 mm (•, ○), 65 mm (▴, ▵) and 130 mm (♦, ⋄) external sodium solution. Note the small amount of reduced availability of AC-modified μ1 channels after conditioning pulses ranging from −140 to −120 mV (arrow). The maximum value of available current in the fitted Boltzmann function was not adjusted to reflect the small reduction in current availability in the negative voltage range. The midpoint voltages (V0.5), slope factors (k) of the fitted Boltzmann function and number of cells (n) for each data set in A and B are listed in Table 2.
Table 2.
V0.5 and k values of steady-state inactivation before (control) and after AC modification in external solutions containing 0, 65, or 130 mM sodium
| Control | AC-modified | ||||
|---|---|---|---|---|---|
| V0.5 (mV) | k (mV) | V0.5 (mV) | k (mV) | ||
| 0 mNa+ | hH1 | −105.2 ± 2.0 | 8.7 ± 0.1 (6) | −111.8 ± 2.3* | 9.0 ± 0.3 (6) |
| μ1 | −87.0 ± 1.1 | 6.5 ± 0.5 (6) | −94.3 ± 1.2* | 8.2 ± 0.6* (5) | |
| 65 mNa+ | hH1 | −104.2 ± 1.1 | 8.8 ± 0.2 (9) | −110.5 ± 1.8* | 9.1 ± 0.4 (7) |
| μ1 | −84.4 ± 1.4 | 6.1 ± 0.2 (5) | −91.6 ± 1.2* | 7.0 ± 0.3 (7) | |
| 130 mNa+ | hH1 | −103.8 ± 1.2 | 8.7 ± 0.4 (6) | −112.0 ± 1.2* | 9.0 ± 0.4 (6) |
| μ1 | −85.0 ± 1.6 | 6.3 ± 0.4 (6) | −92.2 ± 1.4* | 7.8 ± 0.6 (8) | |
P < 0.05 (Student's t test) compared with control data in the same external sodium concentration.
AC modification does not affect the voltage dependence of current decay during open channel inactivation
The data shown in Fig. 1 and Fig. 3 clearly demonstrated that AC modification of hH1 channels reduced open channel inactivation if the external solution contained sodium ions. To examine this further, the decaying phase of hH1 and μ1 currents were measured to determine whether AC modification affected the voltage dependence of channel inactivation. The inset in Fig. 6 shows representative control and AC-modified hH1 current traces (same cell) near the threshold of current activation. Although AC modification reduced the inactivation of hH1 channels, as demonstrated by the sustained inward currents, the fast time constants of current decay during voltage steps to −70 and −60 mV were similar to the decay rates before AC modification. The plot in Fig. 6 shows the mean decay rates of macroscopic hH1 or μ1 currents in 130 mm external sodium solution. The decay of hH1 current was best fitted by the sum of two exponential functions, whereas the decay of μ1 current was best fitted by a single exponential (Wang et al. 1996). AC modification of the channels did not alter the fast or slow time constant of hH1 current inactivation or the single time constant of μ1 current inactivation. Similar results were obtained for both channel subtypes in 0 and 65 mm external sodium solutions (data not shown). The fact that AC modification had little effect on the inactivation rate of μ1 currents was not particularly surprising given that there was no obvious reduction in channel inactivation. In contrast, inactivation of the hH1 current was obviously reduced after AC modification, yet these data indicated that AC modification did not alter hH1 current decay under these conditions.
Figure 6. AC modification of hH1 (▪, □, ▴, ▵) and μ1 (•, ○) does not affect the voltage dependence of macroscopic current decay in external solution containing 130 mm sodium.
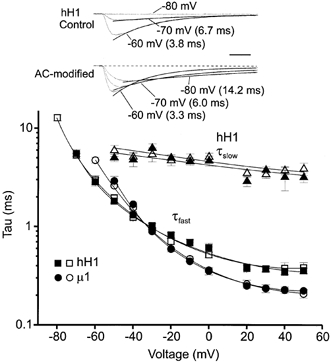
The decay of hH1 current was often best fitted by the sum of two exponentials: y = A1exp(-t/τ1) + A2exp(-t/τ2); the triangles in the plot correspond to the slow time constant of hH1 inactivation. The decay of μ1 current was best fit by a single exponential: y = A1exp(-t/τ1). The decay of macroscopic currents was measured before (filled symbols) and after (open symbols) modification of the channels by 100 μm AC. The traces in the inset are from a representative experiment on hH1 channels. The dashed line in the lower set of traces denotes the baseline current level. Scale, 2 ms.
As an alternative means of examining the reduction of open channel inactivation, peak current amplitudes were measured during the last 14–15 ms of the voltage clamp steps. The 0 mm external sodium data were normalized by dividing the current amplitude at the end of the step commands by the peak outward current evoked during steps to −20 mV (hH1) or −10 mV (μ1). The 65 and 130 mm external sodium data were normalized by dividing the amplitude of the current at the end of the step commands by the peak amplitude of the largest inward current. Before AC modification, the currents at the end of the step commands in all three external solutions were typically ≤ 5 % of the peak outward (Fig. 7A, filled symbols) or peak inward (Fig. 7B and C, filled symbols) current to which they were compared. AC modification did not induce a sustained outward hH1 or μ1 current in external solution containing 0 mm external sodium (Fig. 7A, open symbols). AC modification of μ1 channels in 65 mm external sodium (Fig. 7B) induced a sustained inward current during the voltage step to −60 mV that was ∼10 % of the largest inward current, and in 130 mm external sodium (Fig. 7C) the sustained inward current during the voltage step to −70 mV was ∼12 % of the largest inward current. Although the current records did not obviously indicate that AC modification reduced the inactivation of μ1 channels, these data suggested that there was a small amount of sustained current near the threshold of current activation. AC modification of hH1 channels elicited a more striking reduction in open channel inactivation if the external solution contained sodium. In external solution containing 65 or 130 mm sodium, the mean amplitude of the sustained inward current during the voltage clamp step to −80 mV was ∼25 % of the largest inward current. Thus, an increase in the external sodium concentration from 65 to 130 mm did not enhance the magnitude of the sustained AC-modified hH1 current. These data further confirmed that AC modification elicits a more pronounced reduction in the inactivation of hH1 channels compared with μ1 channels and that external sodium ions influence the inactivation of AC-modified hH1 channels.
Figure 7. Normalized measures of sustained currents in 0 mm (A), 65 mm (B) and 130 mm (C) external sodium solution.
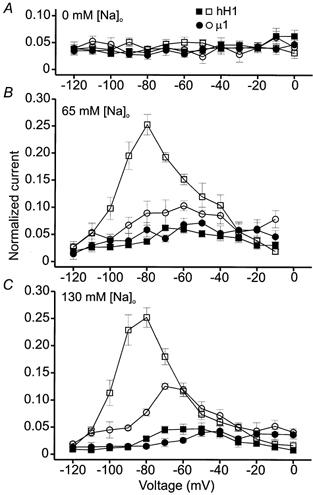
The filled symbols in each panel are the control hH1 (▪) and μ1 (•) data, whereas the open symbols are after AC modification. In A, the current amplitudes during the last millisecond of the 15 ms voltage steps were normalized to the peak current evoked during voltage steps to −20 mV (hH1) or −10 mV (μ1). At these voltages the peak outward current was about 40 % of the normalized maximum current at +50 mV (see Fig. 2). In B and C, the currents at the end of the voltage steps were normalized to the peak amplitude of the largest inward current. A, normalized measures of sustained outward currents in 0 mm external sodium. At all voltages the sustained outward current before and after AC modification was typically ≤5 % of the peak outward current at the test voltage. B, sustained inward currents in 65 mm external sodium. Before AC modification, sustained inward hH1 or μ1 currents were typically ≤ 5 % of the peak inward current. The largest sustained inward current through AC-modified μ1 channels occurred during a voltage step to −60 mV and was ∼10 % of the peak inward current; the largest sustained inward hH1 current occurred during a voltage step to −80 mV and was ∼25 % of the peak inward current. C, sustained inward currents in 130 mm external sodium. As described in B, sustained currents before AC modification were typically ≤ 5 % of the peak inward current. The largest sustained inward current through AC-modified μ1 channels occurred during a voltage step to −70 mV and was ∼12 % of the peak inward current; the largest sustained inward hH1 current occurred during a voltage step to −80 mV and was ∼25 % of the peak inward current. Data are from the same cells used to plot current-voltage relationships in Figs 2 and 3.
High intracellular sodium concentration does not affect the completeness of inactivation
To determine whether the high internal sodium concentration (130 mm) used in these studies prevented a reduction in inactivation of AC-modified μ1 channels, AC modification of the channels using 65 mm external and 10 mm internal sodium solutions was examined (Fig. 8). AC modification under these conditions elicited a sustained hH1 current (Fig. 8A, arrow) near the threshold of current activation but again failed to markedly alter the inactivation of μ1 currents (Fig. 8B). The normalized current amplitudes during the final 14–15 ms of the voltage clamp steps (as described in Fig. 7) were ≤ 5 % of the largest peak inward current before AC modification (Fig. 8C, filled symbols). After AC modification, the normalized amplitudes of sustained inward hH1 and μ1 currents resembled the data acquired when the internal sodium concentration was 130 mm. The largest sustained hH1 currents were ∼28 % of the largest peak inward current and occurred during voltage clamp steps to −80 and −70 mV. The largest sustained μ1 currents were ∼10 % of the largest peak inward current and occurred during the voltage step to −70 mV. These data showed that a high intracellular sodium concentration did not inhibit AC from reducing the inactivation of μ1 channels and that the increase in inward driving force on sodium did not further increase the sustained inward current through AC-modified hH1 channels.
Figure 8. Modification of hH1 and μ1 channels in 65 mm external and 10 mm internal sodium solutions.
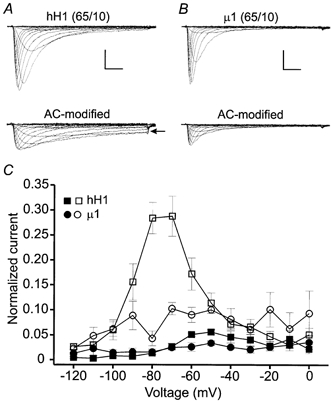
Under these conditions the channels were modified by perfusing the cells with external solution containing 100 μm AC and delivering 100 pulses to −30 mV. A, hH1 current traces before (upper) and after (lower) channel modification by external perfusion of 100 μm AC. AC modification elicited sustained inward currents (arrow) at voltages near the activation threshold. B, μ1 current traces before and after AC modification. Scale in A and B, 2 nA, 2 ms. C, normalized measures of sustained inward currents in 65 mm external and 10 mm internal sodium solutions. The data were normalized as described in Fig. 7B and C. In control saline, the amplitudes of hH1 (n = 4) and μ1 (n = 3) currents at the end of the voltage step commands were ≤ 5 % of the peak inward current. AC modification of hH1 channels (□) elicited a sustained inward current at −80 and −70 mV that was 28 % of the peak inward current (n = 4); the largest sustained inward current of AC-modified μ1 channels (○) was 10 % of the peak inward current and occurred during a voltage step to −70 mV (n = 3).
Reduction of hH1 channel inactivation does not require external sodium during channel modification
The data shown in previous figures indicated that AC modification of hH1 channels elicited a non-inactivating current at voltages near the threshold of current activation only if the external solution contained sodium. To examine the possibility that external sodium must be present during AC modification in order to reduce open channel inactivation, the modification protocol was performed in 0 mm external sodium and the currents of modified channels in 0 mm external sodium and then in 130 mm external sodium were recorded (Fig. 9, n = 4 cells). The activation thresholds of the control and AC-modified hH1 currents were similar to the mean data shown in Fig. 2. That is, the control hH1 current activated near −60 mV (Fig. 9A), whereas the AC-modified current activated near −70 mV (Fig. 9B). As shown in Fig. 9C, subsequent perfusion of the cell with external solution containing 130 mm sodium elicited a sustained inward current during voltage steps ranging from −90 to −70 mV. These data indicated that the presence of external sodium was not required during AC modification in order to induce sustained inward currents in external sodium solutions.
Figure 9. Sustained inward hH1 currents in 130 mm external sodium after AC modification in 0 mm external sodium.
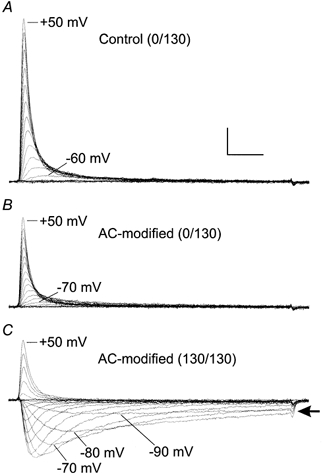
A, control hH1 currents in 0 mm external sodium. B, AC-modified currents in 0 mm external sodium. The channels were modified by perfusing the cell with 100 μm AC (in 0 mm external sodium solution) and delivering 50 pulses to −10 mV. C, AC-modified hH1 currents in 130 mm external sodium solution (same cell). Note the sustained inward currents (arrow) during voltage steps ranging from −90 to −70 mV. Scale, 1 nA, 2 ms.
DISCUSSION
Site 2 neurotoxins modify the kinetics of voltage-gated sodium channels. Although the toxins most probably share a binding domain (Catterall, 1980), the modification of channel kinetics differs from toxin to toxin. In particular, BTX elicits striking shifts in the voltage dependence of channel activation and, as shown in Fig. 1, removes most of the fast inactivation. In contrast, AC modification elicits smaller shifts in channel activation and may or may not affect fast inactivation, suggesting that BTX and AC induce distinct changes in sodium channels. Furthermore, channel modification by BTX or AC seems to induce changes in channel kinetics that are specific for native cardiac and skeletal muscle sodium channels. The results of the present study demonstrated that intracellular or extracellular application of AC modified the kinetics of transiently expressed hH1 and μ1 channels. One of the goals of the study was to compare the effects of AC modification on cardiac and skeletal muscle sodium channels under identical conditions to determine whether the toxin elicits discernible differences in the modification of the two channel subtypes. In general, AC modification of hH1 and μ1 channels produced similar changes in channel kinetics although there were two major differences. First, after AC modification of μ1 channels there was a small reduction in available current during the steady-state inactivation protocol after very negative conditioning pulses, whereas there was no such reduction in available current at AC-modified hH1 channels. Second, AC modification of hH1 channels in external solutions containing sodium reduced inactivation of the currents near the threshold of current activation, whereas AC-modified μ1 currents inactivated nearly completely at all voltages. The most interesting discovery was that AC-modified hH1 channels inactivated completely if the external solution lacked sodium.
Activation of AC-modified channels
Because much of the work on site 2 neurotoxins has been conducted on sodium channels from native tissues, these early studies provided the basis of our understanding about toxins such as BTX and AC. Preparations such as frog node of Ranvier, dissociated heart cells and frog skeletal muscle fibres all require specific experimental procedures and thus increase the difficulty of comparing directly the kinetic changes associated with AC modification of sodium channels in these tissues. Nevertheless, the effects of AC modification on sodium channel activation are qualitatively similar in different preparations in that current activation is shifted toward more hyperpolarized potentials. The largest shifts in current activation of AC-modified sodium channels were ∼-50 mV at frog node of Ranvier (Schmidt & Schmitt, 1974; Mozhaeva et al. 1980) and frog skeletal muscle fibres (Campbell, 1982), whereas the shifts in activation were ∼-30 mV at mouse ventricular cells (Nilius et al. 1986) and neuroblastoma cells (Grishchenko et al. 1983).
In the present study, AC modification of hH1 and μ1 channels elicited similar shifts in current activation. In 0 mm external sodium solution, current activation was shifted by −15 to −20 mV and in 65 or 130 mm sodium solution the shifts were −20 to −30 mV. AC modification in 65 or 130 mm external sodium shifted the V0.5 values of the normalized conductance curves by only −10 to −15 mV and increased the slope values of the fitted Boltzmann functions, perhaps suggesting that the macroscopic currents contained mixtures of modified and unmodified channels. The possibility that some fraction of the channels had not been modified cannot be ruled out. Repetitive depolarizations in the presence of AC reduced the amplitude of the current by > 50 % and channels that had been blocked (Catterall, 1977) rather than modified by AC could have recovered after washout of the toxin. On the other hand, because none of the current-voltage relationships contained a secondary peak inward current after AC modification, which would be indicative of two channel populations (Campbell, 1982), the reduction in current amplitude may simply reflect modification of channel conductance or that a fraction of modified channels had entered fast or slow inactivated states.
In contrast to previously published data (Mozhaeva et al. 1977; Grishchenko et al. 1983; Nilius et al. 1986), AC modification of hH1 or μ1 channels did not cause a large shift in the sodium reversal potential. The probable explanation for the lack of shift in the sodium reversal potential in the present study is that the external and internal solutions contained few ions that became permeable through AC-modified channels. Caesium was the only monovalent cation in the internal solution (CsOH was used to titrate pH) and other studies have demonstrated that caesium permeability through AC-modified channels is relatively small compared with other cations such as potassium or ammonium (Campbell, 1982). The similarities between the activation of hH1 and μ1 currents indicated that AC modification has similar effects on current activation of these sodium channel α subunits.
Inactivation of AC-modified channels
The major differences among sodium channels from different tissues after AC modification are the changes in channel inactivation. Compared with other sodium channels, the steady-state inactivation of sodium channels in frog nodal membrane seems to be most affected by AC modification because the inactivation is incomplete even after conditioning pulses to +50 mV (Schmidt & Schmitt, 1974; Mozhaeva et al. 1980). In contrast, steady-state inactivation of sodium channels progresses to completion in frog skeletal muscle (Campbell, 1982), neuroblastoma cells (Grishchenko et al. 1983), mouse heart (Nilius et al. 1986), or channels cloned from rat brain (Rao & Sikdar, 2000). The hyperpolarizing shift in steady-state inactivation in these preparations ranges from 7 mV in neuroblastoma cells up to 20 mV in frog skeletal muscle fibres.
In this study, external sodium concentration seemed to have little influence on the steady-state inactivation of hH1 or μ1 channels, and the inactivation progressed to completion in control solutions and after AC modification. The hyperpolarizing shifts in steady-state inactivation after AC modification also were not affected by external sodium ions and were only 7–8 mV. Thus, AC modification more strongly affected channel activation compared with steady-state inactivation, thereby increasing the ‘window’ current at the intersection of the activation and inactivation curves (Nilius et al. 1986). Interestingly, there was a slight reduction in the availability of AC-modified μ1 channels after very negative conditioning potentials. Although the mechanism of this reduced availability is unclear, these data could suggest that AC modification elicits some change in μ1 channels at negative membrane potentials that causes a fraction of the channels to become unavailable at the test pulse. The reduced availability after conditioning pulses between −140 and −120 mV could represent the entry of modified channels into a fast or slow inactivated state. Given that AC modification shifts the voltage dependence of channel activation toward more negative potentials, a fraction of modified channels could ‘pre-activate’ at more negative voltages, inactivate, and be unavailable at the test pulse. This possibility is consistent with the kinetic model by Horn et al. (1981) which indicates that (unmodified) sodium channels can inactivate without a prior opening event.
The largest difference among sodium channel subtypes seems to be the degree to which AC modification reduces channel inactivation from the open state, and the voltage dependence of the reduced inactivation differs at virtually every preparation. In most cases, macroscopic current inactivation is incomplete near the activation threshold of the current. At the frog node of Ranvier, AC modification markedly reduces sodium current inactivation during voltage steps to ≤ 0 mV (Mozhaeva et al. 1980). In other preparations, sodium current inactivation becomes complete at ≥ −40 mV (mouse heart; Nilius et al. 1986), at ≥ −50 mV (rat brain IIa; Rao & Sikdar, 2000), or at ≥ −60 mV (frog skeletal muscle; Campbell, 1982). In contrast, sodium currents in neuroblastoma cells inactivate nearly completely at all voltages (Grishchenko et al. 1983).
AC-modified μ1 currents resembled AC-modified currents in neuroblastoma cells in that inactivation was practically complete at all voltages. In addition, the presence or absence of external sodium ions did not influence the completeness of μ1 current inactivation. Unlike AC-modified μ1 currents, the inactivation of AC-modified hH1 currents in external salines containing sodium was incomplete near the threshold of current activation. In contrast to AC modification of rat brain sodium channels (Rao & Sikdar, 2000), the fast and slow time constants of hH1 current decay did not change after AC modification. If AC modification did alter the time constant of current inactivation, then the change may have been masked by the negative shift in the voltage dependence of channel activation, given that channel activation and inactivation are coupled events (O'Leary et al. 1995).
An unexpected result in this study was that AC-modified hH1 channels inactivated completely if the external solution did not contain sodium. To my knowledge, this is the first report to demonstrate that external sodium ions influence the inactivation phenotype of an AC-modified sodium channel. Although the data in the present study cannot address the exact mechanism for the apparent sodium dependence of reduced inactivation, three alternatives include (1) the difference between the activation voltages of hH1 and μ1, (2) specific interactions between AC-modified hH1 channels and extracellular sodium ions, and (3) differences between the slow inactivation phenotypes of hH1 and μ1 channels. The first alternative takes into account the fact that hH1 channel activation occurs at potentials ∼20 mV more negative compared with those of μ1 activation. Thus, the driving force on sodium is larger at the threshold of hH1 current activation compared with that of μ1. This alternative seems unlikely, however, because AC modification reduces the inactivation of frog node of Ranvier (Mozhaeva et al. 1980) and frog skeletal muscle sodium channels (Campbell, 1982) and the thresholds of current activation in these preparations are near to or are more positive than that of μ1. The second alternative incorporates undescribed structural properties of hH1 and μ1 channels. According to this alternative, an interaction between external sodium ions and the outer vestibule (or an internal binding site) of AC-modified hH1 channels modulates the inactivation mechanism. Such interactions would therefore not occur between sodium ions and AC-modified μ1 channels. The third alternative is similar to the second but takes into account the differences between the slow inactivation phenotypes of hH1 and μ1 channels. A previous study has shown that entry of μ1 channels into the slow inactivated state is more rapid and is more voltage dependent compared with that of hH1 channels (O'Reilly et al. 1999), and these differences in the slow inactivation phenotypes of hH1 and μ1 channels could influence channel behaviour after AC modification. Indeed, BTX modification substantially reduced slow inactivation of μ1 channels, but seemed to have less effect on the slow inactivation of hH1 channels (Fig. 1). Another important consideration with regard to the reduced inactivation of AC-modified hH1 channels is the relationship between external sodium ions and slow inactivation. Townsend & Horn (1997) have demonstrated that external sodium inhibits the entry of hH1 channels into the slow inactivated state, whereas the absence of external sodium enhances slow inactivation at negative potentials. Thus, the currents of AC-modified hH1 channels may not exhibit the reduced inactivation phenotype in 0 mm external sodium solution if a significant fraction of modified channels become slow inactivated at the holding potential. Furthermore, the fact that residues on the extracellular surface or within the pore region affect the slow inactivation of μ1 channels (Ong et al. 2000; Hilber et al. 2001) could imply that external sodium ions influence the slow inactivation of μ1 channels as well. In spite of the uncertainties regarding the mechanism responsible for the reduced inactivation of AC-modified hH1 channels, it will be interesting to determine whether external sodium ions influence the reduced inactivation of other sodium channel subtypes after channel modification by AC or other site 2 neurotoxins.
Acknowledgments
I thank Dr Roland Kallen for the hH1 clone, and Drs Sho-Ya Wang and Ging Kuo Wang for the μ1 clone, HEK293t cell line, and CD8-pih3m plasmid. The author also thanks G. K. Wang for a sample of BTX and for helpful comments and a critical reading of the manuscript. This work was supported by grants from the Kentucky Academy of Science and the National Institutes of Health (R15-GM60927).
REFERENCES
- Barnes S, Hille B. Veratridine modifies open sodium channels. Journal of General Physiology. 1988;91:421–443. doi: 10.1085/jgp.91.3.421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell DT. Modified kinetics and selectivity of sodium channels in frog skeletal muscle fibers treated with aconitine. Journal of General Physiology. 1982;80:713–731. doi: 10.1085/jgp.80.5.713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon SC, Strittmatter SM. Functional expression of sodium channel mutations identified in families with periodic paralysis. Neuron. 1993;10:317–326. doi: 10.1016/0896-6273(93)90321-h. [DOI] [PubMed] [Google Scholar]
- Catterall WA. Activation of the action potential Na+ ionophore by neurotoxins. Journal of Biological Chemistry. 1977;252:8669–8676. [PubMed] [Google Scholar]
- Catterall WA. Neurotoxins that act on voltage-sensitive sodium channels in excitable membranes. Annual Review of Pharmacology and Toxicology. 1980;20:15–43. doi: 10.1146/annurev.pa.20.040180.000311. [DOI] [PubMed] [Google Scholar]
- Catterall WA, Trainer V, Baden DG. Molecular properties of the sodium channel: a receptor for multiple neurotoxins. Bulletin de la Societe de Pathologie Exotique. 1992;85:481–485. [PubMed] [Google Scholar]
- Daly JW, Myers CW, Warnick JE, Albuquerque EX. Levels of batrachotoxin and lack of sensitivity to its action in poison-dart frogs (Phyllobates) Science. 1980;208:1383–1385. doi: 10.1126/science.6246586. [DOI] [PubMed] [Google Scholar]
- Gellens ME, George AL, Chen L, Chahine M, Horn R, Barchi RL, Kallen RG. Primary structure and functional expression of the human cardiac tetrodotoxin-insensitive voltage-dependent sodium channel. Proceedings of the National Academy of Sciences of the USA. 1992;89:554–558. doi: 10.1073/pnas.89.2.554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grishchenko II, Naumov AP, Zubov AN. Gating and selectivity of aconitine-modified sodium channels in neuroblastoma cells. Neuroscience. 1983;9:549–554. doi: 10.1016/0306-4522(83)90173-2. [DOI] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Archiv. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Hilber K, Sandtner W, Kudlacek O, Glaaser IW, Weisz E, Kyle JW, French RJ, Fozzard HA, Dudley SC, Todt H. The selectivity filter of the voltage-gated sodium channel is involved in channel activation. Journal of Biological Chemistry. 2001;276:27831–27839. doi: 10.1074/jbc.M101933200. [DOI] [PubMed] [Google Scholar]
- Horn R, Patlak J, Stevens CF. Sodium channels need not open before they inactivate. Nature. 1981;291:426–427. doi: 10.1038/291426a0. [DOI] [PubMed] [Google Scholar]
- Mozhaeva GN, Naumov AP, Khodorov BI. A study of properties of batrachotoxin modified sodium channels. General Physiology and Biophysics. 1986;5:17–46. [PubMed] [Google Scholar]
- Mozhaeva GN, Naumov AP, Negulyaev YuA, Nosyreva ED. The permeability of aconitine-modified sodium channels to univalent cations in myelinated nerve. Biochimica et Biophysica Acta. 1977;466:461–473. doi: 10.1016/0005-2736(77)90339-x. [DOI] [PubMed] [Google Scholar]
- Mozhaeva GN, Naumov AP, Nosyreva ED. Kinetic and steady-state characteristics of sodium channels modified by aconitine. Neirofiziologiya. 1980;12:612–618. [PubMed] [Google Scholar]
- Nilius B, Boldt W, Benndorf K. Properties of aconitine-modified sodium channels in single cells of mouse ventricular myocardium. General Physiology and Biophysics. 1986;5:473–484. [PubMed] [Google Scholar]
- O'Leary ME, Chen L-Q, Kallen RG, Horn R. A molecular link between activation and inactivation of sodium channels. Journal of General Physiology. 1995;106:641–658. doi: 10.1085/jgp.106.4.641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong BH, Tomaselli GF, Balser JR. A structural rearrangement in the sodium channel pore linked to slow inactivation and use dependence. Journal of General Physiology. 2000;116:653–662. doi: 10.1085/jgp.116.5.653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Reilly JP, Wang S-Y, Kallen RG, Wang GK. Comparison of slow inactivation in human heart and rat skeletal muscle Na+ channel chimaeras. Journal of Physiology. 1999;515:61–73. doi: 10.1111/j.1469-7793.1999.061ad.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao S, Sikdar SK. Modification of α subunit of RIIA sodium channels by aconitine. Pflügers Archiv. 2000;439:349–355. doi: 10.1007/s004249900121. [DOI] [PubMed] [Google Scholar]
- Schmidt H, Schmitt O. Effect of aconitine on the sodium permeability of the node of Ranvier. Pflügers Archiv. 1974;349:133–148. doi: 10.1007/BF00586624. [DOI] [PubMed] [Google Scholar]
- Strichartz G, Rando T, Wang GK. An integrated view of the molecular toxinology of sodium channel gating in excitable cells. Annual Review of Neuroscience. 1987;10:237–267. doi: 10.1146/annurev.ne.10.030187.001321. [DOI] [PubMed] [Google Scholar]
- Townsend C, Horn R. Effect of alkali metal cations on slow inactivation of cardiac Na+ channels. Journal of General Physiology. 1997;110:23–33. doi: 10.1085/jgp.110.1.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trimmer JS, Cooperman SS, Tomiko SA, Zhou JY, Crean SM, Boyle MB, Kallen RG, Sheng ZH, Barchi RL, Sigworth FJ. Primary structure and functional expression of a mammalian skeletal muscle sodium channel. Neuron. 1989;3:33–49. doi: 10.1016/0896-6273(89)90113-x. [DOI] [PubMed] [Google Scholar]
- Ulbricht W. Effects of veratridine on sodium currents and fluxes. Reviews of Physiology, Biochemistry, and Pharmacology. 1998;133:1–54. doi: 10.1007/BFb0000612. [DOI] [PubMed] [Google Scholar]
- Wang DW, George AL, Bennett PB. Comparison of heterologously expressed human cardiac and skeletal muscle sodium channels. Biophysical Journal. 1996;70:238–245. doi: 10.1016/S0006-3495(96)79566-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S-Y, Wang GK. Modification of cloned brain Na+ channels by batrachotoxin. Pflügers Archiv. 1994;427:309–316. doi: 10.1007/BF00374539. [DOI] [PubMed] [Google Scholar]
- Wang S-Y, Wang GK. Point mutations in segment I-S6 render voltage-gated Na+ channels resistant to batrachotoxin. Proceedings of the National Academy of Sciences of the USA. 1998;95:2653–2658. doi: 10.1073/pnas.95.5.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright SN, Wang S-Y, Kallen RG, Wang GK. Differences in steady-state inactivation between Na channel isoforms affect local anesthetic binding affinity. Biophysical Journal. 1997;73:779–788. doi: 10.1016/S0006-3495(97)78110-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright SN, Xaio Y-F, Wang S-Y, Wang GK. State-dependent cocaine block of sodium channel isoforms, chimeras, and channels coexpressed with the β1 subunit. Biophysical Journal. 1999;76:233–245. doi: 10.1016/S0006-3495(99)77192-4. [DOI] [PMC free article] [PubMed] [Google Scholar]