

Abstract
The objective of the present study is to examine the potential role of nitric oxide (NO) in short-term potentiation (STP) and long-term facilitation (LTF) of breathing. Experiments were performed in wild-type (WT) and mutant mice deficient in nitric oxide synthase-1 (NOS-1), as well as in WT mice administered the NOS-1 inhibitor 7-nitroindazole (7-NI; 50 mg kg−1; i.p.). Respiratory responses following either single or recurrent episodes of hypoxia (7 % O2, balance N2) were analysed in unanaesthetised animals by body plethysmography along with rate of O2 consumption (V̇O2) and CO2 production (V̇CO2). After a single hypoxic challenge, respiration in WT mice remained elevated for 5 min, suggesting STP in ventilation. Following termination of three consecutive hypoxic challenges, respiration remained elevated during normoxia for as long as 30 min, indicating LTF in breathing under awake conditions. STP and LTF were significantly attenuated or absent in WT mice after 7-NI. A similar attenuation or absence of STP and LTF was also seen in NOS-1 mutant mice. Changes in V̇O2 and V̇CO2 were comparable among mice during the post-hypoxic period, suggesting that the absence of STP and LTF was not due to alterations in body metabolism. These results suggest endogenous NO is an important physiological modulator of ventilatory STP and LTF.
It is being increasingly appreciated that changes in breathing in response to a physiological stimulus often outlast the actual period of stimulation. For instance, hypoxia increases breathing and upon termination of the hypoxic challenge, ventilation remains elevated and returns slowly towards baseline over a span of minutes. This slow return of ventilation is often referred to as [short-term potentiation] or STP of breathing (for references see Eldridge & Millhorn, 1986; Powell et al. 1998). On the other hand, respiratory motor output remains elevated for as long as an hour after repeated hypoxic exposures in anaesthetised animals (Millhorn et al. 1980; see Mitchell et al. 2001 for references). The long lasting respiratory stimulation is unique to episodic hypoxia, because a comparable duration of cumulative hypoxia does not elicit this response (Baker & Mitchell, 2000). The persistence of respiratory stimulation after episodic hypoxia is called [long-term facilitation] or LTF of breathing (see Powell et al. 1998)
STP and LTF have been suggested to play important roles in stabilization of breathing. For instance, STP prevents respiratory oscillations resulting from abrupt changes in blood gases following a hypoxic challenge. Episodic hypoxia occurs in many pathophysiological situations including obstructive sleep apnoeas. LTF of ventilation resulting from episodic hypoxia is thought to preserve the patency of the upper airways (Powell et al. 1998). Both STP and LTF are of central neural origin. Raphé nuclei have been shown to be important for generation of LTF (Eldridge & Millhorn, 1986), whereas pontomedullary and n. tractus solitarii (nTS) regions seem to be critical for the generation of STP (Mifflin, 1997; McCrimmon et al. 1997). Despite their importance in the control of breathing, the cellular mechanisms associated with STP and LTF are not well understood. Available evidence suggests that LTF is a serotonin-dependent process (see Mitchell et al. 2001), whereas changes in Ca2+ conductance in the central synapse might be important for STP (Wagner & Eldridge, 1991; Powell et al. 1998). Whether or not other chemical messengers play a role in the genesis of STP and/or LTF in breathing, however, has not been examined.
Nitric oxide (NO) is a gas molecule generated during the oxidation of arginine by NO synthases (NOS). Recent studies suggest that NO generated by NO synthase-1 (NOS-1) is an important regulator of many physiological processes including regulation of breathing during hypoxia (Kline et al. 1998). Furthermore, studies on hippocampal neurons suggest that NO is critical for eliciting long-term potentiation (LTP) associated with learning and memory (Arancio et al. 1996). Given that NOS-1 is expressed in raphé nuclei (Leger et al. 1998), as well as pontomedullary regions (Vincent & Kimura, 1992), and the recent report that NOS-1 deficient mice exhibit breathing instability during hypoxia (Kline et al. 1998) prompted us to hypothesize that endogenous NO might contribute to the genesis of STP and LTF of breathing. To test this possibility, we analysed STP and LTF in mice deficient in NOS-1 (NOS-1 KO) as well as after pharmacological blockade of NOS-1 by 7-nitroindazole (7-NI) in wild-type (WT) mice. Our results show that STP as well as LTF in breathing are markedly attenuated or absent after NOS-1 inhibitor and also in mutant mice deficient in NOS-1.
METHODS
Experiments were approved by the Institutional Animal Care and Use Committee of Case Western Reserve University and were performed on age-matched, wild-type (WT) mice and NOS-1 mutant mice of either sex. The NOS-1 mice were obtained from Dr P. L. Huang (Huang et al. 1993). Hybrids of the 129/SV and C57BL/6 strains of mice, the parental strains of the mutant mice, were used as wild-type (WT) controls (Kline et al. 1998).
Respiration was monitored in awake mice by a whole body plethysmograph. Briefly, animals were placed in a 600 ml Lucite chamber containing an inlet port for the administration of test gases. The chamber containing the animal and a reference chamber were connected to a high-gain differential pressure transducer (Valydine MP45, Validyne Engineering Corp., Northridge, CA, USA). As the animal breathed, small changes in pressure were converted to a signal representing tidal volume. The signals were amplified and recorded on a strip chart recorder as well as stored in a computer for later analysis. Rates of oxygen consumption (V̇O2) and carbon dioxide production (V̇CO2) were determined by the open-circuit method using Beckman OM-14 and LB-2 analysers, as described previously (Kline et al. 1998).
To examine STP, after recording baseline breathing under room air, mice were challenged with 7 % O2 (balance, nitrogen) for 5 min. Following termination of the hypoxic challenge, breathing was monitored for an additional 5 min and compared with pre-hypoxic controls. To examine LTF, baseline breathing was recorded for 5 min while the animals breathed room air (21 % O2), and then were challenged with three episodes of hypoxia (7 % O2), each lasting 5 min interspersed with 5 min of room air breathing. Following the last episode of hypoxia, animals breathed room air and ventilation, V̇O2 and V̇CO2 were continuously monitored for 60 min. The above protocols were repeated in eight additional WT mice before and after administration of 7-nitroindazole (7-NI; Alexis Corp., San Diego, CA, USA; 50 mg kg−1; i.p.; Kalisch et al. 1996), a putative inhibitor of NOS-1. Following 7-NI, a period of 30 min was allowed before initiation of protocols.
The following variables were analysed: (1) respiratory rate (RR, breaths min−1), (2) inspiratory tidal volume (VT, μl), and (3) minute ventilation (VE, ml min−1, RR × VT). Respiratory variables (RR and VT) were averaged for 15 consecutive breaths every minute during baseline ventilation, as well as during and after hypoxic challenges. Sighs or sniffs were excluded in the analysis. The values of VT and VE were normalized to the body weight of the animal. Metabolic variables were measured at the end of each 5 min inspired O2 challenge.
Each data point in a given animal represents the average of two trials. The data are expressed as means ± s.e.m. Statistical analysis for ventilation and metabolic parameters was performed by one-way ANOVA, and then by Student's t test with Bonferroni correction. P values < 0.05 were considered significant.
RESULTS
Effect of NOS-1 blockade on STP
As a first step towards determining the role of NO in STP, it was necessary to establish whether STP can be elicited in mice under the awake condition. An example of breathing in an awake animal before and during the post-hypoxic period is shown in Fig. 1A (top panel). As can be seen in this example, after terminating the hypoxic challenge breathing did not return immediately to the pre-hypoxic baseline value. Rather, it remained elevated during the 5 min of the post-hypoxic period. Average data of the respiratory variables during the post-hypoxic period are summarized in Fig. 1B. During the post-hypoxic period, minute ventilation (VE) remained significantly elevated at all time points analysed. For instance, VE was +58 ± 17 % and +40 ± 9 % higher during the first and fifth minute of post-hypoxic period, respectively, compared to pre-hypoxic controls (P < 0.05; paired t test). The elevated VE was primarily due to increased respiratory rate (P < 0.05) rather than tidal volume (P > 0.05). To ensure that the observed changes in breathing were not due to flushing of gases, 21 % O2 was substituted for the hypoxic gas (i.e. 7 % O2). Alternate flushing with room air had no effect on breathing (Fig. 1A, second panel, sham exposure). These observations demonstrate awake mice exhibit STP in breathing.
Figure 1. Attenuation of STP in breathing after NOS-1 inhibitor (7-NI) and in NOS-1 mutant mice.
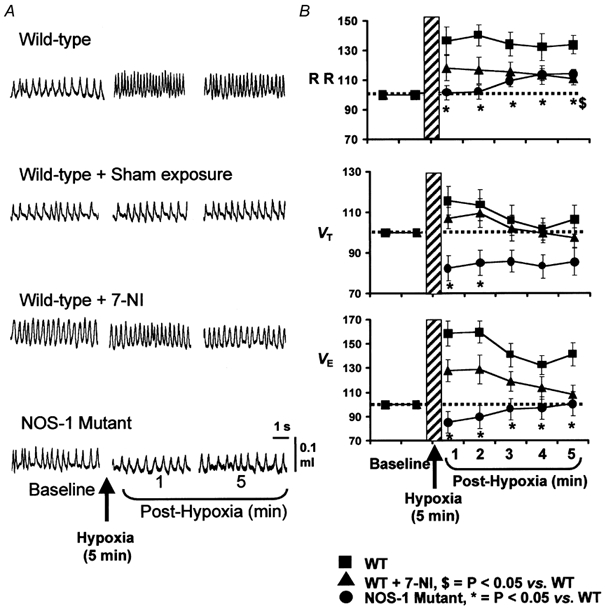
Examples of breathing before and during post-hypoxic period in all three groups of mice and mice after sham exposure are shown in A. Average data (mean ±s.e.m.) of respiratory rate (RR), tidal volume (VT) and minute ventilation (VE) as a percentage of baseline values (pre-hypoxia = 100 %) are shown in B. Note significant attenuation of STP in ventilation in wild-type (WT) mice treated with 7-NI and NOS-1 mutant mice. n = 8 for all mouse groups.
To determine whether endogenous NO from NOS-1 contributes to STP, ventilation was analysed during the post-hypoxic period in WT mice 30 min after administering the NOS-1 inhibitor, 7-NI. The results were compared with untreated controls. Baseline respiratory rate was significantly higher after treatment with 7-NI (7-NI = 204 ± 9 vs. 173 ± 6 breaths min−1, P < 0.05, n = 8). The magnitude of the hypoxic ventilatory response, however, was comparable between both groups of mice (control, 122 ± 14 % vs. 7-NI, 102 ± 15 %; P > 0.05). More importantly, as shown in Fig. 1A and B (example in the left panel and average data in right panel), STP was reduced in 7-NI treated animals at all the time points measured. For example, during the fifth minute of the post-hypoxic period, VE returned to baseline values in 7-NI treated animals, whereas it remained 40 ± 9 % above the pre-hypoxic values in untreated controls (P < 0.05). Changes in respiratory rate as well as VT contributed to the reduction in the STP in 7-NI treated animals.
To further establish the role of NO, STP was examined in mutant mice deficient in NOS-1. Consistent with our previous report (Kline et al. 1998), basal VE and respiratory rate were significantly higher in NOS-1 mutant compared to WT mice (P < 0.05). The magnitude of the hypoxic ventilatory response was comparable with WT controls (control, 122 ± 14 % vs. mutant, 113 ± 7 %; P > 0.05). However, in sharp contrast to WT mice, STP was nearly absent in NOS-1 mutant mice (Fig. 1A and B). In mutant mice, VE showed a 15 % undershoot during the first 2 min of the post-hypoxic period and subsequently returned to baseline values. Respiratory rate remained close to the baseline values, while VT was significantly reduced compared to pre-hypoxic values at all time points measured. These observations suggest that either blockade of NOS-1 or absence of NOS-1 attenuates or abolishes STP in breathing.
Effect of NOS-1 blockade on LTF
An example of ventilation before and after three recurrent episodes of hypoxia (7 % O2) in a WT mouse is depicted in Fig. 2A. As illustrated in this figure, breathing remained elevated above the pre-hypoxic level for up to 30 min. Averaged results are summarized in Fig. 2B. During the post-hypoxic period, VE remained significantly elevated up to 30 min and remained at or near baseline for the remaining period of analysis. The elevation in VE was primarily due to increases in respiratory rate (Fig. 2B). When changes in VE were normalized to changes in V̇CO2, LTF was still evident up to 15 min of post-hypoxic period (119±10 %). Repetitive flushing with 21 % O2 instead of 7 % O2 did not affect breathing (sham controls), suggesting that long-lasting elevation of breathing after episodic hypoxia is not due to flushing of gases. These observations suggest that LTF is expressed in response to recurrent episodes of hypoxia in awake mice.
Figure 2. Attenuation of LTF in breathing after NOS-1 inhibitor (7-NI) and in mutant mice deficient in NOS-1.
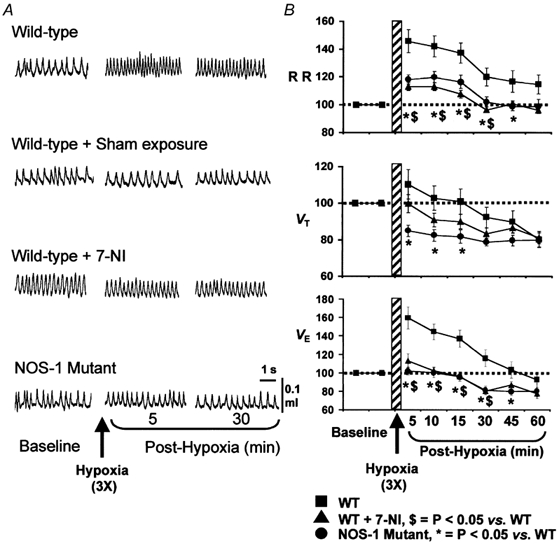
Examples of breathing before and during post-hypoxic period in all three groups of mice and mice after sham exposure are shown in A. Average data (mean ±s.e.m.) of RR, VT and VE as percent of baseline values (pre-hypoxia = 100 %) are shown B. Note significant attenuation of LTF in ventilation in WT mice treated with 7-NI and NOS-1 mutant mice. n = 8 for all mouse groups.
Having established that mice exhibit LTF of breathing, we then tested the effect of 7-NI on LTF. As can be seen from the example depicted in Fig. 2A (third panel), LTF is nearly absent after 7-NI, and ventilation promptly returned to baseline within 5 min after terminating episodic hypoxia. Averaged results showed that VE returned to or near baseline values within the first 5 min of the post-hypoxic period (P > 0.05, paired t test) and remained at this level for 15 min, then showed an undershoot. A decrease in the augmentation of respiratory rate as well as tidal volume contributed to the attenuation of LTF in 7-NI treated animals (Fig. 2B). There was no evidence of LTF after normalizing VE to V̇CO2 in 7-NI treated mice during the post-hypoxic period.
To further confirm that NO from NOS-1 is involved in LTF, and to show that LTF is not due to non-specific effects of 7-NI, experiments were performed on mutant mice deficient in NOS-1. As shown in Fig. 2, LTF could not be elicited in NOS-1 mutant mice following three recurrent episodes of hypoxia. Similar to 7-NI treated animals, respiratory rate returned promptly to baseline values after terminating episodic hypoxic challenge. Whereas, VT showed significant undershoot in the post-hypoxic period. As a result, VE remained close to baseline at all the time points analysed during the post-hypoxic period. Also, LTF was not evident after normalizing VE to V̇CO2 in the NOS-1 mutant. These observations demonstrate that blockade of NOS-1 attenuates or abolishes ventilatory LTF in mice.
STP and LTF are not due to alterations in body metabolism
It is well established that changes in body metabolism affect respiratory responses during hypoxia (Gautier, 1996). We monitored V̇O2 and V̇CO2 to assess whether changes in these variables are related to the absence of STP or LTF in 7-NI treated and NOS-1 mutant mice. However, no significant differences in V̇O2 or V̇CO2 could be seen under basal or after hypoxic episodes in the three animal groups (P > 0.05, ANOVA; Fig. 3). These observations suggest that alterations in V̇O2 and V̇CO2 do not contribute to the attenuated STP and LTF seen in 7-NI treated and NOS-1 mutant mice.
Figure 3. Changes in oxygen consumption (V̇O2) and carbon dioxide production (V̇CO2) before and during post-hypoxic periods.

Data presented are means ±s.e.m. 5′ and 30′ denote minutes. Note changes in V̇O2 and V̇CO2 are comparable between three groups of mice. n = 8 for all mouse groups.
DISCUSSION
In the present study, we used pharmacological and genetic approaches to test the hypothesis that NO generated by NOS-1 contributes to ventilatory STP and LTF. First of all, our results demonstrate that STP and LTF are expressed in unanaesthetised mice. More importantly, STP and LTF are nearly absent in WT mice after exposure to NOS-1 inhibitor as well as in mutant mice deficient in NOS-1. These results suggest that NO generated by NOS-1 is involved in STP and LTF in breathing.
It is clear from the present results that awake mice exhibit both STP and LTF of breathing in response to hypoxia. There have been several reports demonstrating STP and LTF in respiratory motor output in anaesthetised and unanaesthetised animals (see Engwall et al. 1994; Powell et al. 1998; Mitchell et al. 2001; Olson et al. 2001) as well as awake humans (Georgopoulos et al. 1995; Menendez et al. 1999). A notable difference between anaesthetised versus awake animals is that STP and LTF are more robust and long lasting and reflected more in tidal volume in the former than the later. Our results with awake mice are consistent with the earlier observations on unanaesthetised animals in that STP and LTF are primarily due to changes in respiratory rate rather than tidal volume, and LTF lasted only 30 min (or up to 15 min when expressed as VE V̇CO2−1). These observations, especially LTF of breathing, are similar to those reported in awake rats (Olson et al. 2001). We also examined STP and LTF in response to hypoxia in three anaesthetised, vagotomised and mechanically ventilated mice monitoring efferent phrenic nerve activity as an index of respiratory motor output. In all three animals, STP and LTF were reflected in tidal phrenic activity rather than respiratory rate, similar to that reported earlier in anaesthetised preparations (authors' unpublished findings). While it is possible that these differences could reflect different mechanisms, it is likely that the absence of vagal afferent inputs and/or lack of influence from higher brain centres might account for the larger contribution of tidal volume changes to STP and LTF of breathing in anaesthetised animals. Nonetheless, these observations taken together demonstrate that mice express STP and LTF of breathing both in awake and in anaesthetised conditions similar to that reported in other species. The fact that mice exhibit STP and LTF in ventilation is of considerable importance because genetically engineered mice offer an excellent approach for identifying molecular mechanisms associated with respiratory plasticity.
An important observation of the present study is that STP and LTF are markedly attenuated or absent after 7-NI, an inhibitor of NOS-1. Absence of STP and LTF was not due to non-specific effects of 7-NI, such as blockade of NOS-3, because STP and LTF could not be elicited in mutant mice deficient in NOS-1. It has been suggested that the magnitude and duration of STP and LTF are determined by arterial PCO2 and/or the hypoxic ventilatory response (Folgering & Durlinger, 1983; Engwall et al. 1994; Menendez et al. 1999). Hypercapnia augments STP, whereas hypocapnia attenuates STP (Engwall et al. 1994), and the larger the hypoxic ventilatory response the longer the duration of STP (Menendez et al. 1999). The following observations suggest that changes in arterial PCO2 do not account for the absence of STP and LTF in 7-NI treated and NOS-1 mutant mice: (1) when changes in VE are normalized to V̇CO2 (VE V̇CO2−1), LTF of breathing is still pronounced for up to 15 min in the post-hypoxic period in WT but not in 7-NI treated or in mutant mice; (2) changes in V̇CO2 in 7-NI or NOS-1 mutant mice during the post-hypoxic period (an index of arterial PCO2; Hicks et al. 1993) were comparable with control mice. Moreover, we have previously reported that the ventilatory response to hypercapnia in NOS-1 mutant mice is comparable with WT mice (Kline et al. 1998), suggesting that CO2 sensitivity is unaltered by the absence of NOS-1. Thus, it is unlikely that changes in arterial PCO2 would explain the absence of STP or LTF in mutant mice. In addition, the hypoxic ventilatory responses were comparable between the three groups of mice, suggesting that differences in hypoxic ventilatory responses do not account for attenuation of STP in 7-NI treated and NOS-1 mutant mice. In addition, it is unlikely that decreases in body temperature and/or metabolism that occur during hypoxia would account for the absence of STP and LTF in 7-NI treated or NOS-1 mutant mice. Because, changes in body metabolism as evidenced by the analysis of V̇O2 V̇CO2−1 were comparable in the three groups of mice during the post-hypoxic period (Fig. 3), and the changes in body temperature during hypoxia were the same in WT and mutant mice deficient in NOS-1 (Kline et al. 1998). Also, basal as well as changes in arterial blood pressure during hypoxia were comparable in NOS-1 mutant and WT mice (Kline et al. 1998). Hence, changes in arterial blood pressure may not explain the absence of STP and LTF in mutant mice. Rather, we believe that the absence of STP and LTF of breathing in 7-NI treated animals and NOS-1 mutant animals is due to reduced generation of NO from NOS-1 in central neurons responsible for generation of STP and LTF. Consistent with such a notion is our recent observation that hypoxia-induced STP and LTF are not affected in mutant mice (with the same genetic background as NOS-1 mutant mice) deficient in haeme oxygenase-2 (HO-2) (Kline & Prabhakar, 2000). HO-2 generates carbon monoxide (CO), another gas messenger molecule that shares many physiological actions with NO (Dawson & Snyder, 1994). Taken together these observations suggest that NO plays an essential role in ventilatory STP and LTF following hypoxic challenge.
How might NO contribute to generation of STP and LTF in breathing? It has been suggested that intracellular accumulation of calcium increases the synaptic efficacy in respiratory-related neurons in the pontomedullary region that leads to STP in ventilation (Wagner & Eldridge, 1991). Recently, Poon et al. (1999) demonstrated that, in anaesthetised rats, electrical stimulation of the carotid sinus nerve can elicit STP in phrenic nerve activity, and this response could be blocked with an NMDA receptor antagonist. It has been shown that NOS-1 and the NMDA receptors are co-localized in many regions of the central nervous system, and activation of the NMDA receptor increases intracellular calcium levels, which in turn promotes the generation of NO (Christopherson & Bredt, 1997). NO could influence synaptic efficacy either via its interaction with PDZ domains, which are protein-interacting modules that localize components to the synaptic cleft (Tomita et al. 2001), or by altering the ionic conductances in respiratory-related neurons (Ohkuma & Katsura, 2001) and thus contribute to generation of STP in ventilatory output.
With regard to LTF, several studies suggest that it is a serotonin- but not a catecholamine-dependent process and involves raphé neurons in the brainstem, especially the post-synaptic sites (for references see Eldridge & Millhorn, 1986; Mitchell et al. 2001). Furthermore, recent studies suggest that 5-hydroxytryptamine (5-HT) is critical for maintenance rather than initiation of LTF in respiratory motor output (Fuller et al. 2001). NOS-1 and 5-HT are co-localized in many raphé neurons in the rat and mouse (Leger et al. 1998). It has been suggested that NO prolongs the actions of 5-HT within the midbrain periaqueductal grey (PAG) matter (Lovick, 1996; Hamalainen & Lovick, 1997). Thus, NO generated by NOS-1 might contribute to LTF in ventilation by prolonging the actions of 5-HT in raphé neurons and thus amplifying its activity on the post-synaptic respiratory neuron. Alternatively, NO might promote the release of 5-HT and/or inhibit its uptake, thereby amplifying its activity, as it does elsewhere in the central nervous system (Prast & Philippu, 2001). Therefore, it appears that NO generated by NOS-1 may contribute to STP and LTF in ventilation via different mechanisms.
In summary, our results demonstrate that STP and LTF in ventilation are either attenuated or absent after disruption of NOS-1 function. These observations suggest that NO plays a critical and fundamental role in ventilatory dynamics that follow the hypoxic challenge through actions on central neurons.
Acknowledgments
The study is supported by grants from National Institutes of Health, Heart, Lung and Blood Institute, HL-25830, and D.D.K. is supported by training grant T32-HL07887.
REFERENCES
- Arancio O, Kiebler M, Lee CJ, Lev-Ram V, Tsien RY, Kandel ER, Hawkins RD. Nitric oxide acts directly in the presynaptic neuron to produce long-term potentiation in cultured hippocampal neurons. Cell. 1996;87:1025–1035. doi: 10.1016/s0092-8674(00)81797-3. [DOI] [PubMed] [Google Scholar]
- Baker TL, Mitchell GS. Episodic but not continuous hypoxia elicits long-term facilitation of phrenic motor output in rats. Journal of Physiology. 2000;529:215–219. doi: 10.1111/j.1469-7793.2000.00215.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christopherson KS, Bredt DS. Nitric oxide in excitable tissues: physiological roles and disease. Journal of Clinical Investigation. 1997;100:2424–2429. doi: 10.1172/JCI119783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson TM, Snyder SH. Gases as biological messengers: nitric oxide and carbon monoxide in the brain. Journal of Neuroscience. 1994;14:5147–5159. doi: 10.1523/JNEUROSCI.14-09-05147.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eldridge FL, Millhorn DE. Oscillation, gating, and memory in the respiratory control system. In: Cherniack NS, Widdicombe JG, editors. Handbook of Physiology, section 3, The Respiratory System, Control of Breathing. II. Bethesda, MD, USA: American Physiological Society; 1986. pp. 93–114. parts 1 and 2. [Google Scholar]
- Engwall MJ, Smith CA, Dempsey JA, Bisgard GE. Ventilatory afterdischarge and central respiratory drive interactions in the awake goat. Journal of Applied Physiology. 1994;76:416–423. doi: 10.1152/jappl.1994.76.1.416. [DOI] [PubMed] [Google Scholar]
- Folgering H, Durlinger M. Time course of posthyperventilation breathing in humans depends on alveolar CO2 tension. Journal of Applied Physiology. 1983;54:809–813. doi: 10.1152/jappl.1983.54.3.809. [DOI] [PubMed] [Google Scholar]
- Fuller DD, Zabka AG, Baker TL, Mitchell GS. Phrenic long-term facilitation requires 5-HT receptor activation during but not following episodic hypoxia. Journal of Applied Physiology. 2001;90:2001–2006. doi: 10.1152/jappl.2001.90.5.2001. [DOI] [PubMed] [Google Scholar]
- Gautier H. Interactions among metabolic rate, hypoxia, and control of breathing. Journal of Applied Physiology. 1996;81:521–527. doi: 10.1152/jappl.1996.81.2.521. [DOI] [PubMed] [Google Scholar]
- Georgopoulos D, Mitrouska I, Argyropoulou P, Patakas D, Anthonisen NR. Effect of hypoxic sensitivity on decay of respiratory short-term potentiation. Chest. 1995;107:150–155. doi: 10.1378/chest.107.1.150. [DOI] [PubMed] [Google Scholar]
- Hamalainen MM, Lovick TA. Involvement of nitric oxide and serotonin in modulation of antinociception and pressor responses evoked by stimulation in the dorsolateral region of the periaqueductal gray matter in the rat. Neuroscience. 1997;80:821–827. doi: 10.1016/s0306-4522(97)00124-3. [DOI] [PubMed] [Google Scholar]
- Hicks IR, Soni NC, Shephard JN. Comparison of end-tidal and arterial carbon dioxide measurements during anaesthesia with the laryngeal mask airway. British Journal of Anaesthesia. 1993;71:734–735. doi: 10.1093/bja/71.5.734. [DOI] [PubMed] [Google Scholar]
- Huang PL, Dawson TM, Bredt DS, Snyder SH, Fishman MC. Targeted disruption of the neuronal nitric oxide synthase gene. Cell. 1993;75:1273–1286. doi: 10.1016/0092-8674(93)90615-w. [DOI] [PubMed] [Google Scholar]
- Kalisch BE, Connop BP, Jhamandas K, Beninger RJ, Boegman RJ. Differential action of 7-nitro indazole on rat brain nitric oxide synthase. Neuroscience Letters. 1996;219:75–78. doi: 10.1016/s0304-3940(96)13194-3. [DOI] [PubMed] [Google Scholar]
- Kline DD, Prabhakar NR. Endogenous nitric oxide, but not carbon monoxide, contributes to long-term facilitation following episodic hypoxia. American Journal of Respiratory and Critical Care Medicine. 2000;3:484. [Google Scholar]
- Kline DD, Yang T, Huang PL, Prabhakar NR. Altered respiratory responses to hypoxia in mutant mice deficient in neuronal nitric oxide synthase. Journal of Physiology. 1998;511:273–287. doi: 10.1111/j.1469-7793.1998.273bi.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leger L, Charnay Y, Burlet S, Gay N, Schaad N, Bouras C, Cespuglio R. Comparative distribution of nitric oxide synthase- and serotonin-containing neurons in the raphé nuclei of four mammalian species. Histochemistry and Cell Biology. 1998;110:517–525. doi: 10.1007/s004180050313. [DOI] [PubMed] [Google Scholar]
- Lovick TA. Role of nitric oxide in medullary raphé-evoked inhibition of neuronal activity in the periaqueductal gray matter. Neuroscience. 1996;75:1203–1209. doi: 10.1016/0306-4522(96)00325-9. [DOI] [PubMed] [Google Scholar]
- McCrimmon DR, Zuperku EJ, Hayashi F, Dogas Z, Hinrichsen CF, Stuth EA, Tonkovic-Capin M, Krolo M, Hopp FA. Modulation of the synaptic drive to respiratory premotor and motor neurons. Respiration Physiology. 1997;110:161–176. doi: 10.1016/s0034-5687(97)00081-9. [DOI] [PubMed] [Google Scholar]
- Menendez AA, Nuckton TJ, Torres JE, Gozal D. Short-term potentiation of ventilation after different levels of hypoxia. Journal of Applied Physiology. 1999;86:1478–1482. doi: 10.1152/jappl.1999.86.5.1478. [DOI] [PubMed] [Google Scholar]
- Mifflin SW. Short-term potentiation of carotid sinus nerve inputs to neurons in the nucleus of the solitary tract. Respiration Physiology. 1997;110:229–236. doi: 10.1016/s0034-5687(97)00087-x. [DOI] [PubMed] [Google Scholar]
- Millhorn DE, Eldridge FL, Waldrop TG. Prolonged stimulation of respiration by a new central neural mechanism. Respiration Physiology. 1980;41:87–103. doi: 10.1016/0034-5687(80)90025-0. [DOI] [PubMed] [Google Scholar]
- Mitchell GS, Baker TL, Nanda SA, Fuller DD, Zabka AG, Hodgeman BA, Bavis RW, Mack KJ, Olson EB., Jr Invited review: Intermittent hypoxia and respiratory plasticity. Journal of Applied Physiology. 2001;90:2466–2475. doi: 10.1152/jappl.2001.90.6.2466. [DOI] [PubMed] [Google Scholar]
- Ohkuma S, Katsura M. Nitric oxide and peroxynitrite as factors to stimulate neurotransmitter release in the CNS. Progress in Neurobiology. 2001;64:97–108. doi: 10.1016/s0301-0082(00)00041-1. [DOI] [PubMed] [Google Scholar]
- Olson EB, Jr, Bohne CJ, Dwinell MR, Podolsky A, Vidruk EH, Fuller DD, Powell FL, Mitchel GS. Ventilatory long-term facilitation in anaesthetised rats. Journal of Applied Physiology. 2001;91:709–716. doi: 10.1152/jappl.2001.91.2.709. [DOI] [PubMed] [Google Scholar]
- Poon CS, Siniaia MS, Young DL, Eldridge FL. Short-term potentiation of carotid chemoreflex: an NMDAR-dependent neural integrator. Neuroreport. 1999;10:2261–2265. doi: 10.1097/00001756-199908020-00007. [DOI] [PubMed] [Google Scholar]
- Powell FL, Milsom WK, Mitchell GS. Time domains of the hypoxic ventilatory response. Respiration Physiology. 1998;112:123–134. doi: 10.1016/s0034-5687(98)00026-7. [DOI] [PubMed] [Google Scholar]
- Prast H, Philippu A. Nitric oxide as modulator of neuronal function. Progress in Neurobiology. 2001;64:51–68. doi: 10.1016/s0301-0082(00)00044-7. [DOI] [PubMed] [Google Scholar]
- Tomita S, Nicoll RA, Bredt DS. PDZ protein interactions regulating glutamate receptor function and plasticity. Journal of Cell Biology. 2001;153:F19–24. doi: 10.1083/jcb.153.5.f19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent SR, Kimura H. Histochemical mapping of nitric oxide synthase in the rat brain. Neuroscience. 1992;46:755–784. doi: 10.1016/0306-4522(92)90184-4. [DOI] [PubMed] [Google Scholar]
- Wagner PG, Eldridge FL. Development of short-term potentiation of respiration. Respiration Physiology. 1991;83:129–139. doi: 10.1016/0034-5687(91)90098-4. [DOI] [PubMed] [Google Scholar]