

Abstract
Isolated interlobular ducts from the guinea-pig pancreas secrete a HCO3−-rich fluid in response to secretin. To determine the role of Cl− transporters in this process, intracellular Cl− concentration ([Cl−]i) was measured in ducts loaded with the Cl−-sensitive fluoroprobe, 6-methoxy-N-ethylquinolinium chloride (MEQ). [Cl−]i decreased when the luminal Cl− concentration was reduced. This effect was stimulated by forskolin, was not dependent on HCO3− and was not inhibited by application of the anion channel/transporter inhibitor H2DIDS to the luminal membrane. It is therefore attributed to a cAMP-stimulated Cl− conductance, probably the cystic fibrosis transmembrane conductance regulator (CFTR) Cl− channel. [Cl−]i also decreased when the basolateral Cl− concentration was reduced. This effect was not stimulated by forskolin, was largely dependent on HCO3− and was inhibited by basolateral H2DIDS. It is therefore mediated mainly by Cl−/HCO3− exchange. With high Cl− and low HCO3− concentrations in the lumen, steady-state [Cl−]i was 25-35 mm in unstimulated cells. Stimulation with forskolin caused [Cl−]i to increase by approximately 4 mm due to activation of the luminal anion exchanger. With low Cl− and high HCO3− concentrations in the lumen to simulate physiological conditions, steady-state [Cl−]i was 10–15 mm in unstimulated cells. Upon stimulation with forskolin, [Cl−]i fell to approximately 7 mm due to increased Cl− efflux via the luminal conductance. We conclude that, during stimulation under physiological conditions, [Cl−]i decreases to very low levels in guinea-pig pancreatic duct cells, largely as a result of the limited capacity of the basolateral transporters for Cl− uptake. The resulting lack of competition from intracellular Cl− may therefore favour HCO3− secretion via anion conductances in the luminal membrane, possibly CFTR.
Pancreatic duct cells secrete a HCO3−-rich, isotonic fluid in response to stimulation with secretin (Case & Argent, 1993). In previous studies we have investigated the mechanisms of HCO3− secretion across the luminal membrane using interlobular duct segments isolated from guinea-pig pancreas. We have found that in unstimulated cells HCO3− efflux across the luminal membrane is mediated by Cl−/HCO3− exchange, while in stimulated cells luminal HCO3− transport is chiefly mediated by an unidentified HCO3− efflux pathway that is largely independent of luminal Cl− (Ishiguro et al. 1996b, 1998). Furthermore, it appears that the Cl−/HCO3− exchanger is inhibited when the luminal HCO3− concentration reaches physiological values of 125 mm or more (Ishiguro et al. 2000). This property is probably of crucial importance in maintaining the high concentration of HCO3− in pancreatic juice.
Although the cystic fibrosis transmembrane conductance regulator (CFTR) Cl− channel is expressed in the luminal membrane of pancreatic duct cells, its role in HCO3− secretion remains to be determined. CFTR has a lower permeability to HCO3− than to Cl− (Gray et al. 1990) so it is not, therefore, the most obvious candidate for the unidentified HCO3− efflux pathway across the luminal membrane. On the other hand, if Cl− was at electrochemical equilibrium, CFTR could appear to act as a HCO3− channel.
Forskolin-stimulated fluid secretion is markedly reduced in isolated pancreatic ducts from CFTR null (−/−) mice (San-Román et al. 1999). However, pancreatic insufficiency in the phenotypes of different CFTR mutations does not always reflect the defect in the cAMP-dependent Cl− conductance (Fanen et al. 1997). This suggests that CFTR may have regulatory effects on other H+/HCO3− transporters. For example, CFTR has been shown to modulate luminal Cl−/HCO3− exchange in the main duct of the mouse pancreas (Lee et al. 1999). Furthermore, when CFTR mutants are expressed in human embryonic kidney HEK293 cells, it is their ability to stimulate Cl−-HCO3− exchange that appears to correlate with the pancreatic phenotype rather than their Cl− conductance properties (Choi et al. 2001).
It has also been proposed that activation of CFTR indirectly drives basolateral Na+-HCO3− cotransport by membrane depolarization in cultured human pancreatic duct cells (Shumaker et al. 1999). It is possible that changes in intracellular Cl− concentration resulting from CFTR activity have regulatory effects on a variety of key membrane transport proteins, as described in other epithelia (Dinudom et al. 1993; Robertson & Foskett, 1994). In order to understand how the activity of CFTR and the concentration of intracellular Cl− ions are related to HCO3− secretion, it is clearly necessary to measure intracellular Cl− concentration and define the characteristics of the Cl−-transport pathways in these cells.
We recently investigated the permeability characteristics of the basolateral and luminal membranes to HCO3− in microperfused interlobular duct segments by measuring changes in intracellular pH in response to changes in bath and luminal HCO3− concentrations (Ishiguro et al. 2000). Although HCO3− enters the cells readily across the basolateral membrane, largely by Na+-HCO3− cotransport, the luminal membrane is relatively impermeable to extracellular HCO3− and thus presents a significant barrier to the re-entry of secreted HCO3−. To investigate the mechanism of Cl− transport across the pancreatic ductal epithelium, we have now examined the changes in intracellular Cl− concentration that occur when the basolateral or luminal Cl− concentrations are modified in microperfused interlobular duct segments isolated from guinea-pig pancreas.
METHODS
The following study was approved by the Ethical Committee of Nagoya University on Animal Use for Experiments.
Isolation and culture of interlobular ducts
Female Hartley guinea-pigs (350–450 g) were killed by cervical dislocation. The body and tail of the pancreas were removed and interlobular ducts (diameter 100–150 μm, length 800–1200 μm) were isolated and cultured overnight as described previously (Ishiguro et al. 1996a).
Solutions
The standard Hepes-buffered solution contained (mm): 140 NaCl, 5 KCl, 1 CaCl2, 1 MgCl2, 10 d-glucose and 10 Hepes and was equilibrated with 100 % O2. The Cl−-free Hepes-buffered solutions were made by replacing Cl− with glucuronate. The standard HCO3−-buffered solution contained (mm): 115 NaCl, 5 KCl, 1 CaCl2, 1 MgCl2, 10 d-glucose and 25 NaHCO3 and was equilibrated with 95 % O2-5 % CO2. The low Cl−-high HCO3− solution contained 125 mm HCO3− and 24 mmCl−, and was equilibrated with 95 % O2−5 % CO2 (pH ∼8.2). With the exception of the high-HCO3− solution, pH was adjusted to 7.4 at 30 °C.
Microperfusion of isolated ducts
The lumen of isolated interlobular duct segments was microperfused as described previously (Ishiguro et al. 1999) so that the bath and luminal solutions could be modified separately. The bath was continuously perfused at 3 ml min−1 at 30 °C.
Measurement of intracellular Cl−
Intracellular Cl− concentration ([Cl−]i) was estimated by microfluorometry in isolated duct segments loaded with the Cl−-sensitive fluoroprobe, 6-methoxy-N-ethylquinolinium chloride (MEQ). Dihydro-MEQ (diH-MEQ), the membrane-permeant form of MEQ, was synthesized (reduced by sodium borohydride) from MEQ according to the instructions provided by Molecular Probes (Eugene, OR, USA). DiH-MEQ diffuses into the cells and is readily oxidized to the membrane-impermeant MEQ, which remains entrapped in the cytosol (Biwersi & Verkman, 1991). The synthesized diH-MEQ was dissolved at a concentration of 100 mm in dimethyl sulphoxide and stored under N2 at −80 °C for up to 1 month. The duct segments were incubated for 30 min at room temperature (20–23 °C) with diH-MEQ (100 μm) in a Hepes-buffered solution equilibrated with 100 % O2. The ducts were stored at 4 °C until required for use.
Microfluorometry was performed on a small area of the ductal epithelium (10–20 cells) in microperfused ducts which were illuminated at 340 nm. All experiments were performed at 30 °C to minimise the leakage of MEQ from the cells. The fluorescence intensity F was measured at 430 nm and normalized to the value at the beginning of the experiment. Calibration was performed in situ by application of a combination of nigericin (5 μm) and tributyltin chloride (10 μm), a chloride-hydroxyl exchange ionophore. The calibration solutions were prepared by mixing a Cl−-rich solution, containing 150 mm KCl and 10 mm d-glucose, and a Cl−-free solution, containing 150 mm potassium gluconate and 10 mm d-glucose. Unbuffered solutions were chosen because organic pH buffers such as Hepes quench the fluorescence of the Cl−-sensitive dye and Hepes probably enters the cell in the presence of ionophores (Lau et al. 1994). Figure 1A shows representative calibration data and Fig. 1B is a Stern-Volmer plot of the pooled data. The Stern-Volmer constant was 11 m−1. In situ calibration was performed at the end of each experiment by measuring the fluorescence intensity F0 obtained when the duct was exposed to the Cl−-free solution. Changes in F0/F in each experiment were then converted to changes in [Cl−]i using the Stern-Volmer constant obtained from the pooled data.
Figure 1. Calibration of MEQ fluorescence for measurement of intracellular Cl− concentrations in isolated interlobular duct segments.
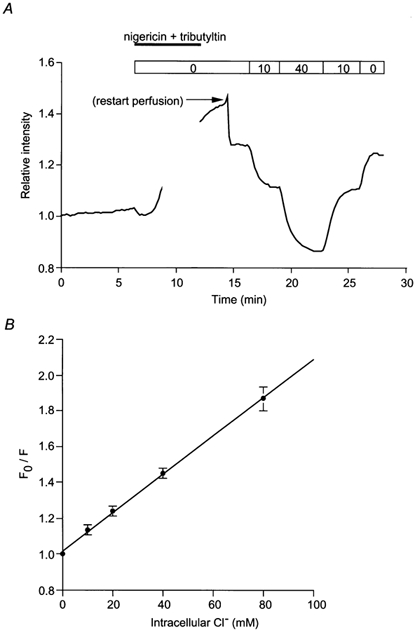
A, a representative experiment showing MEQ calibration performed in situ by exposing the duct to nigericin (5 μm) and tributyltin chloride (10 μm) and by varying the perfusate Cl− concentration (by substitution with gluconate). MEQ fluorescence is plotted as the relative intensity normalised to the value at the beginning of the recording. B, Stern-Volmer plot of the calibration data (means ±s.d., n = 9). F0 and F are the values of the relative intensity in the absence and presence of Cl−, respectively.
Measurement of membrane potential
Membrane potential was measured by impaling the basolateral membrane of microperfused ducts with glass microelectrodes filled with 0.5 m KCl and connected to an electrometer (World Precision Instruments Inc., Sarasota, FL, USA). The intracellular potential (Vm) was measured with reference to the grounded bath. Since one end of the duct was always open to the bath, the apical and basolateral membrane potentials were equal under the conditions of the experiments.
Materials
MEQ and dihydro-4,4′-diisothiocyanatostilbene-2,2′-disulphonic acid (H2DIDS) were obtained from Molecular Probes (Eugene, OR, USA); forskolin, nigericin and dibutyryl cyclic AMP were from Sigma (St Louis, MO, USA); glibenclamide and 5-nitro-2-(3-phenylpropylamino)benzoic acid (NPPB) were from Research Biochemicals International (Natick, MA, USA); tributyltin chloride was from Aldrich Chemical Company (Milwaukee, WI, USA).
Statistics
Averaged data are presented as means ± s.e.m. Statistically significant differences were determined using Student's t test for paired or unpaired data as appropriate.
RESULTS
Changes in [Cl−]i on modifying luminal Cl− concentration
In order to determine the steady-state value of [Cl−]i under physiological conditions, the duct lumen and the bath were perfused initially with a HCO3−-buffered solution containing 124 mmCl− and 25 mm HCO3−. Figure 2A shows changes in MEQ fluorescence intensity in a representative experiment followed by in situ calibration, achieved by exposing the duct to 0, 20 and 40 mmCl− solutions in the presence of nigericin and tributyltin. From such data [Cl−]i was estimated to be 31.2 ± 2.6 mm (n = 5). Under the same conditions, the intracellular potential (Vm) was −56.2 ± 3.3 mV (n = 9). Since a value of 14 mm would be expected if Cl− ions were at electrochemical equilibrium, this suggests that Cl− is accumulated across either the luminal or the basolateral membrane, perhaps by Cl−-HCO3− exchange since this would provide a pathway for electroneutral Cl− entry.
Figure 2. Changes in [Cl−]i on modifying luminal Cl− concentration.
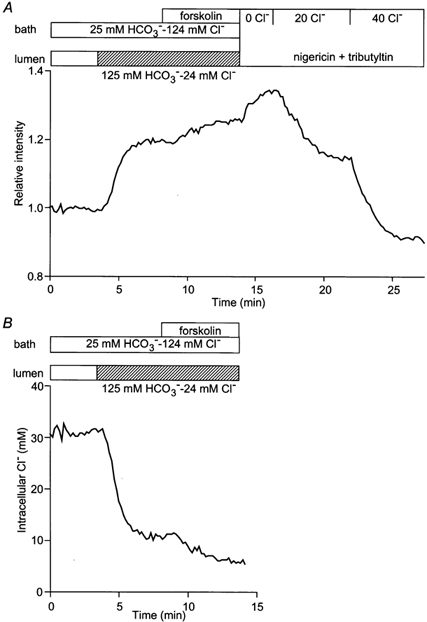
Initially the bath and lumen of the duct segments were perfused with the standard HCO3−-buffered solution containing 124 mmCl− and 25 mm HCO3−. Thereafter, the luminal perfusate was switched to a low Cl−-high HCO3− solution containing 24 mmCl− and 125 mm HCO3−, and then forskolin (1 μm) was applied to the bath. A, changes in relative intensity in a representative experiment followed by in situ calibration by exposing the duct to 0, 20 and 40 mmCl− solution in the presence of nigericin and tributyltin. B, changes in [Cl−]i which were estimated from the calibration equation. One of five experiments.
When the luminal perfusate was switched to a solution more representative of that present in the duct lumen during stimulation under physiological conditions (i.e. containing 24 mmCl− and 125 mm HCO3−), [Cl−]i decreased to 12.5 ± 3.2 mm (n = 5) over a period of 4–5 min (Fig. 2B). This shift to a lower steady-state value is probably due both to the increased gradient for Cl− efflux, via a luminal conductance, and a decrease in Cl− uptake via the luminal anion exchanger. We have shown previously that the latter is inhibited by high luminal HCO3− concentrations (Ishiguro et al. 2000). Under these conditions, basolateral application of forskolin (1 μm), an activator of adenylate cyclase, induced a further decrease in [Cl−]i to 6.7 ± 0.7 mm. This is probably due to an increase in luminal Cl− conductance and the consequent increase in Cl− efflux resulting from the activation of CFTR.
In Fig. 3A forskolin was applied during perfusion of both the lumen and bath with the high Cl− solution (i.e. containing 124 mmCl− and 25 mm HCO3−). Under these conditions, forskolin (1 μm) caused a small increase of 4.3 ± 0.8 mm (n = 7) in the steady-state value of [Cl−]i. Stimulation with forskolin (1 μm) or dibutyryl cyclic AMP (dbcAMP, 0.1–0.5 mm) also caused a small but significant depolarization of Vm from −59.5 ± 3.6 to −50.7 ± 3.2 mV (n = 5, P < 0.05). Since the electrochemical gradient for Cl− would still strongly favour Cl− efflux to the lumen during cAMP stimulation, the increase in steady-state Cl− concentration must indicate a cAMP-evoked increase in Cl− uptake either at the luminal or at the basolateral membrane. When the luminal perfusate was then switched to the high HCO3− solution (24 mmCl− and 125 mm HCO3−), [Cl−]i decreased to 7.7 ± 0.7 mm (n = 7) within 30 s, probably due to an increase in Cl− conductance at the luminal membrane. Again, there was little change in membrane potential, which showed just a small hyperpolarization of 7.1 ± 1.1 mV to −57.8 ± 3.6 mV (n = 5, P < 0.01). [Cl−]i soon returned to its previous value when the luminal perfusate was switched back to the high Cl− solution.
Figure 3. Effects of forskolin on Cl− flux across the luminal membrane in the presence of HCO3−/CO2.
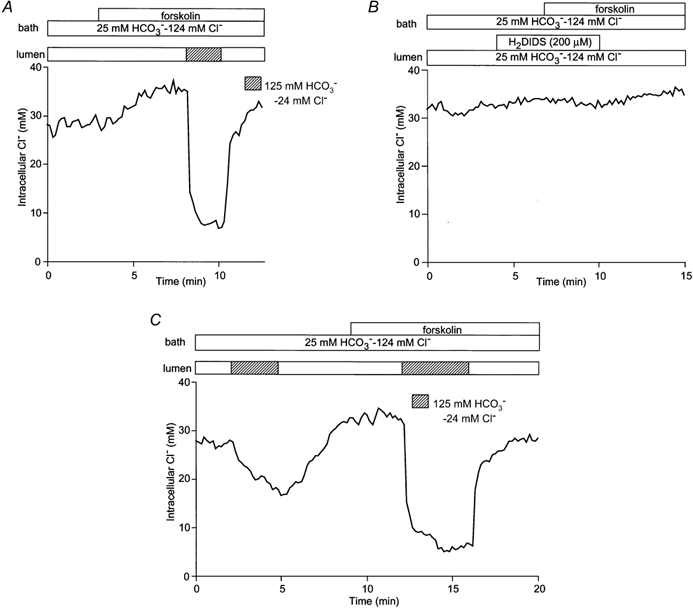
Initially the bath and lumen of the duct segments were perfused with the standard HCO3−-buffered solution. A, forskolin (1 μm) was applied to the bath and then the luminal perfusate was switched to the low Cl−-high HCO3− solution in the continued presence of forskolin. One of seven experiments. B, the luminal membrane was pretreated with H2DIDS (200 μm) and forskolin (1 μm) was applied to the bath. One of four experiments. C, the luminal perfusate was switched to the low Cl−-high HCO3− solution in the absence and presence of forskolin (1 μm). One of five experiments.
To examine the possible role of the luminal membrane anion exchanger in the increase in [Cl−]i induced by forskolin, H2DIDS (200 μm), an established inhibitor of anion channels/transporters, was applied to the luminal membrane (Fig. 3B). Luminal H2DIDS alone had no effect on the resting value of [Cl−]i under these conditions: 29.0 ± 2.2 mm at 3 min compared with the preceding control value of 29.3 ± 2.1 mm (n = 4, n.s.). Upon stimulation with forskolin, [Cl−]i did not change significantly (Δ[Cl−]i = 0.5 ± 0.6 mm, n.s.) indicating that H2DIDS inhibits the rise in [Cl−]i observed in Fig. 3A. This suggests that forskolin stimulates the anion exchanger in the luminal membrane with the result that Cl− uptake occurs in exchange for HCO3− efflux to the lumen. Prior to stimulation, however, the lack of effect of luminal H2DIDS (Fig. 3B) suggests that the basolateral anion exchanger has the dominant role in maintaining [Cl−]i under resting conditions.
Figure 3C shows a direct comparison, in the same duct, of the rates of Cl− efflux across the luminal membrane in the presence and absence of forskolin. In the unstimulated condition, when the luminal Cl− concentration was reduced from 124 mm to 24 mm by replacement with HCO3−, the initial rate of Cl− efflux (measured over the first 30 s) was 0.09 ± 0.02 mm s−1 (n = 5). Forskolin stimulation significantly increased the rate to 0.42 ± 0.08 mm s−1 (P < 0.01). These data indicate that Cl− ions move freely across the luminal membrane according to changes in luminal Cl− and HCO3− concentrations and that the Cl− transport pathway is activated by stimulation with forskolin.
To examine the movement of Cl− across the luminal membrane in the absence of HCO3−, the duct lumen and the bath were perfused with a HCO3−-free, Hepes-buffered solution containing 139 mmCl−. Under these conditions, the steady-state value of [Cl−]i was 22.4 ± 1.8 mm (n = 5) (Fig. 4A). This is significantly lower (P < 0.05) than the mean value of 31.2 ± 2.6 mm obtained in the presence of HCO3− (Fig. 3). It therefore supports our hypothesis that basolateral and/or luminal Cl−-HCO3− exchangers contribute to intracellular Cl− accumulation. When the luminal perfusate was switched to a Cl−-free, Hepes-buffered solution (by substitution with glucuronate), [Cl−]i decreased to 12.4 ± 1.6 mm. This suggests that Cl− can cross the luminal membrane independently of HCO3−, and that there is probably a significant Cl− conductance in the luminal membrane, even in the absence of stimulation. The steady-state value of [Cl−]i thus reflects the balance between basolateral influx and luminal efflux. Stimulation with 1 μm forskolin induced a further decrease in [Cl−]i to 9.9 ± 1.4 mm, presumably due to increased efflux via the luminal Cl− conductance.
Figure 4. Effects of forskolin on Cl− flux across the luminal membrane in the absence of HCO3−/CO2.
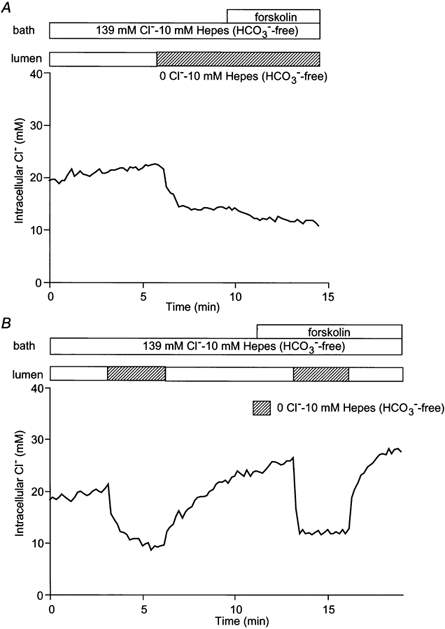
Initially the bath and lumen of the duct segments were perfused with the standard Hepes-buffered solution containing 139 mmCl−. A, the luminal perfusate was switched to the 0 Cl− Hepes-buffered solution and then forskolin (1 μm) was applied to the bath. One of five experiments. B, the luminal perfusate was switched to the 0 Cl− Hepes-buffered solution in the absence and presence of forskolin (1 μm). One of four experiments.
The influence of forskolin on the rate of Cl− efflux in the absence of HCO3−/CO2 is shown in Fig. 4B. When the luminal Cl− concentration was reduced from 139 to 0 mm in the resting duct, the initial rate of Cl− efflux was 0.11 ± 0.02 mm s−1 (n = 4). Forskolin stimulation significantly increased the rate to 0.35 ± 0.07 mm s−1 (P < 0.01). These data indicate that Cl− movement across the luminal membrane is also accelerated by forskolin stimulation in the absence of HCO3−. It seems reasonable to suggest that forskolin-stimulated transport of Cl− across the luminal membrane is mediated by the CFTR Cl− channel. Therefore, glibenclamide (a blocker of the CFTR channel), H2DIDS and NPPB (a non-specific Cl− channel blocker) were each applied to the lumen to test their ability to inhibit the forskolin-stimulated efflux of Cl− across the luminal membrane (Fig. 5). Perhaps surprisingly, the accelerated Cl− efflux due to forskolin stimulation was not inhibited by 100 μm glibenclamide (0.62 ± 0.13 mm s−1 compared with a preceding control value of 0.56 ± 0.10 mm s−1, n = 4, n.s., Fig. 5A) nor by 200 μm H2DIDS (0.47 ± 0.05 mm s−1 compared with 0.49 ± 0.06 mm s−1, n = 4, n.s., Fig. 5B) nor by 10 μm NPPB (0.58 ± 0.13 mm s−1 compared with 0.59 ± 0.14 mm s−1, n = 4, n.s., Fig. 5C).
Figure 5. Effects of luminal inhibitors on forskolin-stimulated Cl− flux across the luminal membrane.
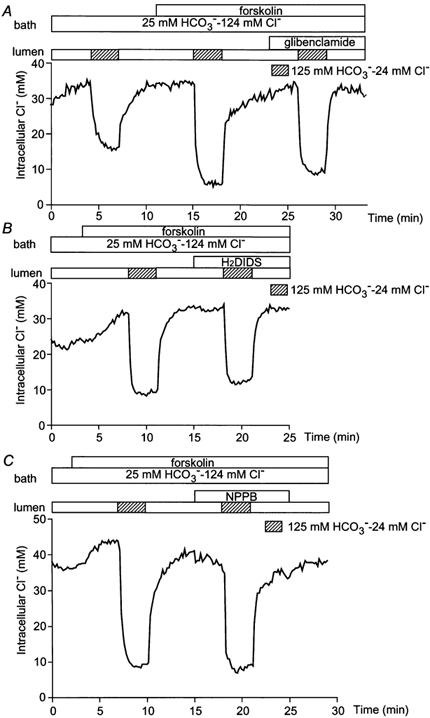
Initially the bath and lumen of the duct segments were perfused with the standard HCO3−-buffered solution and forskolin (1 μm) was applied to the bath. The luminal perfusate was switched to the low Cl−-high HCO3− solution in the absence and presence of 100 μm glibenclamide (A), 500 μm H2DIDS (B), or 10 μm NPPB (C). Each trace is a representative of four experiments.
Changes in [Cl−]i on modifying basolateral Cl− concentration
In order to characterize the transport pathways for Cl− across the basolateral membrane, the duct lumen and bath were again initially perfused with the standard HCO3−-buffered solution containing 124 mmCl− and 25 mm HCO3− (Fig. 6A). When the bath solution was then switched to the low Cl−-high HCO3− solution, [Cl−]i decreased slowly and failed to reach a new steady-state value within 5 min. Also, in contrast to the luminal membrane, the initial rate of Cl− efflux across the basolateral membrane (0.13 ± 0.03 mm s−1, n = 5) was not affected by forskolin stimulation (0.11 ± 0.02 mm s−1, n.s.).
Figure 6. Changes in [Cl−]i on modifying basolateral Cl− concentration.
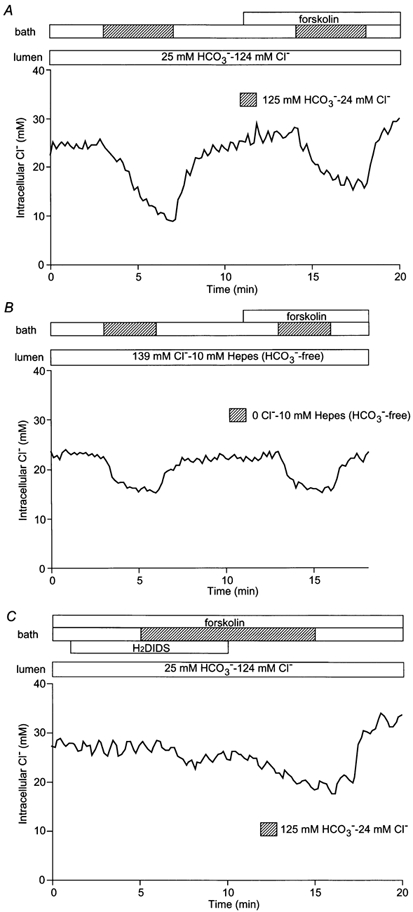
A, initially the bath and lumen of the duct segments were perfused with the standard HCO3−-buffered solution. The basolateral perfusate was then switched to the low Cl−-high HCO3− solution in the absence and presence of forskolin (1 μm). One of five experiments. B, with the bath and lumen initially perfused with the standard Hepes-buffered solution, the luminal perfusate was switched to the 0 Cl− Hepes-buffered solution in the absence and presence of forskolin (1 μm). One of four experiments. C, with the bath and lumen initially perfused with the HCO3−-buffered solution, the basolateral membrane was pretreated with H2DIDS (200 μm) and then the basolateral perfusate was switched to the low Cl−-Ihigh HCO3− solution in the presence of forskolin (1 μm). One of four experiments.
One possible pathway for Cl− movement across the basolateral membrane is the basolateral anion exchanger which we have described previously (Ishiguro et al. 2000). By analogy with the luminal membrane, this exchanger would be expected to contribute to Cl− efflux from the cells when the bath solution is switched from the high Cl− to the high HCO3− solution. To test this hypothesis, the experiment described above was repeated in the nominal absence of HCO3−/CO2. As can be seen in Fig. 6B, the absence of HCO3−/CO2 reduced, but did not completely abolish, the change in [Cl−]i when the concentration of Cl− in the bath was reduced (in this case from 139 mm to zero). The remaining change in [Cl−]i, which was not accelerated by stimulation with forskolin (0.12 ± 0.03 mm s−1 compared with 0.11 ± 0.02 mm s−1 in the unstimulated control, n = 4, n.s.), may perhaps be attributable to a small basolateral Cl− conductance.
Further evidence of basolateral anion exchange is seen in the effect of H2DIDS. Pre-treatment of the basolateral membrane with 200 μm H2DIDS largely abolished the efflux of Cl− when the bath Cl− concentration was reduced during forskolin stimulation (Fig. 6C): the rate of decrease in [Cl−]i was 0.022 ± 0.008 mm s−1 (n = 4) compared with 0.11 ± 0.02 mm s−1 (n = 5, P < 0.05, Fig. 6A) in the absence of the inhibitor. When H2DIDS was subsequently withdrawn, [Cl−]i started to decrease slowly (0.042 ± 0.011 mm s−1). These data indicate that Cl− movement across the basolateral membrane is not stimulated by forskolin, is largely dependent on HCO3−/CO2 and is inhibited by H2DIDS. It is therefore probably mediated by the basolateral anion exchanger.
Effects of basolateral and luminal H2DIDS on steady-state [Cl−]i
The data presented above suggest that both basolateral and luminal Cl−-HCO3− exchangers contribute to Cl− accumulation in pancreatic duct cells. To investigate their relative contributions in unstimulated and forskolin-stimulated cells, we examined the effects of basolateral and luminal H2DIDS on steady-state [Cl−]i (Fig. 7). In each experiment, 200 μm H2DIDS was applied first to the bath and then added to the lumen or vice versa. In all of the experiments, both the bath and lumen were perfused throughout with the high-Cl− solution.
Figure 7. Effects of basolateral or luminal H2DIDS on [Cl−]i.
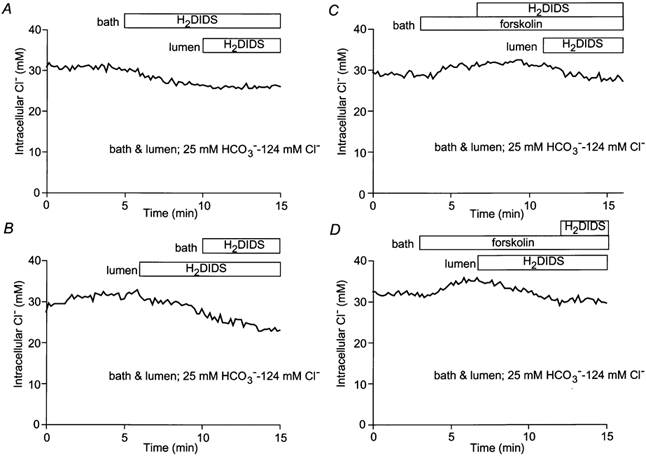
The bath and lumen of the duct segments were perfused with the standard HCO3−-buffered solution. H2DIDS (200 μm) was sequentially applied to the bath and then to the lumen (A and C) or in a reverse order (B and D) in the absence (A and B) or presence (C and D) of forskolin (1 μm). Each trace is a representative of four experiments.
In unstimulated ducts (Fig. 7A and B), basolateral H2DIDS caused [Cl−]i to decrease by 7.1 ± 0.7 mm (n = 4) whereas the decrease was only 1.0 ± 0.7 mm (n = 4) with luminal H2DIDS. This suggests that, in the resting condition, the basolateral Cl−-HCO3− exchanger is more active than the luminal exchanger. This is consistent with our previous intracellular pH (pHi) data where we showed that, in the presence of HCO3−/CO2, substitution of basolateral Cl− caused a larger increase in pHi than substitution of luminal Cl− (Ishiguro et al. 2000).
On the other hand, in forskolin-stimulated ducts (Fig. 7C and D), luminal H2DIDS caused [Cl−]i to decrease by 6.0 ± 0.6 mm (n = 4) while basolateral H2DIDS had no significant effect on [Cl−]i. This suggests that, in forskolin-stimulated ducts, the luminal Cl−-HCO3− exchanger becomes more active and the basolateral exchanger less active. We therefore anticipate that elevation of intracellular cAMP has opposite effects on the activities of the two exchangers.
DISCUSSION
Maximal stimulation of the guinea-pig pancreas with secretin in vivo evokes the production of a juice containing approximately 140 mm HCO3− and only 20 mmCl− (Padfield et al. 1989). This HCO3−-rich fluid probably arises mainly in the pancreatic ducts and we have shown previously that isolated interlobular duct segments can secrete a fluid containing more than 130 mm HCO3− when stimulated with secretin (Ishiguro et al. 1998). This contrasts with the situation in the rat pancreas where the maximal HCO3− concentration in the juice is only about 70 mm and the Cl− concentration correspondingly greater (Sewell & Young, 1975). Why do guinea-pig ducts secrete so much more HCO3−, given that in both species the dominant anion conductance in the luminal membrane is the CFTR Cl− channel, which has a preferential permeability to Cl− over HCO3− (Gray et al. 1990)?
To address this issue, we have investigated the transport pathways for Cl− in microperfused guinea-pig duct segments. Our approach has been to monitor the intracellular Cl− concentration using the Cl−-sensitive fluoroprobe MEQ and to examine the effects of sudden changes in bath and luminal Cl− concentrations, at rest and during stimulation with forskolin, and in the presence and absence of HCO3−. We have also examined the effects of anion transport inhibitors on these changes and on the steady-state intracellular Cl− concentration.
Analysis of the driving forces for Cl− transport requires parallel measurements of membrane potential. Our preliminary measurements using conventional glass microelectrodes indicate that Vm is approximately −60 mV in the resting condition when the lumen is perfused with the high Cl−-low HCO3− solution. Stimulation with cAMP causes Vm to depolarize by about 10 mV, significantly less than the 30 mV depolarization reported previously in rat pancreatic duct cells following stimulation with secretin (Novak & Greger, 1988). Under more physiological conditions, with low Cl− and high HCO3− concentrations in the lumen, Vm remained at approximately −60 mV even during cAMP stimulation. Thus, in guinea-pig pancreatic ducts, Vm remains relatively constant during stimulation with cAMP and following changes in luminal anion composition.
Measurement of intracellular Cl− concentration in pancreatic duct cells
Pancreatic duct cells were easily loaded with diH-MEQ, the membrane-permeant form of the Cl−-sensitive fluoroprobe MEQ. Since MEQ is a single-wavelength, non-ratiometric fluoroprobe, the fluorescence signal tends to decline gradually during an experiment as a result of leakage of the dye from the cells, thus making calibration difficult. Provided that the experiments were performed at 30 °C, however, we found that leakage from the duct cells was negligible and the baseline remained relatively flat during experiments of up to 40 min duration.
Under the conditions of these experiments, steady-state intracellular Cl− concentrations in guinea-pig duct cells were generally in the range from 10 to 30 mm. Although not many data are available for comparison in other epithelia, it is interesting that in salivary acinar cells, which secrete a Cl−-rich primary saliva, [Cl−]i has a substantially higher value of approximately 60 mm (Foskett, 1990). This is maintained largely by a basolateral Na+-K+-2Cl− cotransporter (NKCC) and it ensures that there is a large driving force for Cl− efflux across the luminal membrane during secretion. The lower [Cl−]i values obtained in guinea-pig duct cells may therefore correlate with the fact that these cells preferentially secrete HCO3− ions rather than Cl−. Furthermore it may reflect the absence of the powerful basolateral Cl− uptake pathways, such as the NKCC, which are found in other secretory epithelia.
Cl− transport across the luminal membrane
From experiments in which the luminal Cl− concentration was varied, we conclude that Cl− ions move relatively freely in either direction across the luminal membrane. Furthermore, elevation of intracellular cAMP by application of forskolin accelerated the efflux of Cl− ions to the lumen when the luminal Cl− concentration was subsequently dropped (Fig. 3A and C). This was true whether or not HCO3− was present (Fig. 4), and the efflux of Cl− was not significantly inhibited by luminally applied H2DIDS (Fig. 5B).
The latter observation might appear surprising since an H2DIDS-sensitive Cl−-HCO3− exchanger is known to be present in the luminal membrane. Indeed, Fig. 3B shows that cAMP-induced activation of the luminal membrane anion exchanger contributes to the rise in [Cl−]i following stimulation with forskolin (Fig. 3A). This observation is supported by other evidence suggesting that cAMP activates the luminal anion exchanger in both guinea-pig (Ishiguro et al. 1995) and mouse pancreatic ducts (Lee et al. 1999). The luminal anion exchanger would therefore be expected to carry at least part of the Cl− flux out of the cell when the luminal Cl− concentration was dropped, particularly in experiments where Cl− was substituted by a high concentration of HCO3− (125 mm) since the inward HCO3− gradient would further increase the driving force for anion exchange. The fact that the exchanger did not appear to contribute to Cl− efflux under these conditions is, however, consistent with our previous observation that the luminal exchanger is markedly inhibited by high luminal HCO3− concentrations (Ishiguro et al. 2000).
We therefore suppose that, in experiments such as those shown in Fig. 3, the efflux of Cl− to the lumen was mediated largely by a cAMP-stimulated Cl− conductance, presumably CFTR. However, two recognised CFTR inhibitors, glibenclamide and NPPB, both failed to block the Cl− efflux across the luminal membrane (Fig. 5A and C). On the other hand, their efficacy as CFTR blockers in native tissue is not well documented (Schultz et al. 1999). While effective in airway epithelia (Sheppard & Welsh, 1992), glibenclamide has little or no effect on currents attributed to CFTR in T84 cells (Hongre et al. 1994) or salivary duct cells (Dinudom et al. 1995). It is, however, reported to partially block CFTR currents in single, isolated guinea-pig pancreatic duct cells (O'Reilly et al. 2000) so the lack of effect in our hands is somewhat surprising.
Cl− transport across the basolateral membrane
Cl− ions also move readily in either direction across the basolateral membrane in response to changes in basolateral Cl− concentration. Unlike the flux across the luminal membrane, however, the efflux of Cl− across the basolateral membrane, following a reduction in the bath Cl− concentration, was not stimulated by forskolin (Fig. 6A). Furthermore, it was largely inhibited by H2DIDS (Fig. 6C) and consisted of both HCO3−-dependent and -independent components (Figs. 6B).
The HCO3−-dependent component of the Cl− efflux across the basolateral membrane was probably mediated by an H2DIDS-sensitive Cl−-HCO3− exchanger which, unlike the luminal exchanger, was not activated, and may even have been inhibited, by elevation of intracellular cAMP. This would be consistent with reports that CFTR activates luminal but not basolateral anion exchangers in mouse main pancreatic ducts (Lee et al. 1999) and it suggests that different anion exchanger isoforms may be present at the basolateral and luminal membranes. There was no obvious effect of forskolin stimulation on the HCO3−-independent component of basolateral Cl− efflux (Fig. 6B) which may be due to a small H2DIDS-sensitive anion conductance.
Steady-state intracellular Cl− concentration
As in any epithelium, the intracellular concentration of a transported ion such as Cl− reaches a steady-state value when the driving forces for entry (or exit) across the basolateral membrane and the driving forces for exit (or entry) across the luminal membrane generate identical fluxes across the two membranes. In Fig. 8 we combine the Cl− data from the present study with previous observations in an attempt to interpret the steady-state concentrations and fluxes that exist in four different experimental situations. Figure 8A and B represent, respectively, unstimulated and forskolin-stimulated duct cells bathed on both sides with high-Cl− solutions. The cells in Fig. 8C and D differ only by being bathed on the luminal surface with a high-HCO3− solution, thus more closely resembling the physiological situation during maximal stimulation.
Figure 8. Implications of measured [Cl−]i values for anion channel/transporter activity under different conditions.
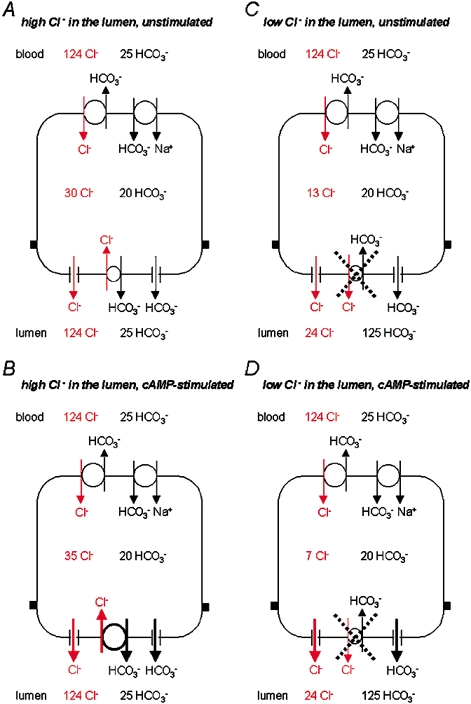
The lumen is perfused either with a high Cl− -low HCO3− solution (A and B) or with a low Cl−-high HCO3− solution (C and D) in either the absence (A and C) or presence (B and D) of a cAMP-mediated secretory stimulus.
With 124 mmCl− and 25 mm HCO3− in both the bath and the lumen, our results indicate that [Cl−]i is approximately 30 mm in the unstimulated duct, and [HCO3−]i is approximately 20 mm since pHi is about 7.2 (Fig. 8A). In this situation, both basolateral and luminal anion exchangers support Cl− uptake since there is a steep inward concentration gradient for Cl− across both membranes, and this is only offset by a much smaller inward HCO3− gradient. The data also suggest that, in the unstimulated duct, the basolateral exchanger is rather more active than the luminal one. Since anion exchange is electrically neutral, the membrane potential is irrelevant and Cl− accumulates in the cell at a concentration above that predicted for electrochemical equilibrium (approximately 14 mm at a membrane potential of −56 mV). The steady-state value for [Cl−]i is thus achieved when the rate of Cl− uptake via the exchangers balances the loss of Cl− through the luminal, and perhaps basolateral, anion conductances. A small flux of Cl− from bath to lumen, combined with HCO3− efflux to the lumen via the luminal anion exchanger, would give rise to the spontaneous, Cl−-dependent secretion that we have observed previously in these conditions (Ishiguro et al. 1998).
Stimulation with forskolin elevates the steady-state value of [Cl−]i by approximately 5 mm (Fig. 8B). This occurs despite the marked increase in luminal membrane Cl− conductance that results from the activation of CFTR, which would otherwise tend to reduce [Cl−]i. There must therefore be a parallel increase in the uptake of Cl−. Our data (Fig. 3) suggest that this is due mainly to an increase in the activity of the luminal membrane Cl−-HCO3− exchanger, which is consistent with previous reports that activation of CFTR can lead to increased anion exchanger activity (Lee et al. 1999; Choi et al. 2001). Combined activation of CFTR and the anion exchanger would result in a marked increase in the net secretion of HCO3−, but not necessarily of Cl−. If, as we suspect, there is relatively little Cl− entry across the basolateral membrane, the efflux of Cl− through CFTR might barely exceed the amount re-entering the cell on the anion exchanger. In contrast, all of the HCO3− secreted to the lumen via the anion exchanger would be rapidly replaced by the HCO3− uptake mechanisms present in the basolateral membrane, specifically the Na+-HCO3− cotransporter and Na+-H+ exchanger (Ishiguro et al. 1996a).
Effects of high luminal HCO3− concentration
The mechanism described above is essentially the same as that originally proposed to account for HCO3− secretion in the rat pancreatic duct (Gray et al. 1988; Novak & Greger, 1988). It depends upon a continuous supply of luminal Cl−, which in the proximal ducts could be derived from the secretion of a small amount of Cl−-rich fluid by the acinar cells. However, while there is evidence for such a secretion in rat (Sewell & Young, 1975), there is no such evidence in guinea-pig (Padfield et al. 1989). Even if there is some acinar Cl− secretion, the net effect in the guinea-pig is the secretion of a HCO3−-rich fluid containing relatively little Cl−. Consequently, as the luminal concentration of HCO3− increases and the concentration of Cl− declines, either with time or distance along the ductal system, the gradients favouring HCO3− secretion by anion exchange at the luminal membrane will gradually diminish and eventually reverse. We have shown previously that the anion exchanger is actually inhibited by high luminal HCO3− concentrations, so this will help to prevent the reabsorption of HCO3− that would otherwise occur once the gradients reverse (Ishiguro et al. 2000).
What therefore is the secretory mechanism operating when the luminal HCO3− concentration is too high, and the Cl− concentration too low, to support HCO3− secretion by luminal membrane anion exchange? There is no doubt that the ducts continue to secrete a HCO3−-rich fluid under these conditions and that the mechanism is largely independent of luminal Cl− (Ishiguro et al. 1996b, 1998).
To simulate this situation, we carried out microperfusion experiments in which the luminal solution contained 125 mm HCO3− and only 24 mmCl−. In the absence of stimulation, the value of [Cl−]i was 10–15 mm, reflecting a balance between uptake via the basolateral anion exchanger and Cl− efflux via the luminal Cl− conductance (Fig. 8C). In contrast to the situation shown in Fig. 8B, forskolin stimulation caused [Cl−]i to fall by approximately a further 5 mm to a very low value (Fig. 8D). This is likely to be the result of an increase in Cl− efflux via the cAMP-activated luminal Cl− conductance but without a compensatory increase in Cl− uptake. The lack of increase in Cl− uptake would be due to (a) the inhibition of the luminal anion exchanger by the high luminal HCO3− concentration and (b) the failure of forskolin stimulation to raise the activity of either the basolateral exchanger or a basolateral Cl− conductance. Indeed, the data in Fig. 7C may even indicate that the basolateral exchanger becomes less active during forskolin stimulation. Because of this, the net secretion of Cl− will be low under these conditions, limited chiefly by the lack of basolateral uptake pathways for Cl−.
Implications for the mechanism of HCO3− transport
While it is not difficult to explain why the pancreatic duct cells of the guinea-pig do not secrete much Cl−, it is still necessary to explain how they secrete HCO3−. Can this be done without invoking the existence of additional transporters in the luminal membrane?
In a recent computer simulation of the pancreatic duct epithelium (Sohma et al. 2000), it was shown that the original model for HCO3− secretion in the rat pancreas, involving Cl− efflux to the lumen via CFTR and its subsequent exchange for HCO3− by anion exchange could, with modification, generate a HCO3−-rich (140 mm) fluid. The additional assumptions required were that high luminal HCO3− concentrations inhibit both the luminal Cl− permeability and also the luminal Cl−-HCO3− exchanger. The first assumption is supported by the demonstration in guinea-pig pancreatic duct cells that extracellular HCO3− reduces the luminal membrane Cl− conductance (O'Reilly et al. 2000). The second assumption is supported by our own observations as discussed above. Inevitably, however, with these additional assumptions, the net secretory flux of HCO3− falls rapidly as the luminal HCO3− concentration rises (Sohma et al. 2000) and it is difficult to see how the secretion of a small volume of HCO3−-rich fluid in the distal ducts could dilute out a larger volume of relatively Cl−-rich fluid from the proximal ducts. Furthermore, our previous work on isolated pancreatic ducts from the guinea-pig has demonstrated clearly that vigorous secretin-evoked secretion of HCO3−-rich fluid can occur with less than 8 mmCl− in the lumen (Ishiguro et al. 1996b, 1998).
One possibility is that, because of the very low intracellular concentration of Cl− that exists in the physiological situation illustrated in Fig. 8D, there is little competition between Cl− and HCO3− ions for efflux via a luminal membrane anion conductance. If a channel equally permeable to Cl− and HCO3− were expressed at the luminal membrane, a low value for [Cl−]i compared with [HCO3−]i would inevitably favour the secretion of a HCO3−-rich fluid. Interestingly, such an anion conductance has been reported both in CAPAN-1 cells (a cell line derived from human pancreatic duct) (Mahiew et al. 1994) and in rat choroid plexus (Kibble et al. 1996) although its molecular identity has not yet been established. On the other hand, a possible role for CFTR as a HCO3− conductance has also been proposed in several epithelia. For example, in a human airway epithelial cell line, CFTR is believed to work both as a Cl− channel and as a HCO3− channel (Devor et al. 1999). In this case, the anionic composition of the secreted fluid, whether Cl−-rich or HCO3−-rich, is thought to depend simply on whether HCO3− uptake by Na+-HCO3− cotransport or Cl− uptake by Na+-K+-2Cl− cotransport is the dominant mechanism operating at the basolateral membrane.
Applied to the guinea-pig pancreas, the combination of a low intracellular Cl− concentration and a significant luminal HCO3− conductance could explain the Cl− independence of HCO3− transport in this species. Our preliminary measurements of membrane potential certainly indicate that the electrochemical gradient for HCO3− remains directed towards the lumen, even during stimulation in the presence of a high luminal HCO3− concentration. Finally, it is tempting to suggest that the fundamental difference between the pancreatic duct cells of the guinea-pig and those of the rat may simply be that the duct cells of the rat possess a basolateral Cl− uptake pathway, possibly a Na+-K+-2Cl− cotransporter, which the guinea-pig cells lack.
Acknowledgments
This study was supported by the Ministry of Education, Science, and Culture (Japan), the Ministry of Health and Welfare (Japan), the Uehara Memorial Foundation and the Cystic Fibrosis Trust (UK). We thank Dr Y. Sohma for helpful discussions.
REFERENCES
- Biwersi J, Verkman AS. Cell-permeable fluorescent indicator for cytosolic chloride. Biochemistry. 1991;30:7879–7883. doi: 10.1021/bi00246a001. [DOI] [PubMed] [Google Scholar]
- Case RM, Argent BE. Pancreatic duct cell secretion: control and mechanisms of transport. In: Go VLW, Dimagno EP, Gardner JD, Lebenthal E, Reber HA, Scheele GA, editors. The Pancreas: Biology, Pathophysiology, and Disease. 2. New York: Raven Press; 1993. pp. 301–350. [Google Scholar]
- Choi JY, Muallem D, Kiselyov K, Lee MG, Thomas PJ, Muallem S. Aberrant CFTR-dependent HCO3− transport mutations associated with cystic fibrosis. Nature. 2001;410:94–97. doi: 10.1038/35065099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devor DC, Singh AK, Lambert LC, Deluca A, Frizzell RA, Bridges RJ. Bicarbonate and chloride secretion in Calu-3 human airway epithelial cells. Journal of General Physiology. 1999;113:743–760. doi: 10.1085/jgp.113.5.743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinudom A, Komwatana P, Young JA, Cook DI. A forskolin-activated Cl− current in mouse mandibular duct cells. American Journal of Physiology. 1995;268:G806–812. doi: 10.1152/ajpgi.1995.268.5.G806. [DOI] [PubMed] [Google Scholar]
- Dinudom A, Young JA, Cook DI. Na+ and Cl− conductances are controlled by cytosolic Cl− concentration in the intralobular duct cells of mouse mandibular glands. Journal of Membrane Biology. 1993;135:289–295. doi: 10.1007/BF00211100. [DOI] [PubMed] [Google Scholar]
- Fanen P, Labarthe R, Garnier F, Benharouga M, Goossens M, Edelman A. Cystic fibrosis phenotype associated with pancreatic insufficiency does not always reflect the cAMP-dependent chloride conductive pathway defect. Analysis of C225R-CFTR and R1066C-CFTR. Journal of Biological Chemistry. 1997;272:30563–30566. doi: 10.1074/jbc.272.48.30563. [DOI] [PubMed] [Google Scholar]
- Foskett JK. [Ca2+]i modulation of Cl− content controls cell volume in single salivary acinar cells during fluid secretion. American Journal of Physiology. 1990;259:C998–1004. doi: 10.1152/ajpcell.1990.259.6.C998. [DOI] [PubMed] [Google Scholar]
- Gray MA, Greenwell JR, Argent BE. Secretin-regulated chloride channels on the apical plasma membrane of pancreatic duct cells. Journal of Membrane Biology. 1988;105:131–142. doi: 10.1007/BF02009166. [DOI] [PubMed] [Google Scholar]
- Gray MA, Pollard CE, Harris A, Coleman L, Greenwell JR, Argent BE. Anion selectivity and block of the small-conductance chloride channel on pancreatic duct cells. American Journal of Physiology. 1990;259:C752–761. doi: 10.1152/ajpcell.1990.259.5.C752. [DOI] [PubMed] [Google Scholar]
- Hongre AS, Baro I, Berthon B, Escande D. Effects of sulphonylureas on cAMP-stimulated Cl− transport via the cystic fibrosis gene product in human epithelial cells. Pflügers Archiv. 1994;426:284–287. doi: 10.1007/BF00374783. [DOI] [PubMed] [Google Scholar]
- Ishiguro H, Lindsay ARG, Steward MC, Case RM. Secretin stimulation of Cl−-HCO3− exchange in interlobular ducts isolated from guinea-pig pancreas. Journal of Physiology. 1995;482P:22P. [Google Scholar]
- Ishiguro H, Naruse S, Kitagawa M, Hayakawa T, Case RM, Steward MC. Luminal ATP stimulates fluid and HCO3− secretion in guinea-pig pancreatic duct. Journal of Physiology. 1999;519:551–558. doi: 10.1111/j.1469-7793.1999.0551m.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishiguro H, Naruse S, Kitagawa M, Suzuki A, Yamamoto A, Hayakawa T, Case RM, Steward MC. CO2 permeability and bicarbonate transport in microperfused interlobular ducts isolated from guinea-pig pancreas. Journal of Physiology. 2000;528:305–315. doi: 10.1111/j.1469-7793.2000.00305.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishiguro H, Naruse S, Steward MC, Kitagawa M, Ko SBH, Hayakawa T, Case RM. Fluid secretion in interlobular ducts isolated from guinea-pig pancreas. Journal of Physiology. 1998;511:407–422. doi: 10.1111/j.1469-7793.1998.407bh.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishiguro H, Steward MC, Lindsay ARG, Case RM. Accumulation of intracellular HCO3− by Na+-HCO3− cotransport in interlobular ducts from guinea-pig pancreas. Journal of Physiology. 1996a;495:169–178. doi: 10.1113/jphysiol.1996.sp021582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishiguro H, Steward MC, Wilson RW, Case RM. Bicarbonate secretion in interlobular ducts from guinea-pig pancreas. Journal of Physiology. 1996b;495:179–191. doi: 10.1113/jphysiol.1996.sp021583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kibble JD, Trezise AE, Brown PD. Properties of the cAMP-activated Cl− current in choroid plexus epithelial cells isolated from the rat. Journal of Physiology. 1996;496:69–80. doi: 10.1113/jphysiol.1996.sp021666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau KR, Evans RL, Case RM. Intracellular Cl− concentration in striated intralobular ducts from rabbit mandibular salivary glands. Pflügers Archiv. 1994;427:24–32. doi: 10.1007/BF00585938. [DOI] [PubMed] [Google Scholar]
- Lee MG, Choi JY, Luo X, Strickland E, Thomas PJ, Muallem S. Cystic fibrosis transmembrane conductance regulator regulates Cl−/HCO3− exchange in mouse submandibular and pancreatic ducts. Journal of Biological Chemistry. 1999;274:14670–14677. doi: 10.1074/jbc.274.21.14670. [DOI] [PubMed] [Google Scholar]
- Mahiew I, Becq F, Wolfensberger T, Gola M, Carter N, Hollande E. The expression of carbonic anhydrase II and IV in the human pancreatic cancer cell line (Capan 1) is associated with bicarbonate ion channels. Biology of the Cell. 1994;81:131–141. doi: 10.1016/s0248-4900(94)80004-9. [DOI] [PubMed] [Google Scholar]
- Novak I, Greger R. Properties of luminal membrane of isolated rat pancreatic ducts: effect of cyclic AMP and blockers of chloride transport. Pflügers Archiv. 1988;411:546–553. doi: 10.1007/BF00582376. [DOI] [PubMed] [Google Scholar]
- O'Reilly CM, Winpenny JP, Argent BE, Gray MA. Cystic fibrosis transmembrane conductance regulator in guinea-pig pancreatic duct cells: Inhibition by bicarbonate ions. Gastroenterology. 2000;118:1187–1196. doi: 10.1016/s0016-5085(00)70372-6. [DOI] [PubMed] [Google Scholar]
- Padfield PJ, Garner A, Case RM. Patterns of pancreatic secretion in the anaesthetised guinea pig following stimulation with secretin, cholecystokinin octapeptide, or bombesin. Pancreas. 1989;4:204–209. doi: 10.1097/00006676-198904000-00009. [DOI] [PubMed] [Google Scholar]
- Robertson MA, Foskett JK. Na+ transport pathways in the secretory acinar cells: membrane cross talk mediated by [Cl−]i. American Journal of Physiology. 1994;267:C146–156. doi: 10.1152/ajpcell.1994.267.1.C146. [DOI] [PubMed] [Google Scholar]
- San Román JI, Colledge WH, Evans MJ, Case RM, Steward MC. Fluid secretion in ducts isolated from the pancreas of CFTR null (−/−) mice. Journal of Physiology. 1999;517P:23P. doi: 10.1007/s00424-009-0704-9. [DOI] [PubMed] [Google Scholar]
- Schultz BD, Singh AK, Devor DC, Bridges RJ. Pharmacology of CFTR chloride channel activity. Physiological Reviews. 1999;79(suppl. 1):109–144. doi: 10.1152/physrev.1999.79.1.S109. [DOI] [PubMed] [Google Scholar]
- Sewell WA, Young JA. Secretion of electrolytes by the pancreas of the anaesthetized rat. Journal of Physiology. 1975;252:379–396. doi: 10.1113/jphysiol.1975.sp011149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheppard DN, Welsh MJ. Effect of ATP-sensitive K+ channel regulators on cystic fibrosis transmembrane conductance regulator chloride currents. Journal of General Physiology. 1992;100:573–591. doi: 10.1085/jgp.100.4.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shumaker H, Amlal H, Frizzell R, Ulrich Ii CD, Soleimani M. CFTR drives Na+-nHCO3− cotransport in pancreatic duct cells: a basis for defective HCO3− secretion in CF. American Journal of Physiology. 1999;276:C16–25. doi: 10.1152/ajpcell.1999.276.1.C16. [DOI] [PubMed] [Google Scholar]
- Sohma Y, Gray MA, Imai Y, Argent BE. HCO3− transport in a mathematical model of the pancreatic duct epithelium. Journal of Membrane Biology. 2000;176:77–100. doi: 10.1007/s00232001077. [DOI] [PubMed] [Google Scholar]