

Abstract
In non-excitable cells, the major Ca2+ entry pathway is the store-operated pathway in which emptying of intracellular Ca2+ stores activates Ca2+ channels in the plasma membrane. In many cell types, store-operated influx gives rise to a Ca2+-selective current called ICRAC (Ca2+ release-activated Ca2+ current). Using both the whole-cell patch clamp technique to measure ICRAC directly and fluorescent Ca2+ imaging, we have examined the role of the lipo-oxygenase pathway in the activation of store-operated Ca2+ entry in the RBL-1 rat basophilic leukaemia cell-line. Pretreatment with a variety of structurally distinct lipo-oxygenase inhibitors all reduced the extent of ICRAC, whereas inhibition of the cyclo-oxygenase enzymes was without effect. The inhibition was still seen in the presence of the broad protein kinase blocker staurosporine, or when Na+ was used as the charge carrier through CRAC channels. The lipo-oxygenase blockers released Ca2+ from intracellular stores but this was not associated with subsequent Ca2+ entry. Lipo-oxygenase blockers also reduced both the amount of Ca2+ that could subsequently be released by the combination of thapsigargin and ionomycin in Ca2+-free solution and the Ca2+ influx component that occurred when external Ca2+ was re-admitted. The inhibitors were much less effective if applied after ICRAC had been activated. This inhibition of ICRAC could not be rescued by dialysis with 5(S)-hydroxyperoxyeicosa-6E,8Z,11Z,14Z, tetraenoic acid (5-HPETE), the first product of the 5-lipo-oxygenase pathway. Our findings indicate that exposure to pharmacological tools that inhibit the lipo-oxygenase enzymes all decrease the extent of activation of the current. Our results raise the possibility that a lipo-oxygenase might be involved in the activation of ICRAC. Alternative explanations are also discussed.
In non-excitable cells, depletion of intracellular inositol 1,4,5-trisphosphate (InsP3)-sensitive Ca2+ stores results in the activation of Ca2+ influx through the store-operated Ca2+ entry pathway (Putney, 1986; Parekh & Penner, 1997). Although several distinct store-operated Ca2+ entry pathways have been described, the best characterized and most widely distributed is the Ca2+ release-activated Ca2+ current (ICRAC; Hoth & Penner, 1992). Ca2+ entry through CRAC channels is necessary not only for refilling the InsP3-sensitive Ca2+ stores, but also provides the trigger Ca2+ for driving a host of kinetically distinct processes including secretion, gene transcription and cell growth and proliferation (reviewed in Parekh & Penner, 1997).
The mechanism whereby CRAC channels, and indeed other types of store-operated Ca2+ channels, are activated remains contentious. Three major models have been proposed: (i) conformational coupling in which InsP3 receptors on the intracellular Ca2+ stores directly interact with store-operated Ca2+ channels in the plasma membrane (Irvine, 1990; Berridge, 1995; Rosado & Sage, 2000a), (ii) diffusible messengers that are released from the stores or generated/activated in the cytosol and which then open the channels (Randriamampita & Tsien, 1993; Csutora et al. 1999) and (iii) vesicular fusion in which the store-operated Ca2+ channels are somehow inserted into the plasma membrane by regulated exocytosis following store depletion (Yao et al. 1999). In spite of intense research by many laboratories, there is no accepted mechanism for store-operated Ca2+ influx.
In rat basophilic leukaemia (RBL) cells, a popular system for studying store-operated Ca2+ entry, secretion is strongly dependent on Ca2+ influx (Beaven et al. 1984; Mohr & Fewtrell, 1987). Using the capacitance technique to record vesicular fusion in single cells, we have found that quite marked store emptying is required for exocytosis to occur (Artalejo et al. 1998). In spite of robust Ca2+ release, InsP3 is unable to promote vesicular fusion unless store refilling is compromised by inhibiting the SERCA pumps. This is strikingly similar to the activation of ICRAC in weak intracellular Ca2+ buffer. Here, InsP3 generally fails to activate any detectable macroscopic current unless store refilling is suppressed by inhibition of the SERCA pumps (Broad et al. 1999; Fierro & Parekh, 2000). These findings lead us to speculate that there may be a link between RBL cell degranulation and the activation of CRAC channels. Some signal, associated with degranulation, might also increase the activity of CRAC channels such that a large macroscopic Ca2+ current, required to drive exocytosis, develops.
Eicosanoids, produced by intracellular metabolism of arachidonic acid, are very important messengers from mast cells and basophils where they activate other cells of the immune system to release products that feedback to sustain mast cell and basophil degranulation (Samuelsson, 1983). The effects of eicosanoids on the properties of ICRAC have not been investigated. We have therefore examined the consequences of interfering with eicosanoid production on the activation of ICRAC in RBL-1 cells. We have found that inhibition of the lipo-oxygenase family of enzymes reduces the extent of ICRAC, an effect not mimicked by interefering with the cyclo-oxygenase enzymes. Our results raise the possibility that the lipo-oxygenase pathway might be an important regulator of Ca2+ influx in these non-excitable secretory cells.
METHODS
Rat basophilic leukaemia cells (RBL-1) cells, which were bought from the Cell Bank at the Sir William Dunn School of Pathology, Oxford University, were cultured as previously described (Parekh et al. 1997; Fierro & Parekh, 2000).
Patch-clamp experiments were conducted in the tight-seal whole-cell configuration at room temperature (20–25 °C) (Hamill et al. 1981; Fierro & Parekh, 2000). Sylgard-coated, fire-polished pipettes had DC resistances of 2.8–4.0 MΩ when filled with standard internal solution that contained (mm): caesium glutamate 145, NaCl 8, MgCl2 1, EGTA 10, Hepes 10, adjusted to pH 7.2 with CsOH. In some experiments (described in text), cells were dialysed with a pipette solution in which Ca2+ was strongly buffered at 120 nm (10 mm EGTA, 4.1 mm CaCl2). A correction of +10 mV was applied for the subsequent liquid junction potential that arose from this glutamate-based internal solution. Extracellular solution contained (mm): NaCl 145, KCl 2.8, CaCl2 10, MgCl2 2, CsCl 10, glucose 10, Hepes 10, adjusted to pH 7.4 with NaOH. Divalent ion-free external solution contained (mm): NaCl 155, KCl 2.8, CsCl 10, EDTA 2, glucose 10, Hepes 10, adjusted to pH 7.4 with NaOH. ICRAC was measured by applying voltage ramps (−100 to +100 mV in 50 ms) at 0.5 Hz from the holding potential of 0 mV as previously described (Fierro & Parekh, 2000). Currents were filtered using an 8-pole Bessel filter at 2.5 kHz and digitized at 100 μs. Currents were normalized by dividing the amplitudes (measured from the voltage ramps at −80 mV) by the cell capacitance. Capacitative currents were compensated before each ramp by using the automatic compensation of the EPC 9–2 amplifier. All leak currents were subtracted by averaging the first one to three ramp currents, and then subtracting this from all subsequent currents. Data are presented as means ± s.e.m., and statistical evaluation was carried out using Student's unpaired t test.
Ca2+ imaging experiments were carried out at room temperature using the IMAGO system from TILL Photonics (Bakowski, et al. 2001). Cells were alternately excited at 356 and 380 nm (30 ms exposures) and images were acquired using the TILL Vision software once every 4 s. The images were analysed off-line using IGOR Pro for Windows (Wavemetrics, OR, USA). Cells were loaded with Fura 2-AM (1 μm) for 40 min at room temperature in external solution containing (mm): NaCl 145, KCl 2.8, CaCl2 2, MgCl2 2, glucose 10, Hepes 10, adjusted to pH 7.4 with NaOH, as previously described (Bakowski et al. 2001). After loading, cells were washed three times in the above solution and then left for 15 min to allow for further de-esterification. In some experiments, after loading, cells were pre-incubated with drugs in the presence of 10 mm Ca2+ (described in text). Drugs were applied locally to the cell in Ca2+-free external solution by means of an application pipette placed within 20 μm of the cell. Results are presented as ΔF/F0, where F0 denotes the ratio (356 nm/380 nm) prior to stimulation (averaged over 10 s) and ΔF represents the ratio as a function of time. Each image was corrected for background fluorescence.
Nordihydroguaiaretic acid, indomethacin, 5,8,11,14-eicosatertaynoic acid (ETYA), cinnamyl-3,4-dihydroxy- α-cyanocinnamate (CDC) and gossypol were all dissolved in DMSO (50 mm stock solutions) and freshly prepared each day. Final DMSO concentration was < 0.05% except when 50 μm NDGA was used (0.1%). The stocks were stored at −20 °C and new aliquots were used for each coverslip. 5-HPETE was dissolved in ethanol as a 145 mm stock. All stock solutions (except 5-HPETE) were wrapped in aluminium foil and recordings were carried out with the microscope light intensity dimmed and using red light (patch clamp) or with the light switched off (fluorescence). Thapsigargin was from Alomone Laboratories. 5-HPETE, gossypol, ETYA and CDC were from Biomol (see text for full names). All other chemicals were purchased from Sigma.
RESULTS
Inhibition of the lipo-oxygenase enzymes with nordihydroguaiaretic acid (NDGA) prevents the activation of ICRAC
Figure 1A plots the amplitude of ICRAC (normalized to cell capacitance) against the time after the onset of whole-cell recording. The current was evoked by dialysing the cell with 30 μm InsP3 and 10 mm EGTA (Fig. 1Ai, open circles). These concentrations activate ICRAC to its maximal extent (Parekh et al. 1997; Glitsch & Parekh, 2000). Within 2 s of breaking into the cell, ICRAC started to activate and developed with a mono-exponential time course (time constant of 26 s for this cell) peaking after almost 100 s. Figure 1B shows the I–V relationship, once the current amplitude had reached steady state. The key features of ICRAC are readily apparent (voltage independent, inwardly rectifying, reversal potential > +70 mV). Mean kinetic properties of the current are summarized in Fig. 1C–E (10 cells).
Figure 1. The lipo-oxygenase inhibitor NDGA interferes with the activity of ICRAC.
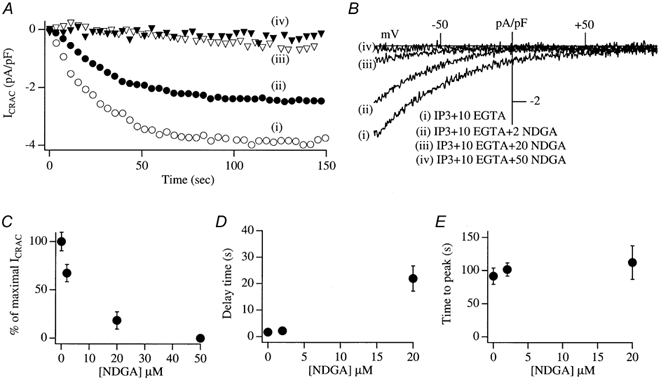
A, the development of ICRAC (measured at −80 mV from the voltage ramps) in control conditions (trace i, open circles) and then following pre-incubation (> 15 min) with different concentrations of NDGA. B, I–V relationships taken when the currents had reached steady state (80–100 s). Stores were emptied by dialysis with InsP3+ 10 mm EGTA. C, the relationship between the concentration of NDGA and inhibition of ICRAC is shown. D, the delay before ICRAC activates versus concentration of NDGA. E, time to peak versus NDGA concentration. In this, and all subsequent figures, the data points are means ± s.e.m.
To examine the effects of interfering with the lipo-oxygenase pathway on ICRAC, we pre-incubated cells for at least 15 min with different concentrations of the membrane-permeable lipo-oxygenase inhibitor nordihydroguaiaretic acid (NDGA). Three different lipo-oxygenases are known: the 5-, 12- and 15-enzymes, which oxidize polyunsaturated fatty acids that contain a 1,4-cis, cis-pentadiene system to generate a 1, hydroperoxy-2, 4-trans, cis-pentadiene product (Needleman et al. 1986). These enzymes are expressed in RBL cells (Van der Donk et al. 1991; Wong et al. 1992) and all three enzymes are inhibited by NDGA, albeit with slightly different potencies (Hope et al. 1983; Salari et al. 1984). Pretreatment with a low concentration of NDGA (2 μm) reduced the amplitude of ICRAC, when the latter was triggered by InsP3 and 10 mm EGTA (trace ii, filled circles in Fig. 1A and B; mean amplitude from 8 cells plotted in Fig. 1C, P < 0.05 relative to controls). However, neither the delay before the current activated (Fig. 1D) nor the time to peak (Fig. 1E) were significantly affected by this concentration of NDGA (P > 0.3 for both parameters). The time constant of activation of ICRAC was also similar between NDGA-treated cells and the controls (29.1 ± 4.3 versus 24.2 ± 2.8 s, respectively; P > 0.3). Raising the NDGA concentration to 20 μm had a stronger effect on ICRAC in that the amplitude of the current was substantially reduced (trace iii, open triangles in Fig. 1A and B; mean data from 12 cells shown in Fig. 1C, P < 0.01 compared with controls) and the delay before the current activated was increased almost 5-fold (Fig. 1D, P < 0.01). However, once the current started to activate, its time to peak was not significantly different from the controls (Fig. 1E). In the presence of 50 μm NDGA, ICRAC could not be detected at all (Fig. 1A–C; 4 cells).
Inhibition of the cyclo-oxygenase class of enzymes does not affect the activation of ICRAC
The lipo-oxygenase enzymes compete with the prostaglandin endoperoxide synthase complex for the same substrate, namely arachidonic acid. Inhibition of the lipo-oxygenase pathway could therefore lead to the build-up of arachidonic acid, and it is possible that this compound is responsible for the inhibition of ICRAC observed above. If this is true, then it predicts that inhibition of any pathway that metabolizes arachidonic acid should prevent ICRAC from activating, since the net effect would be a build-up in arachidonic acid levels. The prostaglandin endoperoxide synthase complex is composed of two distinct activities: the cyclo-oxygenase enzymes which oxidize arachidonic acid to form prostaglandin (PG)G2 and then a peroxidase which reduces PGG2 to PGH2 (Needleman et al. 1986). We blocked the prostaglandin endoperoxide synthase by inhibiting the cyclo-oxygenase enzyme with indomethacin (at maximally effective concentrations of either 20 or 50 μm). However, this failed to reduce the size of ICRAC compared with control recordings (Fig. 2A and B). Similarly, neither the time constant for activation of ICRAC (Fig. 2C) nor the time to peak (Fig. 2D) were affected by indomethacin compared with control recordings.
Figure 2. Structurally distinct lipo-oxygenase blockers all reduce the extent of ICRAC.
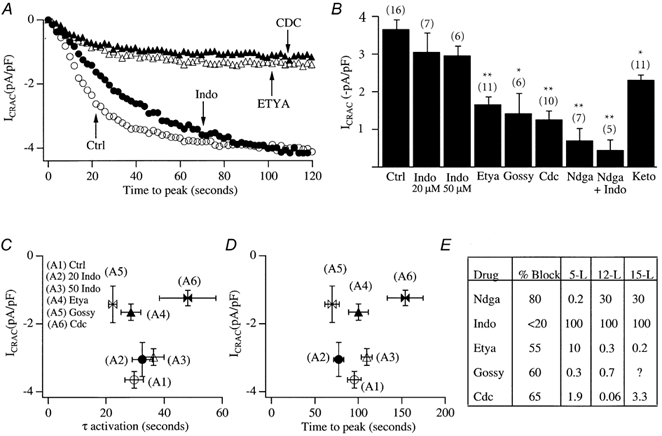
A, the effect of various inhibitors on the activation of ICRAC. Ctrl (open circles) denotes control (0.1% DMSO-treated cells), Indo (filled circles) denotes indomethacin (50 μm). B, the histogram summarizes mean data from several cells. Gossy denotes gossypol, Keto denotes ketoconazole. 20 and 50 Indo represent 20 and 50 μm Indo, respectively. All inhibitors were tested at a concentration of 20 μm (except gossypol which was used at 5 μm), and pre-incubated for at least 15 min. *P < 0.05 and **P < 0.01. C, amplitude of ICRAC against activation time constant. D, amplitude of ICRAC against time to peak. E, table summarizing the percentage block of ICRAC for the various inhibitors together with reported IC50 values for the different lipo-oxygenases, taken from the literature. Data for Ndga are from Salari et al. (1984) and Hope et al. (1983); data for Indo are from Salari et al. (1984) and Shen & Winter (1977); data for Etya are from Salari et al. (1984), Taylor et al. (1985) and Bokoch & Reed, 1981; data for Gossy were from Hamasaki & Tai (1985); and data for Cdc were from Cho et al. (1991).
We considered the further possibility that a product of the cyclo-oxygenase pathway was still responsible for inhibiting ICRAC, but the flux through the pathway was modest under resting conditions. Hence interference with the cyclo-oxygenase alone would have no effect on ICRAC. However, block of the lipo-oxygenase pathway by NDGA would increase arachidonic acid flux through the cyclo-oxygenase pathway, and this might then result in generation of an inhibitory factor. If this scenario is true, then simultaneous block of the cyclo-oxygenase and lipo-oxygenase enzymes should prevent the loss of ICRAC that is seen with inhibition of the lipo-oxygenase pathway alone. To test this, we treated cells with both indomethacin (20 μm) and NDGA (20 μm). However, ICRAC was still impeded, and to an extent not significantly different from that seen in the presence of NDGA alone (Fig. 2B, marked Ndga+Indo). Taken together, these results indicate that the inhibition of ICRAC is specific to NDGA and therefore presumably to the lipo-oxygenase pathway, rather than being a secondary effect due to increased substrate flux through the cyclo-oxygenase pathway.
Effects of NDGA are mimicked by other lipo-oxygenase inhibitors
If the inhibition of the activation of ICRAC by NDGA were due to an action on the lipo-oxygenase family of enzymes, then one would expect that other drugs which share this property should also reduce the extent of the current. Specific inhibitors of the lipo-oxygenase enzymes are lacking and NDGA is in fact considered the most potent and diagnostic. However, 5,8,11,14-eicosatertaynoic acid (ETYA), and cinnamyl-3,4-dihydroxy- α-cyanocinnamate (CDC) both interfere with lipo-oxygenase activity, although with less potency and selectivity than NDGA. Gossypol has also been found to be an effective 5- and 12-lipo-oxygenase inhibitor in RBL-1 cells (Hamaski & Tai, 1985). We found that these structurally distinct inhibitors were all able to significantly reduce the size of ICRAC compared with control cells (Fig. 2A and B). ETYA (20 μm) and gossypol (5 μm) did not affect either the time to peak (Fig. 2D) or the time constant of activation of ICRAC (Fig. 2C), whereas both were increased slightly, but significantly, by CDC (20 μm).
At high concentrations (100 μm), NDGA has been reported to inhibit cytochrome P-450, an enzyme initially suggested to be involved in the regulation of store-operated Ca2+ influx (Alvarez et al. 1991; Graier et al. 1995). Although a lower concentration of NDGA was effective in interfering with ICRAC (Fig. 1), we nevertheless tested the effects of ketoconazole (20 μm), a more potent and specific inhibitor of cytochrome P-450. ICRAC was reduced slightly following pre-incubation with ketoconazole, but to an extent significantly less than that observed in the presence of NDGA or CDC (Fig. 2B).
Figure 2E summarizes the extent of block of ICRAC for the inhibitors used (all at 20 μm except gossypol which was used at 5 μm) as well as the known IC50 values of these inhibitors for the 5-, 12- and 15-lipo-oxygenases. Although it is hard to relate our results to these IC50 values because the latter often involve studies on the purified enzyme, the pharmacological profile of the 5-lipo-oxygenase enzyme would appear most compatible with our data.
The inhibition of ICRAC is not rescued by thapsigargin, intracellular ATP or staurosporine
We entertained the possibility that pre-incubation with the lipo-oxygenase blockers altered the activity of either InsP3 receptors or SERCA pumps such that it became harder for InsP3 to deplete the stores sufficiently for ICRAC to activate. However, because dialysis of RBL cells with 10 mm EGTA alone depletes the stores to such an extent that ICRAC is activated maximally (Fierro & Parekh, 1999a; Bakowski et al. 2001), and that this passive depletion is independent of InsP3-mediated Ca2+ release (Fierro & Parekh, 1999a; Bakowski et al. 2001), we feel it unlikely that the combination of both InsP3 and 10 mm EGTA should fail to deplete the stores in the presence of the inhibitors. Nevertheless, after exposure to either 20 μm NDGA or CDC, we compared the size of ICRAC in cells dialysed with InsP3 together with 10 mm EGTA and 2 μm thapsigargin (a sarco-endoplasmic reticulum Ca2+-ATPase) versus that seen in InsP3 and 10 mm EGTA alone. Despite the inclusion of thapsigargin, the current was still substantially reduced with both inhibitors (Fig. 3A and B).
Figure 3. Lipo-oxygenase inhibition of ICRAC persists in the presence of thapsigargin, ATP and staurosporine.
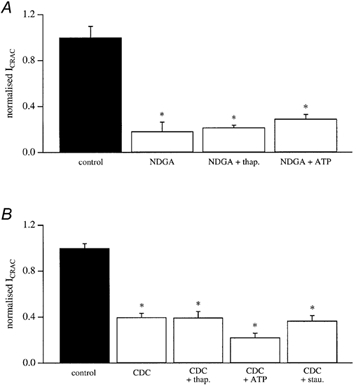
A, the size of ICRAC in control cells, in those pre-exposed to 20 μm NDGA, and then after either 2 mm Mg-ATP or 2 μm thapsigargin have been included in the pipette solution following pre-exposure to NDGA. B, the size of ICRAC in control cells, after exposure to 20 μm CDC, and then after inclusion of either Mg-ATP or thapsigargin in the pipette solution following CDC pretreatment. Also included are data taken from cells first exposed to staurosporine and then CDC. Note that CDC still reduces ICRAC even in the presence of the kinase blocker. Each bar is the mean of 4–7 cells.
In some experiments, we included 2 mm Mg-ATP in the recording pipette but ICRAC was still reduced following pretreatment with either 20 μm NDGA or CDC compared with controls (Fig. 3A and B).
Protein kinase C accelerates the inactivation of ICRAC (Parekh & Penner, 1995). In case the lipo-oxygenase blockers stimulated this enzyme, which would explain the reduction in ICRAC, we examined whether the broad kinase blocker staurosporine could prevent the inhibition of the current evoked by CDC. Staurosporine suppresses the ability of protein kinase C to inactivate ICRAC (Parekh & Penner, 1995). However, CDC still reduced the current even in the presence of staurosporine (Fig. 3B).
NDGA pretreatment reduces store-operated Ca2+ influx in intact cells
Although NDGA inhibited the activation of ICRAC in whole-cell recording, it was necessary to confirm that this was also the case for intact cells. To this end, we loaded cells with the fluorescent calcium dye fura-2 (see Methods). Stores were depleted by local application of thapsigargin (2 μm) together with the calcium ionophore ionomycin (100 nm) in Ca2+-free external solution (containing 0.2 mm EGTA; Bakowski et al. 2001). Note that, with these low concentrations of ionomycin, Ca2+ influx is entirely due to store depletion and not to calcium transport into the cell directly by the ionophore (Morgan & Jacob, 1994). A typical control recording is shown in Fig. 4A. The combination of thapsigargin and ionomycin resulted in robust, but transient, Ca2+ release due to store emptying. The mean increase in cytosolic Ca2+ due to release from the stores, is summarized in Fig. 4C. Readmission of external Ca2+ now resulted in further elevation of intracellular Ca2+ since Ca2+ influx occurred through the open store-operated Ca2+ channels (Fig. 4A). Although the size of this response is not an accurate assessment of the extent of Ca2+ influx (since the activity of other Ca2+ removal mechanisms also contribute), it is nevertheless a rough estimate of the amount of store-operated Ca2+ influx. We therefore measured the peak amplitude of this signal, as well as the time to peak. Pooled data for Ca2+ influx from 13 control cells is summarized in Fig. 4D and E. The pattern of the Ca2+ signal was dramatically altered by pre-exposure to NDGA (20–50 μm). Now, the combination of ionomycin and thapsigargin was generally unable to trigger any detectable Ca2+ release (Fig. 4B and D) and the subsequent Ca2+ entry was substantially reduced and developed at a very slow rate (Fig. 4B and E, 10 cells). These results are consistent with our whole-cell patch clamp recordings, and indicate that NDGA pretreatment can reduce store-operated Ca2+ influx in intact cells. However, the observation that NDGA pretreatment suppressed calcium release to the combination of ionomycin and thapsigargin indicated that NDGA itself was depleting the intracellular Ca2+ stores.
Figure 4. NDGA reduces store-operated Ca2+ influx to thapsigargin and ionomycin.
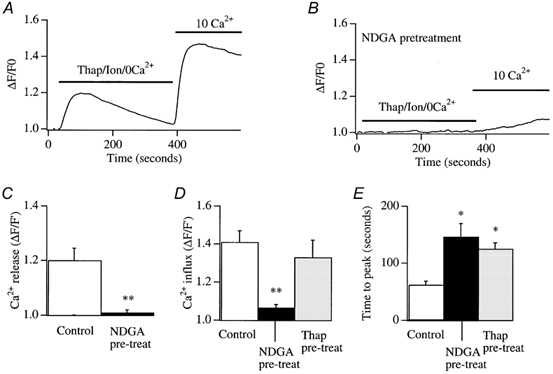
A, Ca2+ release by the combination of ionomycin (100 nm) and thapsigargin (2 μm) is followed by store-operated Ca2+ influx when external Ca2+ is readmitted. B, pretreatment with NDGA (> 20 min) abolishes Ca2+ release to ionomycin/thapsigargin and subsequent Ca2+ entry is substantially reduced. C, pooled data for the extent of Ca2+ release for control (i.e. where thapsigargin and ionomycin were applied) cells and for those pre-exposed to NDGA. D, the extent of store-operated Ca2+ entry for control cells and after pre-treatment with NDGA. Included in the bar chart are the results from cell pre-exposed to thapsigargin for 15 min, perfused with Ca2+-free solution, and then external Ca2+ was re-admitted (see text for details). E, the time to peak of the store-operated Ca2+ entry signal for the different conditions indicated. The Thap pre-treatment data are discussed in a later section.
NDGA releases Ca2+ in a concentration-dependent manner
To test whether NDGA mobilized intracellular Ca2+, we applied NDGA in Ca2+-free solution and monitored the Ca2+ response. We found that NDGA concentrations as low as 5 μm were able to generate a Ca2+ signal (14/15 cells, Fig. 5Aa) and, because no external Ca2+ was present, this reflected Ca2+ release from intracellular stores. We applied different concentrations of NDGA in Ca2+-free solution and constructed a dose-response curve (Fig. 5C, open circles), where the response was obtained by integrating the entire Ca2+ signal over a defined time window (20–300 s). To see whether NDGA was also able to trigger Ca2+ influx, we constructed a dose-response curve but now in the continuous presence of 2 mm external Ca2+. A typical recording in external Ca2+ is shown in Fig. 5Ab, where 5 μm NDGA was applied, and pooled data is included in Fig. 5C (filled circles). The dose-response curves overlapped for all concentrations tested, indicating that NDGA-induced Ca2+ release was not associated with significant Ca2+ influx. To monitor Ca2+ entry arising from maximal store depletion, we applied the combination of ionomycin and thapsigargin in 2 mm external Ca2+. A typical recording is shown in Fig. 5B and the integrated fluorescence signals obtained following exposure to ionomycin and thapsigargin in either 0 (open triangles) or 2 mm external Ca2+ (filled triangles) are included in the graph of Fig. 5C. The integrated signal in Ca2+-free solution to ionomycin and thapsigargin was similar to that evoked by 50 μm NDGA, consistent with the notion that NDGA can deplete the intracellular Ca2+ stores. The integrated signal in external Ca2+ elicited by ionomycin and thapsigargin (Fig. 5C, filled triangle) was substantially larger than that evoked by high NDGA concentrations in external Ca2+ (open circles, Fig. 5C), or by ionomycin and thapsigargin in Ca2+-free solution (open triangle, Fig. 5C; P < 0.01), the difference reflecting store-operated Ca2+ entry.
Figure 5. NDGA releases Ca2+ in a concentration-dependent manner.
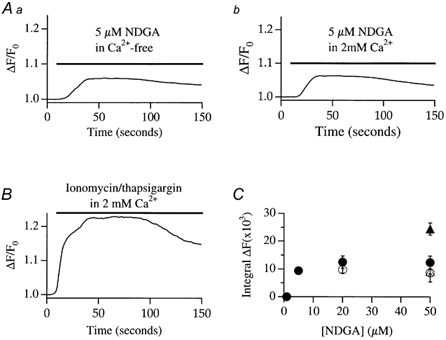
Aa, the time course of Ca2+ release from a cell exposed to 5 μm NDGA. b depicts a similar experiment but now in the presence of external Ca2+. B, time course of the Ca2+ response in a cell in which stores were fully depleted (combination of ionomycin and thapsigargin) in the presence of external Ca2+. C, dose-response curve to NDGA in the absence (open circles) and presence (filled circles) of external Ca2+. Included in the graph are the responses to ionomycin+ thapsigargin in the absence (open triangle) and presence (filled triangle) of external Ca2+. These latter two points are included in the graph for comparative purposes, but the cells were not exposed to NDGA.
We also tried a different approach, namely to readmit Ca2+ to cells that had been exposed to NDGA in Ca2+-free solution. However, results with this were much more variable. Some cells showed a clear increase in intracellular Ca2+ upon Ca2+ readmission whereas others showed no increase at all. Unexpectedly, we found that simply perfusing cells with Ca2+-free solution alone for a similar time was sufficient to elicit a calcium response when external Ca2+ was readmitted (7/7 cells), although its size was quite variable. This would lead us to overestimate the extent of NDGA-induced Ca2+ entry and we therefore abandoned this approach.
Application of indomethacin failed to evoke any consistent increase in intracellular Ca2+ (data not shown).
Release of intracellular Ca2+ by other lipo-oxygenase blockers
To see whether the other lipo-oxygenase blockers were also able to release Ca2+, we repeated the above experiments but applied different inhibitors. Results are summarized in Fig. 6. Unlike NDGA, ETYA was much less effective in mobilizing Ca2+ (Fig. 6Aa and b) and again, there was no significant difference between the integrated signals in the absence and presence of external Ca2+ (Fig. 6Ac). CDC and gossypol both triggered a rapid decrease in the fluorescence ratio (Fig. 6B), associated with a change in the 356 nm signal. These drugs are brightly coloured and probably interfere directly with the fluorophore.
Figure 6. Effects of other lipo-oxygenase blockers on Ca2+ release.
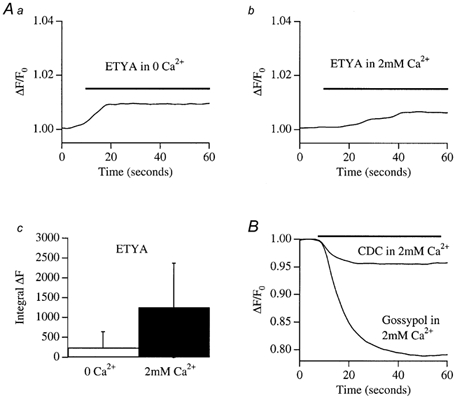
A, ETYA (20 μm) evoked a small Ca2+ increase both in the absence (a) and presence (b) of external Ca2+. The integrated signals were not significantly different (c). B, CDC and gossypol both caused a fall in the fluorescence ratio, due to a decrease in the 356 nm signal.
Ca2+ release by lipo-oxygenase blockers does not prevent ICRAC from activating
Because Ca2+-dependent inactivation of ICRAC channels has been extensively described (Zweifach & Lewis, 1995; Parekh, 1998; Fierro & Parekh, 1999b), we considered that the increase in intracellular Ca2+ following the release of stored Ca2+ by the lipo-oxygenase blockers might inactivate CRAC channels such that Ca2+ entry does not occur when whole-cell recordings are started. However, the following arguments can be raised against this. First, we have recently succeeded in recording ICRAC even when cells are dialysed with very weak Ca2+ buffer and high cytosolic Ca2+ (Fierro & Parekh, 2000). Although Ca2+ -dependent inactivation can reduce the overall extent of the current, it does not prevent the current from being detected. Second, we pre-incubated cells with thapsigargin (2 μm) in 10 mm external Ca2+ (conditions which gave a very large increase in intracellular Ca2+ concentration) and then broke in with a pipette solution containing InsP3 and 10 mm EGTA (Fig. 7A). In all cells we were able to record a clear ICRAC (around −2.8 pA pF−1), although the current did have a slightly reduced amplitude (by 25%) compared with controls from the same preparations and developed more slowly (Fig. 7B and C). Third, we carried out identical fluorescence experiments to those used for the lipo-oxygenase blockers, but now with thapsigargin instead. After loading with fura-2, cells were exposed to thapsigargin in the presence of 10 mm external Ca2+ for 15 min. We then briefly perfused the cells with Ca2+-free solution and then readmitted external Ca2+. Robust Ca2+ increases were seen (Fig. 4D and FE, columns marked Thap pre-treat).
Figure 7. Pretreatment with thapsigargin does not prevent ICRAC from developing.
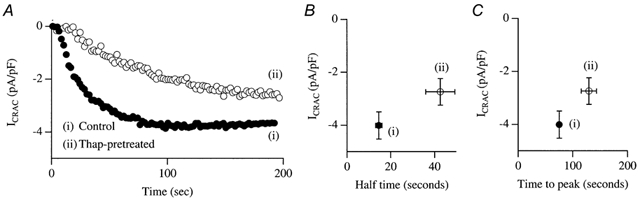
A, recordings taken from a control cell and from a cell that had been exposed to 2 μm thapsigargin in 10 mm external Ca2+ for 20 min. B, size of ICRAC against half-time for control and thapsigargin-treated cells. C, size of the current against the time to peak for the two conditions.
Na+ permeation through CRAC channels is suppressed by NDGA and CDC
In the absence of external divalent cations, CRAC channels become permeable to Na+ (Hoth & Penner, 1993) and large inward Na+ currents can be detected in RBL-1 cells (Fierro et al. 2000). Under these conditions, increases in intracellular Ca2+ which arise from Ca2+ influx clearly do not occur. Hence both Ca2+ entry-dependent fast and slow inactivation of CRAC channels (Zweifach & Lewis, 1995; Parekh, 1998; Fierro & Parekh, 1999b) would be suppressed when Na+ is the conducting cation. If the actions of the lipo-oxygenase blockers were due to an acceleration of Ca2+ entry-dependent inactivation, then one would predict that the Na+ current should not be affected by pretreatment with these drugs. Figure 8Aa and Ba compare the time course of the Na+ current through CRAC channels from control cells (filled circles) and from one pre-exposed to 20 μm NDGA or CDC, respectively. The Na+ current was largely suppressed by pretreatment with either inhibitor. Corresponding ramp I–Vs are shown in Fig. 8Ab and Bb. Bar charts summarizing mean data from four control and four NDGA-pretreated cells is shown in Fig. 8Ac and the corresponding one for CDC is shown in Fig. 8Bc. The Na+ current through CRAC channels was almost abolished by either NDGA or CDC, indicating that the underlying inhibition does not arise from Ca2+-dependent inactivation of CRAC channels.
Figure 8. Pretreatment with either NDGA or CDC suppresses the Na+ current through CRAC channels in divalent ion-free external solution.
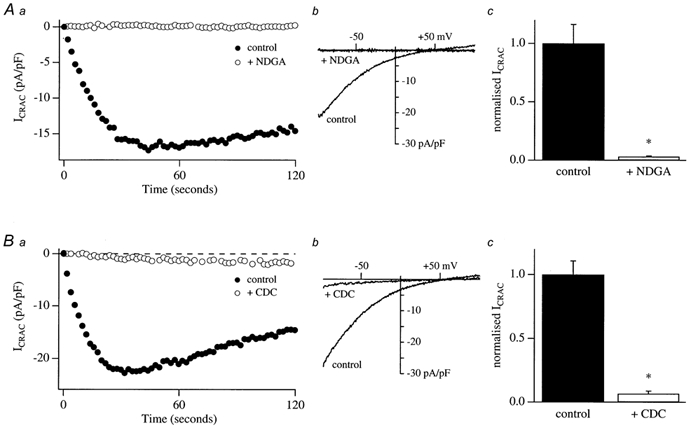
Aa, comparison of the time course of the monovalent Na+ current for a control cell and that for a cell which had been pre-incubated with 20 μm NDGA for around 15 min. b, the ramp I–V relationships are shown for the cells in a, taken at 60 s for both conditions. c, mean data from four control cells and four cells pre-exposed to NDGA. Ba, time course of a control cell and a cell pre-incubated with 20 μm CDC. b, corresponding ramp I–V. c, mean data from four control cells and four cells pre-exposed to CDC prior to break-in. Data have been normalized to the amplitude of the controls.
Do the lipo-oxygenase inhibitors directly block CRAC channels?
One possible explanation for the ability of the lipo-oxygenase blockers to inhibit activation of ICRAC is that the drugs block CRAC channels directly. To test this, we activated ICRAC by dialysis with InsP3 and 10 mm EGTA and, once the current had reached its maximum extent and was stable, we then applied the inhibitors to the cell. For more than 150 s of NDGA application, very little decline in the current was observed in 6/9 cells (a representative recording is given in Fig. 9Ai). The ramp I–V for this cell is shown in the right panel. The lack of effect of NDGA over this time frame is unlikely to reflect a diffusion problem because local application of La3+ or 2-aminoethoxydiphenyl borate (2-APB) block ICRAC with time constants of 10–20 s under similar conditions (Bakowski et al. 2001). In three other cells, however, a rapid decline was seen when NDGA was applied and this had a time course similar to that observed previously with 2-APB (Fig. 9Aii; ramp I–V shown in right panel). We compared the extent of inhibition of ICRAC when cells were either pretreated with NDGA or when the drug was applied after the current had developed. Pooled data for the two conditions are plotted in Fig. 9D. The block by NDGA was significantly less when applied after ICRAC had activated, but there was considerable variability in the extent of block (all 9 cells to which NDGA was applied have been included). Application of ETYA after ICRAC had developed (Fig. 9Bi) resulted in a significantly smaller block than was seen when cells were first exposed to the drug prior to store depletion (Fig. 9Bii). Mean data are summarized in Fig. 9D. The greatest difference was seen with CDC. Here, application of the drug had little effect on ICRAC once it had activated (Fig. 9Ci), but strongly reduced the development of the current if applied prior to the emptying of the stores (Fig. 9Cii; mean data in Fig. 9D). Hence these inhibitors are less effective if applied after ICRAC has developed.
Figure 9. Effect of application of the lipo-oxygenase inhibitors after ICRAC had developed.
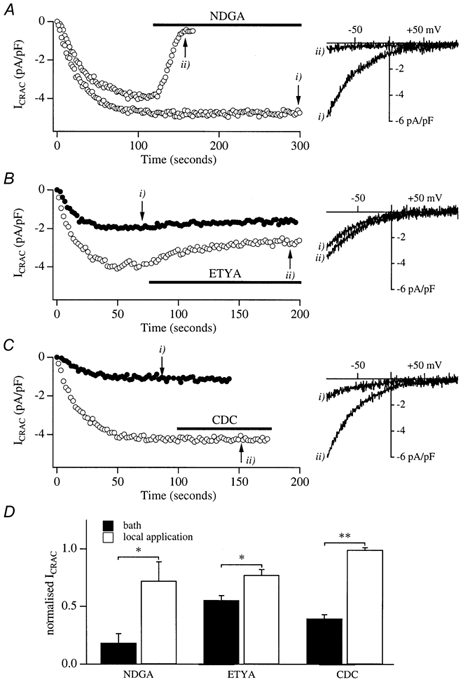
A, the effects of applying 50 μm NDGA after ICRAC had developed in two cells. Ramp I–V relations are shown on the right. B, the effects of applying ETYA either before (filled circles) or after (open circles) ICRAC had activated. C, comparison of the ability of CDC to interfere with ICRAC when applied before (filled circles) or after (open circles) ICRAC had activated. D, summary of the effects of the three inhibitors on ICRAC. The filled columns represent the inhibition seen when cells were pre-incubated with each drug and the open columns reflect the inhibition seen following exposure to each drug after ICRAC had developed. For cells pre-exposed to inhibitors, recordings were normalized to controls taken from the same preparations in the absence of inhibitors. For those experiments where drugs were applied after ICRAC had developed, the steady-state level in the presence of inhibitors was normalized to the peak amplitude reached before the inhibitors were applied. These latter currents were not significantly different from the controls used for normalizing the currents obtained following pretreatment with the lipo-oxygenase blockers. Each column represents the mean of 6–11 cells. In all cases, inhibition was significantly less if the drug was applied after store depletion than before.
Effects of 5-HPETE on ICRAC
If the lipo-oxygenase blockers interfere with the activation of ICRAC through a mechanism involving 5-lipo-oxygenase (as suggested by the pharmacological profile of the inhibitors summarized in Fig. 2E), one might expect 5(S)-hydroxyperoxyeicosa-6E,8Z,11Z,14Z,tetraenoic acid (5-HPETE), the first product of the 5-lipo-oxygenase pathway, to regulate ICRAC.
To test this, we dialysed control cells with 1 μm 5-HPETE in the presence of buffered Ca2+ and ATP (in order to prevent passive depletion of stores). As shown in Fig. 10A, 5-HPETE did not activate any inward current (4/4 cells). After 180 s of dialysis, we applied thapsigargin to see whether ICRAC could still be activated in the presence of intracellular 5-HPETE. The current developed normally and reached an amplitude similar to that evoked by thapsigargin in control cells (Fig. 10A and B). Hence 5-HPETE did not activate ICRAC itself nor did it seem to interfere with the ability of thapsigargin to evoke the current.
Figure 10. Effects of 5-HPETE on ICRAC.
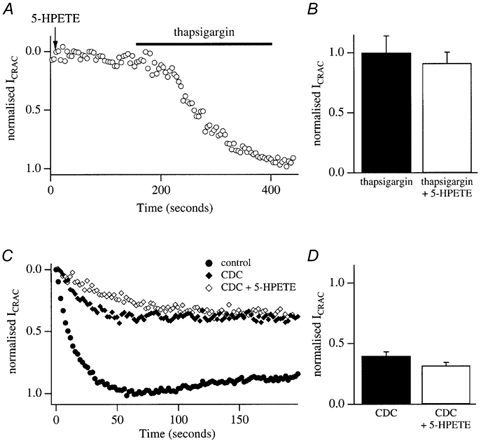
A, representative whole-cell recording from a cell dialysed with 5-HPETE in 120 nm buffered Ca2+. ICRAC did not activate. After 180 s, thapsigargin was applied and ICRAC subsequently developed. B, size of ICRAC following thapsigargin application in control cells, and in those dialysed with 5-HPETE. There was no significant difference. C, control recording (filled circles), recording taken after pre-exposure to CDC (diamonds) and recording after pre-exposure to CDC but with 5-HPETE in the pipette solution. The recording pipette contained InsP3+ 10 mm EGTA. Data have been normalized to control recordings. D, pooled data indicating that the reduction in ICRAC amplitude following pretreatment with CDC cannot be rescued by inclusion of 5-HPETE in the pipette.
We then examined whether 5-HPETE could rescue ICRAC in cells pre-exposed to CDC. A typical experiment is depicted in Fig. 10C. We first obtained control recordings for each preparation, evoked by dialysing cells with InsP3+ thapsigargin+10 mm EGTA (shown as filled circles). We then incubated cells in 20 μm CDC and confirmed that ICRAC was reduced (filled triangles in Fig. 10C). We then examined whether inclusion of 5-HPETE in the pipette (together with InsP3+thapsigargin+10 mm EGTA) could rescue ICRAC in CDC-treated cells. However, in spite of the presence of 5-HPETE, ICRAC could not be increased in size (Fig. 10C, filled circles; mean data in Fig. 10D). 5-HPETE also could not rescue ICRAC following exposure to NDGA (data not shown).
5-HPETE is an unstable compound. Although we used 5-HPETE shortly after its arrival and followed the procedures described by the supplier, we do not have a positive control that it was indeed active. We were also unable to test 12- and 15-HPETEs, alone or in combination with 5-HPETE, because of the cost of these compounds.
DISCUSSION
Our central finding is that a variety of structurally distinct lipo-oxygenase blockers all share the ability to reduce the activation of ICRAC. The key question is whether this reflects a requirement for the lipo-oxygenase enzymes in the elusive activation mechanism of ICRAC or whether it involves an action on either an inhibitory pathway or on a step not related to lipo-oxygenase block.
A lipo-oxygenase-sensitive step could easily be accommodated into each of the three major mechanisms proposed to account for the activation of store-operated Ca2+ influx. It could be important in maintaining close proximity of InsP3 receptors and CRAC channels, as required by the conformational-coupling model. Our recent results impose some constraints on this model in that, if it is applicable to RBL-1 cells, then the coupling must already be preformed prior to store depletion and involve a novel InsP3 receptor (Bakowski et al. 2001). Perhaps lipo-oxygenase enzymes maintain the integrity of this tightly formed complex. On the other hand, the lipo-oxygenase pathway might generate a diffusible metabolite which then activates CRAC channels. Finally, the lipo-oxygenase pathway might be required for fusion of vesicles containing CRAC channels with the plasma membrane upon store depletion. The fact that we could not rescue ICRAC with 5-HPETE in cells already exposed to lipo-oxygenase blockers is a major problem for a model that proposes that lipo-oxygenases are involved in the activation of ICRAC. But interpretation of this result is not straightforward for several reasons. First, we do not have a positive control for 5-HPETE. Although we used it shortly after its arrival and ordered it twice, it is a relatively unstable compound and we cannot be sure that it was fully active. Second, the products arising from the activity of one lipo-oxygenase enzyme can regulate fluxes through the others (Needleman et al. 1986). It is possible that a combination of metabolites are required rather than just 5-HPETE. Third, certain lipo-oxygenases translocate to the plasma membrane in RBL-1 cells upon stimulation. The site of arachidonic acid production might therefore be critical, and this may not be suitably mimicked by dialysing cells with 5-HPETE from a patch pipette. Finally, lipo-oxygenase activity might be involved in the activation of ICRAC but without the requirement for a subsequent metabolite. This would be similar to the putative role of phospholipase C, an enzyme which hydrolyses phosphatidylinositol 4,5-bisphosphate, in the activation of ICRAC in RBL-1 cells. Here, an inhibitor of the enzyme suppressed ICRAC but neither hydrolysis product (InsP3 or diacylglycerol) could rescue the current (Broad et al. 2001).
It is unlikely that a known mechanism that inactivates ICRAC becomes more prominent in the presence of lipo-oxygenase block and that this accounts for the reduction in the current. In RBL cells, ICRAC can be inactivated by fast Ca2+-dependent inactivation (reflecting a build-up of Ca2+ in the vicinity of each open channel; Fierro & Parekh, 1999b) and Ca2+-dependent slow inactivation (Parekh, 1998), which both arise from Ca2+ influx, Ca2+-dependent store refilling (Bakowski, Glitsch & Parekh, 2001) and protein kinase C (Parekh & Penner, 1995). Although some lipo-oxygenase blockers elevated intracellular Ca2+, we do not think Ca2+-dependent inactivation is involved because first NDGA and CDC were still effective in the absence of external Ca2+ under conditions where Na+ was the charge carrier, and second both store-operated Ca2+ influx (using Fura-2 in intact cells) and ICRAC were still substantial in spite of pretreating cells with thapsigargin in external Ca2+, a condition which elevates intracellular Ca2+ levels significantly. Because inclusion of thapsigargin in the recording pipette failed to prevent the loss of ICRAC following exposure to the lipo-oxygenase blockers, it is unlikely that store refilling is the mechanism of inhibition. A role for protein kinase C in the inhibition of ICRAC by lipo-oxygenase blockers is also unlikely because staurosporine, which prevents kinase-mediated inactivation of the current (Parekh & Penner, 1995) was not able to suppress the inhibition brought about by CDC. However, we cannot rule out the possibility that a novel inhibitory mechanism exists, which is both Ca2+ and protein kinase independent, and is activated following inhibition of the lipo-oxygenase pathway.
The lipo-oxygenase blockers could, in spite of their structural divergence, all share an action unrelated to lipo-oxygenase inhibition. This is always a concern when utilizing a pharmacological approach. The CRAC channels are possibly themselves one such site. In this scenario, the inhibitors would function as direct channel blockers. Indeed, it has been reported that NDGA can directly inhibit voltage-gated Ca2+ channels independently of any actions on lipo-oxygenases (Korn & Horn, 1990) and NDGA evoked rapid inhibition of whole-cell ICRAC in some cells, which would be consistent with a direct channel block. But it is not clear why most cells were relatively resistant to NDGA application after ICRAC had activated. We do not know why local application of NDGA had such kinetically distinct effects. The Ca2+ channels that are activated by epidermal growth factor stimulation of the 5-lipo-oxygenase pathway in human A431 and murine P19 embryonal carcinoma cells can be inhibited following only a 2 min pre-exposure to NDGA (Peppelenbosch et al. 1992). Moreover, application of NDGA to outside-out patches completely abolished channel activity within 60 s (Peppelenbosch et al. 1992). Hence inhibition of lipo-oxygenases can be fast and rapid kinetics of block in whole-cell recording (time constant of 20–35 s) does not necessarily demonstrate direct channel block. Interestingly, NDGA does not block the drosophila TRP protein (Chyb et al. 1999), and certain TRP homologues are thought to encode the CRAC channels (Zhu & Birnbaumer, 1998). Furthermore, direct CRAC channel block is not an entirely satisfactory explanation for the other drugs. ETYA and CDC in particular were much less effective when applied after ICRAC had activated than before. This kind of result is interpreted as evidence that first the drugs are not direct channel blockers and second, once activated, store-operated influx becomes independent of the initial signal (Rosado & Sage, 2000b; Broad et al. 2001). Our results could most easily be interpreted along these lines. However, it is possible that the inhibitors still block the CRAC channels directly but in a state-dependent manner. In this admittedly speculative scheme, CDC may bind to the resting but not open state of the CRAC channels and hence would be a more effective blocker if administered prior to store depletion. ETYA would bind significantly more favourably to the resting than the open state whereas NDGA would have to show only a modest preference for the resting state in some cells but bind very favourably to this state in other cells.
Hence we cannot rule out the possibility that the inhibitors interact with CRAC channels in a complex and state-dependent manner. One way to resolve this would be to record CRAC channel activity in an excised patch and then test whether the lipo-oxygenase blockers suppress the current and with what time course. Single-channel Na+ currents, thought to reflect monovalent permeation through CRAC channels in jurkat T-lymphocytes, have already been reported (Kerschbaum & Cahalan, 1999). But we have consistently failed to see such single-channel currents in RBL-1 cells and the properties of the Na+ current through CRAC channels differ substantially between the two cell types (Bakowski & Parekh, 2002).
In summary, our results show that lipo-oxygenase blockers all reduce the activation of ICRAC in RBL-1 cells. One possibility is that the activation step encompasses a lipo-oxygenase enzyme. Although we can eliminate other possible explanations, we cannot rule out a slow, state-dependent direct channel block by the inhibitors we have used. We are currently trying to resolve this using a molecular biological approach.
Acknowledgments
M.D.G. was supported by the Wellcome Trust (Grant no. 034204). D.B. is a British Heart Foundation Prize Student. A.B.P. is a Lister Institute Senior Research Fellow and holds the Amersham Fellowship in Medical Cell Biology at Keble College, Oxford.
REFERENCES
- Alvarez J, Montero M, Garcia-Sancho J. Cytochrome P-450 may link intracellular Ca2+ stores with plasma membrane Ca2+ influx. Biochemical Journal. 1991;274:193–197. doi: 10.1042/bj2740193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakowski D, Glitsch MD, Parekh AB. An examination of the secretion-like coupling model for the activation of the Ca2+ release-activated Ca2+ current ICRAC in RBL-1 cells. Journal of Physiology. 2001;532:55–71. doi: 10.1111/j.1469-7793.2001.0055g.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barowski D, Parekh AB. Monovalent cation permeability and Ca2+ block of the store-operated Ca2+ current ICRAC in rat basophilic leukemia cells. Pflügers Archiv. 2002 doi: 10.1007/s00424-001-0775-8. (in the Press) [DOI] [PubMed] [Google Scholar]
- Beaven MA, Rogers J, Moore JP, Hesketh TR, Smith GA, Metcalfe JC. The mechanism of the calcium signal and correlation with histamine release in 2H3 cells. Journal of Biological Chemistry. 1984;259:7129–7136. [PubMed] [Google Scholar]
- Berridge MJ. Capacitative Ca2+ entry. Biochemical Journal. 1995;312:1–11. doi: 10.1042/bj3120001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bokoch GM, Reed PW. Evidence for inhibition of leukotriene A4 synthesis by 5,8,4,11-eicosatetraynoic acid in guinea pig polymorphonuclear leucocytes. Journal of Biological Chemistry. 1981;256:4156–4159. [PubMed] [Google Scholar]
- Broad LM, Armstrong DL, Putney JW., Jr Role of the inositol 1,4,5-trisphosphate receptor in Ca2+ feedback inhibition of calcium release-activated calcium current (I(crac)) Journal of Biological Chemistry. 1999;274:32881–32888. doi: 10.1074/jbc.274.46.32881. [DOI] [PubMed] [Google Scholar]
- Broad LM, Braun F,-J, Lievremont J,-P, Bird G, St J, Putney JW., Jr Role of the phospholipase C-inositol 1,4,5-trisphosphate pathway in calcium release-activated calcium current and capacitative calcium entry. Journal of Biological Chemistry. 2001;276:15945–15952. doi: 10.1074/jbc.M011571200. [DOI] [PubMed] [Google Scholar]
- Cho H, Ueda M, Tamaoka M, Hamaguchi M, Aisaka K, Kiso Y, Inoue T, Ogino R, Tatsuoka T, Ishihara T, et al. Novel caffeic acid derivatives: extremely potent inhibitors of 12-lipoxygenase. Journal of Medical Chemistry. 1991;34:1503–1505. doi: 10.1021/jm00108a039. [DOI] [PubMed] [Google Scholar]
- Chyb S, Raghu P, Hardie RC. Polyunsaturated fatty acids activate the Drosophila light-sensitive channels TRP and TRPL. Nature. 1999;397:255–259. doi: 10.1038/16703. [DOI] [PubMed] [Google Scholar]
- Csutora P, Su Z, Kim HY, Bugrim A, Cunningham KW, Nuccitelli R, Keizer JE, Hanley MR, Blalock JE, Marchase RB. Calcium influx factor is synthesized by yeast and mammalian cells depleted of organellar Ca2+ stores. Proceedings of the National Academy of Sciences of the USA. 1999;96:121–126. doi: 10.1073/pnas.96.1.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fierro L, Lund P-E, Parekh AB. Comparison of the activation of the Ca2+-release-activated Ca2+ current ICRAC to InsP3 in jurkat T-lymphocytes, pulmonary artery endothelia and RBL-1 cells. Pflügers Archiv. 2000;440:580–587. doi: 10.1007/s004240000336. [DOI] [PubMed] [Google Scholar]
- Fierro L, Parekh AB. Substantial depletion of the intracellular calcium stores is required for macroscopic activation of ICRAC in RBL-1 cells. Journal of Physiology. 2000;522:247–257. doi: 10.1111/j.1469-7793.2000.t01-1-00247.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fierro L, Parekh AB. On the characterisation of the mechanism underlying passive activation of the Ca2+ realeseactivated Ca2+ current ICRAC. Journal of Physiology. 1999a;520:407–416. doi: 10.1111/j.1469-7793.1999.00407.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fierro L, Parekh AB. Fast Calcium-dependent inactivation of calcium release-activated calcium current (CRAC) in RBL-1 cells. Journal of Membrane Biology. 1999b;168:9–17. doi: 10.1007/s002329900493. [DOI] [PubMed] [Google Scholar]
- Glitsch MD, Parekh AB. Ca2+ store dynamics determines pattern of activation of the store-operated Ca2+ current ICRAC to InsP3. Journal of Physiology. 2000;523:283–290. doi: 10.1111/j.1469-7793.2000.t01-2-00283.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graier WF, Simecek S, Sturek M. Cytochrome P450 mono-oxygenase-regulated signalling of Ca2+ entry in human and bovine endothelial cells. Journal of Physiology. 1995;482:259–274. doi: 10.1113/jphysiol.1995.sp020515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamasaki Y, Tai HH. Gossypol, a potent inhibitor of arachidonate 5- and 12-lipoxygenases. Biochemica et Biophysica Acta. 1985;834:37–41. doi: 10.1016/0005-2760(85)90173-0. [DOI] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth F. Improved patch-clamp tecnhiques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Archiv. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Hope WC, Welton AF, Fiedler-Nagy C, Batula-Bernardo C, Coffey JW. In vitro inhibition of the biosynthesis of slow reacting substance of anaphylaxis (SRS-A) and lipoxygenase activity by quercetin. Biochemical Pharmacology. 1983;32:367. doi: 10.1016/0006-2952(83)90569-5. [DOI] [PubMed] [Google Scholar]
- Hoth M, Penner R. Depletion of intracellular calcium stores activates a calcium current in mast cells. Nature. 1992;355:353–356. doi: 10.1038/355353a0. [DOI] [PubMed] [Google Scholar]
- Hoth M, Penner R. Calcium release-activated calcium current in rat mast cells. Journal of Physiology. 1993;465:359–386. doi: 10.1113/jphysiol.1993.sp019681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irvine RF. Quantal’ Ca2+ release and the control of Ca2+ entry by in ositol phosphates—a possible mechanism. FEBBS Letters. 1990;283:524–530. doi: 10.1016/0014-5793(90)80692-c. [DOI] [PubMed] [Google Scholar]
- Kerschbaum H, Cahalan MD. Single-channel recording of a store-operated Ca2+ channel in Jurkat T lymphocytes. Science. 1999;283:836–839. doi: 10.1126/science.283.5403.836. [DOI] [PubMed] [Google Scholar]
- Korn SJ, Horn R. Nordihydroguaiaretic acid inhibits voltage-activated Ca2+ currents independently of lipoxygenase inhibition. Molecular Pharmacology. 1990;38:524–530. [PubMed] [Google Scholar]
- Mohr FC, Fewtrell C. Depolarization of rat basophilic leukemia cells inhibits calcium uptake and exocytosis. Journal of Cell Biology. 1987;104:783–792. doi: 10.1083/jcb.104.3.783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan AJ, Jacob R. Ionomycin enhances Ca2+ influx by stimulating store-regulated cation entry and not by a direct action at the plasma membrane. Biochemical Journal. 1994;300:665–672. doi: 10.1042/bj3000665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Needleman P, Turk J, Jakschik BA, Morrison AR, Leftkowitz JB. Arachidonic acid metabolism. Annual Review of Biochemistry. 1986;55:69–102. doi: 10.1146/annurev.bi.55.070186.000441. [DOI] [PubMed] [Google Scholar]
- Parekh AB. Slow feedback inhibition of calcium-release activated calcium current by calcium entry. Journal of Biological Chemistry. 1998;273:14925–14932. doi: 10.1074/jbc.273.24.14925. [DOI] [PubMed] [Google Scholar]
- Parekh AB, Fleig A, Penner R. The store-operated calcium current ICRAC: Non-linear activation by InsP3 and possible dissociation from calcium release. Cell. 1997;89:973–980. doi: 10.1016/s0092-8674(00)80282-2. [DOI] [PubMed] [Google Scholar]
- Parekh AB, Penner R. Depletion-activated Ca2+ current is inhibited by protein kinase in RBL-2H3 cells. Proceedings of the National Academy of Sciences of the USA. 1995;92:7907–7911. doi: 10.1073/pnas.92.17.7907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parekh AB, Penner R. Store-operated calcium influx. Physiological Reviews. 1997;77:901–930. doi: 10.1152/physrev.1997.77.4.901. [DOI] [PubMed] [Google Scholar]
- Peppelenbosch MP, Tertoolen LG, Den Hertog J, De Laat SW. Epidermal growth factor activates calcium channels by phospholipase A2/5-lipoxygenase-mediated leukotriene C4 production. Cell. 1992;69:295–303. doi: 10.1016/0092-8674(92)90410-e. [DOI] [PubMed] [Google Scholar]
- Putney JW. A model for receptor-regulated calcium entry. Cell Calcium. 1986;7:1–12. doi: 10.1016/0143-4160(86)90026-6. [DOI] [PubMed] [Google Scholar]
- Randriamampita C, Tsien RY. Emptying of intracellular Ca2+ stores releases a novel small messenger that stimulates Ca2+ influx. Nature. 1993;364:809–813. doi: 10.1038/364809a0. [DOI] [PubMed] [Google Scholar]
- Rosado JA, Sage SO. The actin cytoskeleton in store-mediated calcium entry. Journal of Physiology. 2000a;526:221–229. doi: 10.1111/j.1469-7793.2000.t01-2-00221.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosado JA, Sage SO. Phosphoinositides are required for store-mediated calcium entry in human platelets. Journal of Biological Chemistry. 2000b;275:9110–9113. doi: 10.1074/jbc.275.13.9110. [DOI] [PubMed] [Google Scholar]
- Salari H, Braquet P, Borgeat P. Comparative effects of indomethacin, acetylenic acids, 15-HETE, nordihydroguaiaretic acid and BW755C on the metabolism of arachidonic acid in human leukocytes and platelets. Prostaglandins and Leukotrienes, and Medicine. 1984;13:53–60. doi: 10.1016/0262-1746(84)90102-1. [DOI] [PubMed] [Google Scholar]
- Samuelsson B. Leukotrienes: mediators of immediate hypersensitivity reactins and inflammation. Science. 1983;220:568–575. doi: 10.1126/science.6301011. [DOI] [PubMed] [Google Scholar]
- Shen TY, Winter CA. Chemical and biological studies on indomethacin, sulindac and their analogs. Advances in Drug Research. 1977;12:90–245. [PubMed] [Google Scholar]
- Taylor AS, Morrison AR, Russell JH. Incorporation of 5,8,11,14-eicosatetraynoic acid (ET-1A) into cell lipids: competition with arachidonic acid for esterification. Prostaglandins. 1985;29:449–458. doi: 10.1016/0090-6980(85)90102-9. [DOI] [PubMed] [Google Scholar]
- Van der Donk EMM, Dubois GR, Verhagen J, Veldink GA, Vliegenhart JFG. Improved purification of 12-lipoxygenase from rat basophilic leukemia cells and conditions for optimal enzyme activity. Biochemica et Biophysica Acta. 1991;1074:443–447. doi: 10.1016/0304-4165(91)90098-2. [DOI] [PubMed] [Google Scholar]
- Wong A, Cook MN, Hwang SM, Sarau HM, Foley JJ, Crooke ST. Stimulation of leukotriene production and membrane translocation of 5-lipoxygenase by cross-linking of the IgE receptors in RBL-2H3 cells. Biochemistry. 1992;31:4046–4053. doi: 10.1021/bi00131a021. [DOI] [PubMed] [Google Scholar]
- Yao Y, Ferrer-Montiel AV, Montal M, Tsien RY. Activation of store-operated Ca2+ current in Xenopus oocytes requires SNAP-25 but not a diffusible messenger. Cell. 1999;98:475–485. doi: 10.1016/s0092-8674(00)81976-5. [DOI] [PubMed] [Google Scholar]
- Zhu X, Birnbaumer L. Calcium channels formed by mammalian Trp homologues. News in Physiological Sciences. 1998;13:211–217. doi: 10.1152/physiologyonline.1998.13.5.211. [DOI] [PubMed] [Google Scholar]
- Zweifach A, Lewis RS. Slow calcium-dependent inactivation of depletion-activated calcium current. Store-dependent and independent mechanisms. Journal of Biological Chemistry. 1995;270:14445–14451. doi: 10.1074/jbc.270.24.14445. [DOI] [PubMed] [Google Scholar]