

Abstract
Intracellular [Na+] ([Na+]i) is centrally involved in regulation of cardiac Ca2+ and contractility via Na+-Ca2+ exchange (NCX) and Na+-H+ exchange (NHX). Previous work has indicated that [Na+]i is higher in rat than rabbit ventricular myocytes. This has major functional consequences, but the reason for the higher [Na+]i in rat is unknown. Here, resting [Na+]i was measured using the fluorescent indicator SBFI, with both traditional calibration and a novel null-point method (which circumvents many limitations of prior methods). In rabbit, resting [Na+]i was 4.5 ± 0.4 mm (traditional calibration) and 4.4 mm (null-point). Resting [Na+]i in rat was significantly higher using both the traditional calibration (11.1 ± 0.7 mm) and the null-point approach (11.2 mm). The rate of Na+ transport by the Na+ pump was measured as a function of [Na+]i in intact cells. Rat cells exhibited a higher Vmax than rabbit (7.7 ± 1.1 vs. 4.0 ± 0.5 mm min−1) and a higher Km (10.2 ± 1.2 vs. 7.5 ± 1.1 mm). This results in little difference in pump activity for a given [Na+]i below 10 mm, but at measured resting [Na+]i levels the pump-mediated Na+ efflux is much higher in rat. Thus, Na+ pump rate cannot explain the higher [Na+]i in rat. Resting Na+ influx rate was two to four times higher in rat, and this accounts for the higher resting [Na+]i. Using tetrodotoxin, HOE-642 and Ni2+ to block Na+ channels, NHX and NCX, respectively, we found that all three pathways may contribute to the higher resting Na+ influx in rat (albeit differentially). We conclude that resting [Na+]i is higher in rat than in rabbit, that this is caused by higher resting Na+ influx in rat and that a higher Na+,K+-ATPase pumping rate in rat is a consequence of the higher [Na+]i.
The concentration of free intracellular Na+ ([Na+]i) is very important in modulating the contractile and electrical activity of the heart. Via Na+−Ca2+ exchange (NCX) and Na+−H+ exchange (NHX), [Na+]i is involved in controlling the intracellular Ca2+ and H+ concentration, and both these ions are important factors in excitation-contraction coupling (ECC) (Bers, 2001). The Na+ electrochemical gradient is maintained by the Na+,K+-ATPase, which uses the energy derived from ATP hydrolysis to exchange intracellular Na+ for extracellular K+.
Traditionally, [Na+]i has been measured by using ion-selective microelectrodes, which offer advantages over isotope tracer or atomic absorption experiments, such as direct and continuous monitoring of free [Na+]i, and relatively direct calibration (e.g. Shattock & Bers, 1989). However, this technique has limitations. The cell has to be impaled with a Na+-selective microelectrode and the membrane potential (Em) must be measured simultaneously by a second microelectrode. Another potential problem is that following impalement, the bath solution or the electrode contents might leak into the cytosol (Desilets & Baumgarten, 1986) thus altering the cytosolic ionic composition.
The preferred method for measuring [Na+]i in recent years is the use of the fluorescent indicator SBFI. The dye can be calibrated intracellularly by equilibrating the intra- with the extracellular [Na+] using Na+ ionophores. However, this method also has limitations, due to the relatively high degree of compartmentalization of the dye when loaded as the acetoxymethylester (AM) form (Donoso et al. 1992; Levi et al. 1994). Indicator compartmentalization can affect the intracellular SBFI calibration. This may explain the relatively low values reported for resting [Na+]i (4–5 mm) obtained in rabbit myocardium with SBFI (Levi et al. 1994; Yao et al. 1998) as compared to the values (∼10 mm) obtained by using ion-selective microelectrodes (Shattock & Bers, 1989). SBFI has also been reported to bind to intracellular proteins (Baartscheer et al. 1997; Despa et al. 2000) and this results in lower sensitivity to [Na+]i. Since the absolute value of [Na+]i has a strong effect on cellular Ca2+ transport, it is especially valuable to have more than one way to measure resting [Na+]i. To this end, we complemented traditional SBFI measurements with a novel null-point method to measure resting [Na+]i.
Because all available methods for measuring [Na+]i have some drawbacks, interspecies comparisons of [Na+]i are best performed using the same method, similar conditions and preferably in the same laboratory. Studies using either ion-selective microelectrodes (Shattock and Bers, 1989) or SBFI (Levi et al. 1994) showed that the resting [Na+]i is higher in rat than in rabbit myocytes. The different [Na+]i of rat and rabbit myocytes has important implications for the function of NCX in each cell type (Levi et al. 1994; Bers, 2001). Shattock & Bers (1989) suggested that the higher [Na+]i in rat explains why rest potentiation is observed in rat ventricle whereas rest decay is found in rabbit ventricle (and many other mammalian species). [Na+]i is determined by the balance between inward Na+ fluxes and outward pumping. While it seems clear that [Na+]i in rat ventricle is much higher than in rabbit ventricle, it is not known whether this reflects a lower rate of Na+ extrusion by the Na+−K+ pump or a higher rate of Na+ entry (e.g. by Na+ channels, NCX or NHX).
The aim of this study was threefold. First, we devised a new null-point approach to measure resting [Na+]i, which circumvents compartmentalization issues. Second, we sought to confirm the [Na+]i difference between rabbit and rat ventricle, using SBFI and both traditional calibrations and the null-point approach. Third, we aimed to determine the cause of the higher [Na+]i in rat than in rabbit myocytes. We used a Na+ loading/recovery protocol to derive the rate of Na+ efflux and the Vmax and [Na+]i dependence for Na+,K+-ATPase in these species. We found that both Vmax and Km are higher in rat than in rabbit myocytes. Surprisingly, Na+,K+-ATPase rate at resting [Na+]i is actually higher in rat. This alone implies that the higher [Na+]i in rat is a result of greater Na+ influx in rat than in rabbit. Our measurements of Na+ influx confirmed this.
METHODS
Cell isolation and fluorescence measurements
All experiments were carried out according to the guidelines laid down by the Loyola University Chicago animal welfare committee. The procedure for isolation of ventricular myocytes has been described previously (Bassani et al. 1994). Briefly, rabbits were anaesthetized by i.v. injection of pentobarbital sodium (50–70 mg kg−1) and rats were anaesthetized by i.p. injection of nembutal (∼1 mg g−1). Hearts were excised quickly and placed on a Langendorff perfusion apparatus. Hearts were perfused for 5–7 min with nominally Ca2+-free Tyrode solution, then perfusion was switched to the same solution containing 1 mg ml−1 collagenase and for rat hearts also 0.02 mg ml−1 protease. When the heart became flaccid, the ventricles were dispersed and filtered. The cell suspension was washed several times with a gradual increase in the [Ca2+] up to 2 mm for rabbit and 1 mm for rat.
Isolated myocytes were plated on laminin-coated glass coverslips mounted in perfusion chambers. Myocytes were loaded with 10 μm SBFI AM for 90 min, at room temperature, in the presence of the non-ionic surfactant Pluronic F-127 (0.05 % w/v). After washing out the external dye, we allowed SBFI AM to de-esterify for 20 min before proceeding with [Na+]i measurements. The perfusion chamber was mounted on a Nikon TMD epifluorescence microscope. Fluorescence was elicited by illumination with a 75 W xenon lamp. Dual excitation measurements (at 340 and 380 nm; F340 and F380; alternating at 100 Hz) were performed using a PTI Alphascan system equipped with a chopper (PTI, Monmouth Junction, NJ, USA). The emitted fluorescence was recorded at 535 ± 20 nm. The emission field was restricted to a single cell with a rectangular diaphragm. A linear regression of autofluorescence at each wavelength versus two-dimensional projection of cell area for 10 unloaded cells of the same preparation was used to predict the cell size-dependent background, which was subtracted from the individual wavelength signals in SBFI-loaded cells. All the measurements were at room temperature.
Traditional [Na+]i calibration
In vivo calibration of SBFI was accomplished by exposing the myocytes to various extracellular [Na+] ([Na+]o) in the presence of 10 μm gramicidin D and 100 μm strophanthidin. The solutions with various [Na+]o were prepared by mixing, in different proportions, two solutions of equal ionic strength. One solution contained 145 mm Na+ (30 mmNaCl, 115 mm sodium gluconate) and no K+, while the other one had 145 mm K+ (30 mm KCl, 115 mm potassium gluconate) and was Na+ free. Both calibration buffers also contained 10 mm Hepes, 10 mm glucose and 2 mm EGTA and the pH was adjusted to 7.2 with Tris base. A calibration was performed at the end of each experiment.
Null-point method to assess resting [Na+]i
The strategy here was to determine the [Na+]o which does not alter [Na+]i when the cell is suddenly made leaky (the null point). During rest the superfusate was quickly switched (∼2 s) to solutions of various [Na+]o, comparable with anticipated [Na+]i (0–10 mm for rabbit, 0–20 mm for rat). These solutions were identical to those used for traditional calibration, except that 1.2 mm CaCl2 was added (resulting in free [Ca2+] ∼100 nm). After a few seconds in these solutions, the cell was quickly poked a few times with a micropipette containing the same external solution, or exposed abruptly to 10 μm digitonin to damage the external membrane. We monitored the fluorescence ratio R and determined whether it increased or decreased following sarcolemmal permeabilization. An increase in R indicates that [Na+]i was lower than [Na+]o, whereas a decrease in R means that [Na+]i was, on the contrary, higher than [Na+]o. Based on the direction of change in R (ΔR), tests on subsequent cells used a lower or higher [Na+]o, until reaching the null point where ΔR = 0. This means that no net Na+ movement occurred at this [Na+]o upon cell rupture. Thus, this [Na+]o corresponds to the resting [Na+]i. The null-point approach (unlike the SBFI calibration procedure) uses abrupt membrane damage and initial changes in R, and does not depend on full equilibration of [Na+]i with [Na+]o (which also minimizes effects of compartmentalized SBFI). Measurements of ΔR were restricted to the time before SBFI leaked out of the cell (see Results).
Na+−K+ pump activity
To determine the [Na+]i dependence of the Na+−K+ pump, we measured the time course of Na+ efflux in quiescent Na+-loaded cells. Then the rate of Na+ efflux (−d[Na+]i/dt) was plotted as a function of [Na+]i and we fitted these data with a Hill expression: Jpump = Vmax/(1 + (Km/[Na+]i)n), where Vmax is maximal Na+ transport rate, Km is the [Na+]i for half-maximal stimulation and n is the Hill coefficient.
Resting Na+ influx
Na+ influx was derived from the slope of the initial increase in the [Na+]i (+d[Na+]i/dt) following abrupt Na+ pump inhibition in K+-free solution during quiescence. Note that [Na+]o and [Ca2+]o were kept at normal levels to better estimate physiological Na+ influx rate.
Solutions and chemicals
The normal Tyrode solution (NT) contained (mm): 140 NaCl, 4 KCl, 1 MgCl2, 10 Hepes, 10 glucose and 2 (rabbit) or 1 (rat) CaCl2 (pH 7.4 with Tris base). The K+-free solution used to Na+-load the cells for subsequent Na+−K+ pump measurements contained (mm): 145 NaCl, 2 EGTA, 10 Hepes and 10 glucose (pH 7.4 with Tris base). Na+ efflux was measured in a Na+-free solution with the following composition (mm): 140 TEA, 4 KCl, 2 EGTA, 10 Hepes and 10 glucose (pH 7.4 with Tris base). Resting Na+ influx was measured in a solution identical to NT but with K+ omitted.
SBFI and Pluronic F-127 were from Molecular Probes (Eugene, OR, USA). Gramicidin D, strophanthidin and digitonin were from Sigma (St Louis, MO, USA). TTX was from Alomone Labs (Jerusalem, Israel) and HOE 642 was kindly provided by Dr J. Punter from Aventis Pharma (Frankfurt, Germany).
Statistical analysis
Data are expressed as means ± s.e.m. Statistical discriminations were performed with Student's unpaired t test. Values of P < 0.05 were considered significant.
RESULTS
Resting [Na+]i
Intracellular SBFI calibration
Traditional SBFI calibration was done by superfusing the cells with calibration solutions with various [Na+]o (with [Na+]o + [K+]o = 145 mm) and no divalent cations. To equilibrate the intra- with extracellular [Na+], we used the Na+ ionophore gramicidin D (10 μm) and we blocked the Na+−K+ pump with strophanthidin (100 μm). Such a calibration was performed at the end of each experiment. Figure 1A shows a typical experiment for SBFI in a rabbit myocyte. First, we monitored the F340/F380 ratio during physiological manipulations of [Na+]i (see below). Then, we applied the calibration procedure. The SBFI ratio increased with increasing [Na+]i. Figure 1B shows that the calibration curve is linear for [Na+]i between 0 and 30 mm. At higher, non-physiological [Na+]i this curve tends toward saturation (not shown). This method resulted in resting [Na+]i values of 4.5 ± 0.4 mm (n = 34) for rabbit and 11.1 ± 0.7 mm (n = 35) for rat myocytes (Table 1).
Figure 1. Traditional in vivo SBFI calibration.
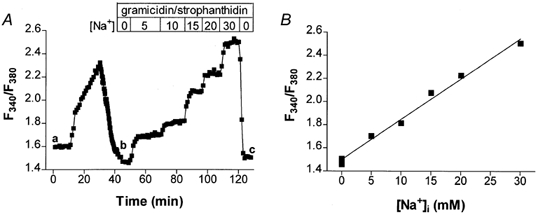
A, representative experiment in a rabbit cell. At the end of a physiological protocol (region a-b) the cells were perfused with external solutions with various [Na+]o in the presence of 10 μm gramicidin and 100 μm strophanthidin (region b-c). The F340/F380 ratio increased with increasing [Na+]. B, linear fit of the calibration points in the same cell.
Table 1.
Summary of results
| Rabbit | (n) | Rat | (n) | |
|---|---|---|---|---|
| Resting [Na+]i (mm) | ||||
| Traditional | 4.5 ± 0.4 | (34) | 11.1 ± 0.7* | (35) |
| Null-point† | 4.4 | 11.2 | ||
| Na+,K+-ATPase | ||||
| Vmax (mmmin−1) | 4.0 ± 0.5 | (6) | 7.7 ± 1.1* | (8) |
| Km(mm) | 7.5 ± 1.1 | (6) | 10.2 ± 1.2 | (8) |
| Hill coefficient | 3.6 ± 0.4 | (6) | 4.0 ± 0.5 | (8) |
| Pump efflux at resting [Na+]i (mmmin−1) | 0.55 ± 0.40 | (6) | 4.5 ± 1.6* | (8) |
| Na+ influx (mm min−1) | ||||
| Total | 0.70 ± 0.18 | (7) | 1.71 ± 0.25* | (7) |
| TTX sensitive | 0.14 ± 0.06 | (6) | 0.68 ± 0.09* | (7) |
| HOE-642 sensitive | 0.12 ± 0.10 | (6) | 0.43 ± 0.19* | (5) |
| Ni2+ sensitive | 0.28 ± 0.09 | (6) | 0.64 ± 0.10* | (6) |
| Ni2++TTX+HOE-642 sensitive | 0.50 ± 0.05 | (4) | 1.18 ± 0.07* | (6) |
Minimum 10 cells for each [Na+] as in Fig. 3.
Significant difference between rat and rabbit ventricular myocytes.
Null-point method
Intracellular SBFI loaded as the AM derivative has been reported to compartmentalize in internal organelles (Donoso et al. 1992; Levi et al. 1994) and this might affect [Na+]i measurements. Gramicidin selectively permeabilizes the sarcolemma, and therefore we expected that the relationship between [Na+] in internal organelles and [Na+]i would not be affected. If the ionophore used also controls mitochondrial [Na+] this would further complicate the calibration. Thus, our in vivo calibration should reflect mainly [Na+]i. However, because we are interested in absolute values of [Na+]i, we sought to determine the resting [Na+]i in these species by a second method, a null-point approach.
The principle of the null-point method is illustrated in Fig. 2A. If sarcolemmal damage causes Na+ efflux and a decline in the SBFI ratio R (left), this indicates that [Na+]i is higher than [Na+]o. When, on the contrary, the R increases following membrane rupture (right), [Na+]i must be lower than [Na+]o. When membrane permeabilization results in no change in R (centre, null-point), [Na+]i = [Na+]o. For these experiments, fluorescence was measured first in NT, then the perfusion was switched abruptly to different [Na+]o values in the range anticipated for [Na+]i (indicated by arrows in Fig. 2B and C). The sarcolemma was permeabilized either by adding 10 μm digitonin (concurrent with altered [Na+]o) or by poking the cell with a micropipette containing the same bath solution. After membrane damage, the F340 signal, which is nearly [Na+]i independent in ventricular myocytes (Borzak et al. 1992; Donoso et al. 1992), remained relatively constant for 20–30 s, but then decreased as SBFI gradually leaked out of the cells (Fig. 2B-C). Because Na+ diffuses faster (two to three times) than a molecule the size of SBFI (Push & Neher, 1988), initial changes in [Na+]i occur much faster than dye depletion (and [Na+] equilibration is not required).
Figure 2. Evaluation of resting [Na+]i using SBFI and the null-point approach.
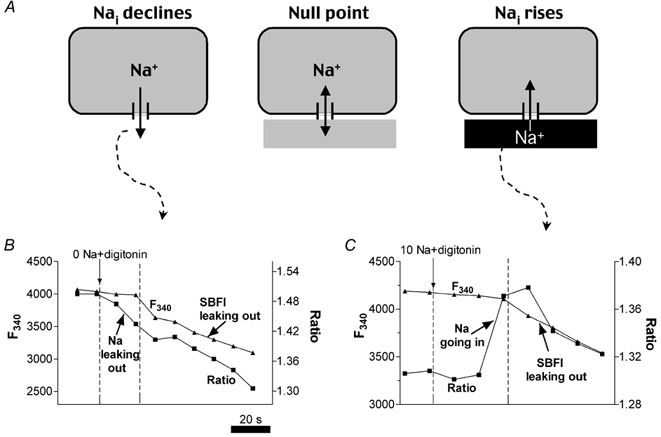
A, principle of the method. When a cell was perfused with a solution with [Na+]o < [Na+]i (left), membrane damage (with digitonin or by poking the cell with a pipette) resulted in Na+ exit from the cell. Therefore the SBFI ratio decreased (see B). If the cells were bathed with a solution with [Na+]o > [Na+]i and the membrane was damaged (right), external Na+ entered the cell, and thus the SBFI ratio increased (see C). When [Na+]o=[Na+]i (middle), the SBFI ratio remained unchanged (null point). B and C, changes in the F340 fluorescence signal and the F340/F380 ratio (R) in rabbit myocytes upon membrane permeabilization with digitonin (10 μm), in the absence of external Na+ (B) and in a solution with 10 mm Na+ (C). Vertical arrows indicate the solution switch from NT to the low [Na+]o solution. Note that R changed before F340 changed.
For rabbit myocytes, R decreased when [Na+]o was 0 mm (Fig. 2B), but increased when [Na+]o was 10 mm (Fig. 2C), indicating that the [Na+]o that would produce no fluorescence change (i.e. the null point; Fig. 2A) is between these two values. In both species, R decreased as SBFI leaked out, possibly indicating lower [Na+] in the internal organelles than in the cytosol. It is also possible that fluorescence ratios at identical [Na+] are lower for compartmentalized SBFI than for SBFI in the cytosol (Borzak et al. 1992).
Figure 3 shows average data for the null-point approach. Because very similar results were obtained when the membrane was damaged with a pipette or permeabilized with digitonin, all the data were pooled in Fig. 3. It is clear that the null point (and therefore the resting [Na+]i) for rabbit myocytes was between 3 (F340/F380 increased when [Na+]o = 3 mm) and 5 mm (F340/F380 decreased when [Na+]o = 5 mm), whereas for rat cells it was between 10 and 15 mm. The Y-intercept of the line between the closest points to the null point indicated a resting [Na+]i of 4.4 mm in rabbit and 11.2 mm in rat.
Figure 3. Summary of null-point results.
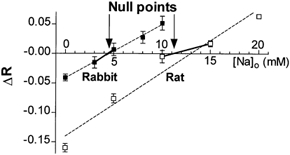
Average changes in SBFI ratio vs.[Na+]o(n≥ 10 for each point). Considering only the first points above and below the null point (arrows), resting [Na+]i was found to be 4.4 mm for rabbit and 11.2 mm for rat ventricle. The dotted line shows linear regressions for all the points for rabbit and rat (the overall regression would give resting [Na+]i of 4.5 mm for rabbit and 13.4 mm for rat).
The results described so far indicate, first, that traditional calibration and the null-point method provide almost identical values for resting [Na+]i, giving us confidence that these numbers are accurate. Second, these results confirm prior work indicating that resting [Na+]i is higher in rat than in rabbit myocytes (Shattock & Bers, 1989; Levi et al. 1994).
[Na+]i dependence of the Na+−K+ pump activity
The higher [Na+]i in rat than in rabbit myocytes could be explained by a lower Vmax and/or affinity for intracellular Na+ of the Na+−K+ pump in rat (i.e. both could cause lower Na+ efflux in rat). To test this hypothesis, we determined the [Na+]i dependence of the Na+−K+ pump by measuring the Na+ efflux from Na+-loaded myocytes (Negulescu & Machen, 1990). Cells were Na+ loaded by incubation in K+-free solution (to block Na+,K+-ATPase) in the absence of divalent cations (Fig. 4A). We then removed extracellular Na+ and added back K+ to activate the Na+,K+-ATPase. Under these conditions, Na+ efflux is mediated by both the Na+,K+-ATPase and passive mechanisms (outward leak). To account for the leak, the experiment was repeated, but this time Na+ efflux was measured with the Na+,K+-ATPase blocked (by 100 μm strophanthidin for rabbit, 10 mm ouabain for rat). We then numerically differentiated the [Na+]i decline and plotted d[Na+]i/dt as a function of [Na+]i (Fig. 4B). When the leak component is subtracted from the total, one can derive the Na+−K+ pump-mediated Na+ efflux as a function of [Na+]i. Figure 4C shows average data for the [Na+]i dependence of the Na+−K+ pump in rabbit and rat myocytes. We fitted the experimental points with a Hill expression to derive the Vmax, Km and Hill coefficient (n) of the pump (Table 1). Both Vmax and Km were higher for rat than for rabbit Na+−K+ pump, although the difference in Km was not statistically significant.
Figure 4. Na+ efflux measurement.
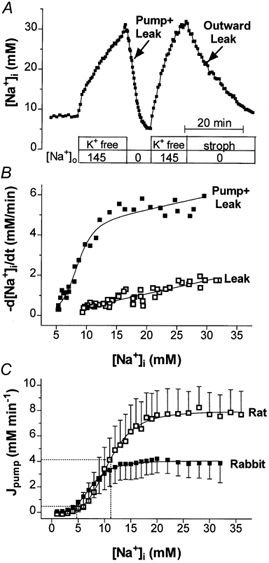
A, a rabbit myocyte was first exposed to K+-free NT without divalent cations to cause Na+ loading of the cell. Then the cell was superfused with Na+-free solution (containing 4 mm K+) in the presence (outward leak) and absence (pump + leak) of 100 μm strophanthidin (10 mm ouabain was used for rat). The time course of [Na+]i decline was used to calculate d[Na+]i/dt. B, dependence of the rate of [Na+]i decrease on [Na+]i in the presence (leak) and absence (pump + leak) of strophanthidin. The difference between these two curves is the Na+ pump-mediated Na+ efflux. The outward leak depended linearly on [Na+]i with a slope of 0.036 ± 0.002 min−1 (n = 6) in rabbit and 0.069 ± 0.004 min−1 (n = 6) in rat myocytes. C, [Na+]i dependence of the Na+ efflux attributable to the Na+−K+ pump in rabbit (n = 6) and rat (n = 8) myocytes.
An important conclusion from the results in Fig. 4 is that Na+,K+-ATPase is not appreciably lower in rat (at any [Na+]i), so lower Na+,K+-ATPase cannot explain the elevated [Na+]i in rat (dispelling that hypothesis). Moreover, the Vmax for Na+,K+-ATPase in rat was about twofold higher than in rabbit.
Resting Na+ influx
Figure 4C shows that at resting [Na+]i (4.5 and 11.1 mm for rabbit and rat, respectively), the Na+ efflux mediated by the Na+−K+ pump is several times higher in rat than in rabbit myocytes. Since Na+ influx and efflux must be equal and opposite at true steady state, this suggests that the resting Na+ influx is also higher in rat; and this could be the cause of the higher [Na+]i in rat. To test this hypothesis, we determined the Na+ influx as the initial rate of [Na+]i increase upon abrupt Na+−K+ pump inhibition (Fig. 5A). In contrast to the Na+-loading procedure used in Fig. 4A, in this case the K+-free solution contained Mg2+ (1 mm) and Ca2+ (2 mm for rabbit, 1 mm for rat) to better simulate physiological Na+ influx conditions. NT superfusion was restored when spontaneous contraction was observed. As expected, Na+ influx was higher in rat (1.71 ± 0.25 mm min−1, n = 7) than in rabbit myocytes (0.70 ± 0.18 mm min−1, n = 7) (Fig. 5B and Table 1). Considering the surface to cytosol volume ratios as measured by Satoh et al. (1996), Na+ influx per unit membrane area is 1.8 ± 0.4 pmol cm−2 s−1 in rabbit and 2.8 ± 0.4 pmol cm−2 s−1 in rat.
Figure 5. Resting Na+ influx in rabbit and rat myocytes.
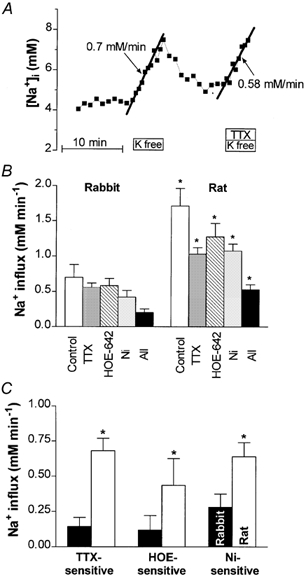
A, representative experiment in a rabbit cell. Na+ influx was calculated as the slope of the initial increase in [Na+]i following abrupt pump inhibition with K+-free solution. Na+,K+-ATPase was blocked in the absence or presence of various inhibitors of Na+ entry (30 μm TTX in this case). B, average data for rabbit and rat myocytes. Shown are the total influx (Control) and the influx measured in the presence of 30 μm TTX (to block Na+ channels), 2 μm HOE 642 (to block Na+/H+ exchange), 5 mm Ni2+ (to block NCX), and also in the presence of all three blockers together (All). *P < 0.05 in rat vs. rabbit. C, TTX-, HOE 642- and Ni2+-sensitive Na+ influx (estimated as the difference between the total resting influx and the influx measured in the presence of the respective drug).
In the next series of experiments, we sought to investigate different Na+ entry pathways. Figure 5B shows the average Na+ influx measured in the presence of 30 μm TTX (to block Na+ channels), 2 μm HOE 642 (to block Na+-H+ exchange), 5 mm Ni2+ (to block Na+-Ca2+ exchange) and when all three blockers were applied together. While each of these inhibitors reduced Na+ influx in both species, the influx remained significantly higher in rat than in rabbit in each series of experiments. Figure 5C shows the TTX-, HOE 642- and Ni2+-sensitive Na+ entry, i.e. the difference between the total Na+ influx and the influx in the presence of the respective inhibitor. In rabbit, resting Na+ influx via NCX is about twice as large as via Na+ channels or NHX, whereas in rat Na+ channels and NCX contribute equally to Na+ entry, with a smaller influx via NHX. Figure 5C indicates that the greater Na+ influx in rat than in rabbit cannot be attributed to a single pathway, but is the result of elevated Na+ entry through Na+ channels, NHX and NCX. Notably, the TTX-sensitive Na+ influx in rat is 4.5 times larger than in rabbit, whereas Ni2+-sensitive influx is only 2.3 times larger in rat.
In rabbit, 75 % of the resting Na+ influx occurs via the sum of TTX-, HOE 642- and Ni2+-sensitive pathways, and this agrees very well with the data obtained with simultaneous use of all three blockers (Table 1). In rat myocytes the sum of TTX, HOE 642 and Ni2+ would be expected to block Na+ entry completely. However, when the three blockers were applied together only 71 % of the total Na+ influx was inhibited. This difference might be explained by either an additional influx pathway or incomplete block by one of these agents.
Frequency dependence of [Na+]i
Na+ influx is significantly higher in contracting myocytes than at rest. Analogous to estimates by Shivkumar et al. (1997), we infer an excitation-dependent Na+ influx of ∼37 μm per beat (7 μm via Na+ channels and 30 μm via Na+-Ca2+ exchange), yielding 2.2 mm min−1 at 1 Hz. In these conditions one expects a rise in [Na+]i to a new steady state where the increased influx again equals the efflux. Figure 6A shows a representative experiment where we followed the time course of [Na+]i in a rat myocyte during stimulation. [Na+]i increased during the first 2–4 min of stimulation and then reached a plateau. When the stimulation was stopped, [Na+]i returned to the resting level. The increase in [Na+]i depended on the stimulation frequency in both species (Fig. 6B-C). [Na+]i rose more linearly as a function of stimulation frequency in rat than with a more saturable relationship in rabbit.
Figure 6. Frequency-dependent increase in [Na+]i upon field stimulation.
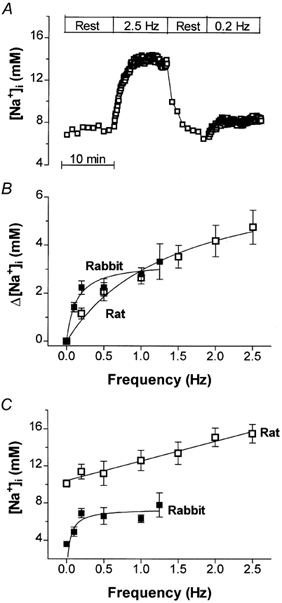
A, representative experiment in a rat cell. [Na+]i increased during the first 2–4 min of stimulation and then reached a plateau. When stimulation was stopped, [Na+]i returned to the resting level. B, frequency dependence of [Na+]i increase in rabbit and rat myocytes. A minimum of six cells were studied at each frequency. C, the actual values measured for [Na+]i at different frequencies.
DISCUSSION
We monitored [Na+]i in rabbit and rat ventricular myocytes using SBFI and a traditional in vivo calibration (using gramicidin and strophanthidin) and we also measured resting [Na+]i via a novel null-point method. These two methods yielded comparable values for the resting [Na+]i in each species and confirmed previous reports of higher resting [Na+]i in rat than in rabbit myocytes (Shattock & Bers, 1989; Levi et al. 1994). To determine the mechanism underlying this difference, we measured Na+ influx and the rate of Na+ extrusion via the Na+−K+ pump in both species. The results suggest that the higher resting [Na+]i in rat than in rabbit ventricle is due to a greater Na+ influx in rat.
Resting [Na+]i in rabbit and rat myocytes
Methodological aspects
Relatively small changes in [Na+]i can have a major effect on [Ca2+]i, contractile force and intracellular pH, because of ionic shifts via NCX and Na+-H+ exchange (Bers, 2001). In some cases, one may only need information about relative [Na+]i changes. However, absolute values for [Na+]i are necessary for a more quantitative understanding of how NCX functions and alters Ca2+ transients.
All the currently available methods for measuring [Na+]i have drawbacks. Fluorescent measurements with SBFI require intracellular indicator calibration because of binding to proteins and compartmentalization into internal organelles (Donoso et al. 1992; Levi et al. 1994; Baartscheer et al. 1997). This in vivo calibration is typically accomplished by exposing the cells to various [Na+]o in the presence of ionophores that equilibrate the intra- with extracellular [Na+]. However, calibration protocols vary among groups with respect to the Na+ ionophores used and their concentration, whether the Na+,K+-ATPase is blocked and whether divalent cations are in the calibration solutions. Levi et al. (1994) performed a detailed investigation of SBFI calibration in ventricular myocytes. Here we used a similar method where calibration solutions were free of both Mg2+ and Ca2+ (to allow rapid Na+ movement through Ca2+ channels and to prevent NCX from altering [Na+]i), Na+,K+-ATPase was blocked and gramicidin was used as the Na+ ionophore (but at higher concentration). We did not use monensin because it may also distribute to internal membranes. Since SBFI compartmentalizes into mitochondria in ventricular myocytes (Donoso et al. 1992), the in vivo calibration is faithful only if mitochondrial [Na+] and [SBFI] maintain the normal relationship with cytoplasmic [Na+] and [SBFI] during calibration (which might not occur if ionophore enters the mitochondrial membrane or SBFI distribution changes). We find that ∼45 % of the SBFI is compartmentalized (based on eventual F340 decline upon digitonin permeabilization), and our results suggest that mitochondrial [Na+] is lower than [Na+]i. This lower mitochondrial [Na+] agrees with prior studies using either isolated heart mitochondria (respiring and non-respiring; Jung et al. 1992) or ventricular myocytes (Donoso et al. 1992). While our calibration cannot eliminate compartmentalization problems completely, it should minimize them.
Because of the issues described above, we also determined resting [Na+]i by an independent null-point approach. If [Na+]o differs from [Na+]i the fluorescence ratio changes rapidly upon membrane damage, before the cellular [SBFI] changes (assessed by the F340 signal). It is important to appreciate that this method does not rely on full equilibration of [Na+]i and [Na+]o. Rather, the direction of ΔR indicates whether [Na+]i is higher or lower than [Na+]o, allowing us to approach the correct ‘null’ [Na+]. So, in the final analysis the amplitude of ΔR is less important than its direction. The null-point approach is less prone to artefacts because of its ultimate simplicity. That is, if one chooses [Na+]o = [Na+]i when damaging the membrane then [Na+]i and SBFI ratio cannot immediately change.
[Na+]i and Na+/Ca2+ exchanger function in rabbit and rat myocytes
Using both the null-point method and the traditional calibration we found resting [Na+]i to be ∼4.5 mm in rabbit myocytes. This is within the range of [Na+]i values reported by others for rabbit or guinea-pig ventricle (Levi et al. 1994; Satoh et al. 1994; Yao et al. 1998). With both methods resting [Na+]i was much higher in rat myocytes, thus confirming the previous reports of Shattock & Bers (1989) and Levi et al. (1994). Harrison et al. (1992b) also found that [Na+]i is higher in rat than in guinea pig ventricular myocytes. The resting [Na+]i found here for rat ventricle is also consistent with several reported values (Borzak et al. 1992; Donoso et al. 1992; Levi et al. 1994; Baartscheer et al. 1997).
The level of [Na+]i in rabbit and rat myocytes has important implications for the NCX function both at rest and during stimulation. Assuming a [Ca2+]i = 100 nm, the reversal potential of NCX (ENCX) in quiescent rabbit and rat myocytes is +11 mV and −43 mV, respectively. Since ENCX is positive to resting Em, Ca2+ extrusion is favoured at rest, especially in rabbit where the thermodynamic driving force favouring Ca2+ efflux (ENCX - Em) is large (91 mV). This contributes to resting loss of cellular and sarcoplasmic reticulum Ca2+ and consequent rest decay in rabbit. When [Na+]i is elevated ENCX may approach Em and rabbit ventricle exhibits rest potentiation (Sutko et al. 1986; Mubagwa et al. 1997). Rest potentiation is typically seen in rat ventricle, due partly to reduced Ca2+ extrusion (or even Ca2+ gain) via NCX during rest because of high [Na+]i (Shattock & Bers, 1989; Bers, 2001).
During stimulation [Na+]i rose by several millimolar over 2–4 min in both rabbit and rat ventricular myocytes (as seen by Frampton et al. 1991; Harrison et al. 1992b). Figure 7 shows how ENCX and INCX may be expected to vary during a steady state action potential (AP) in rabbit and rat myocytes, using values of [Na+]i measured here. Em exceeds ENCX only briefly during the initial AP upstroke and overshoot. Thus outward INCX (Ca2+ influx) is only expected for a brief period. The rise in [Ca2+]i quickly shifts INCX toward Ca2+ efflux (inward INCX). Furthermore, there is compelling evidence that the local [Ca2+]i sensed by the NCX during sarcoplasmic reticular Ca2+ release is much higher than that sensed by global cytosolic Ca2+ indicators (Trafford et al. 1995). This would curtail outward INCX even earlier than illustrated in Fig. 7, so there may be very little (or no) Ca2+ influx via NCX during the ventricular AP.
Figure 7. Expected ENCX and NCX current during action potentials and Ca2+ transients in rabbit (A) and rat ventricle (B).
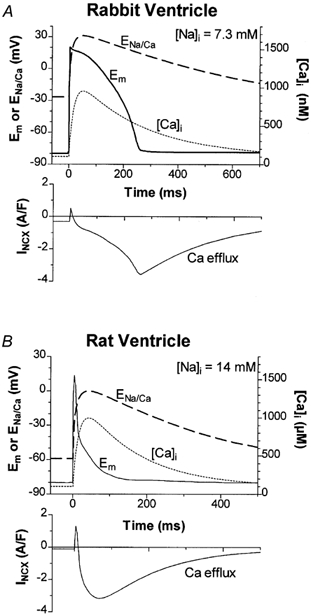
Simulated Ca2+ transients rose with an exponential time constant (τ= 20 ms), peak at ∼1 μm and decline with τ= 250 ms (rabbit) and 150 ms (rat). We assumed a stoichiometry of 3:1 for NCX (so ENCX= 3ENa - 2ECa, where ENa and ECa are the equilibrium potentials for Na+ and Ca2+, respectively). [Na+]o is 140 mm and [Ca2+]o is 2 (rabbit) or 1 (rat) mm. Action potentials (Em) were recorded from rabbit and rat ventricle. INCX was calculated using the equation described by Weber et al. (2001) using the same parameters (as reported there) for both rabbit and rat. [Na+]i values indicated correspond to measured values at steady state stimulation of 1–2 Hz (as in Fig. 6C).
Mechanisms underlying [Na+]i differences between rabbit and rat ventricle
[Na+]i is determined by the balance between Na+ influx and efflux. Therefore higher [Na+]i in rat than in rabbit myocytes might be explained by either lower Na+,K+-ATPase Vmax and/or [Na+]i sensitivity, or by greater Na+ influx. While the [Na+]i affinity of Na+,K+-ATPase was indeed lower in rat than in rabbit ventricle, the Vmax of Na+,K+-ATPase was nearly twice as high in rat. The overall result is that there is relatively little difference in [Na+]i dependence of Na+ pump rate for [Na+]i below 11 mm. Moreover, the pump-mediated Na+ efflux at the appropriate resting [Na+]i (4.5 mm for rabbit, 11.1 mm for rat) is much greater in rat (> 5-fold; Table 1 and Fig. 4C). It is unlikely that other Na+ efflux mechanisms could be substantial. That is, the non-pump-mediated Na+ efflux (Fig. 4B) was at most five times smaller than that mediated by the Na+,K+-ATPase. Indeed, our conditions overestimate the importance of non-pump-mediated Na+ efflux, because Na+ efflux was measured without [Na+]o. The actual Na+ efflux via Na+-H+ exchange or passive leak would be much less (or even reversed) with the physiological [Na+] gradient. The Na+ efflux measured here does not include outward INCX because the exchanger was prevented by 0 Na+, 0 Ca2+ conditions. Furthermore, as illustrated in Fig. 7, NCX strongly favours Ca2+ efflux and Na+ influx under physiological conditions. Borzak et al. (1992) also showed that NCX has only a small contribution to the Na+ efflux in rat myocytes. Since Na+ efflux rate is higher in rat, the higher [Na+]i in rat ventricle must be due to a higher [Na+]i influx. Our results show that resting Na+ influx in rat is more than twice as high as that in rabbit, confirming this conclusion.
For rabbit myocytes, the measured Na+ influx compares very well with the pump rate at resting [Na+]i (Table 1). However, for rat the pump rate is higher than the influx. This difference might be due to the Na+-free solution used for Na+−K+ pump measurements, because external Na+ can inhibit steady-state Na+,K+-ATPase current (Nakao & Gadsby, 1989). However, our approach does have the advantage over pump current measurements of using more physiological conditions (i.e. cells can control freely their intracellular content, without interference from the pipette solution). Another advantage is that we could derive the whole pump rate vs.[Na+]i curve in the same cell, whereas only a limited number of points can be obtained by direct measurements of pump current because of the difficulties in changing the pipette solutions. Although we did not control Em here, possible Em changes are unlikely to affect the results because Na+,K+-ATPase is voltage insensitive at [Na+]o = 0 (Nakao & Gadsby, 1989). A last methodological remark is related to the pH sensitivity of SBFI. Harrison et al. (1992a) estimated that a reduction of pHi by 0.2 units in rat myocytes mimics an apparent decrease in [Na+]i of 1 mm. In 0 Na+ solution NHX could lower pHi. However, Semb & Sejersted (1997) did not observe any change in pHi during a similar protocol in Purkinje fibres. Even if such an effect occurred here, it would only affect the Na+−K+ pump curves and would have caused an underestimation of the Km for [Na+]i by 1–2 mm, and the effect would be similar in both species (and would not affect our conclusions).
The Na+−K+ pump in rat heart is known to have lower ouabain sensitivity as compared to other mammalian species. However, functional interspecies comparisons are lacking. Here we showed that the biggest difference between rabbit and rat myocytes in the pump's ability to extrude Na+ resides in Vmax, which is almost two times larger in rat. Satoh et al. (1996) measured the surface to cytosol volume ratio in rat and rabbit myocytes. Using these values, we estimate that the maximal pump current density under our experimental conditions is 0.33 and 0.41 A F−1 in rabbit and rat, respectively (at 23 °C). Various studies have estimated the cardiac Na+−K+ pump current at [Na+]i ∼10 mm to be 0.25–0.4 A F−1 at 32–36 °C (Shattock et al. 1994; Gao et al. 1995), which is in agreement with our results for both species (accounting for temperature differences, Fig. 4C). The values for the half-maximal activation of the pump by [Na+]i were also within the range previously reported for cardiac cells (Glitsch et al. 1989; Nakao & Gadsby, 1989; Gao et al. 1995; Barmashenko et al. 1999).
The higher Na+,K+-ATPase Vmax in rat may indicate a higher pump protein expression level in rat than in rabbit. We speculate that this might be an adaptation to a chronically higher [Na+]i, and there is precedent for such [Na+]i-dependent upregulation of Na+,K+-ATPase expression levels (Liu & Songu-Mize, 1998). We estimate that at rest the Na+−K+ pump is only running at ∼15 % of Vmax in rabbit ventricle, vs. ∼60 % in rat (Fig. 4C and Table 1). Therefore rabbit cells might have a greater reserve to cope with an increase in Na+ influx brought about by higher stimulation frequency or by pathological conditions. This might be why [Na+]i continues to rise progressively with frequency in rat (vs. rabbit cells, Fig. 6).
Several pathways are involved in Na+ influx in quiescent ventricular myocytes. The increase in [Na+]i during the perfusion of K+-free solution was reduced by TTX, HOE 642 and Ni2+ in both species. Since Na+ influx sensitive to each blocker was larger in rat, it is not possible to identify a single influx pathway responsible for higher [Na+]i in rat. The presence of all these blockers in the K+-free solution did not abolish the influx, suggesting that some other mechanisms may also contribute to Na+ entry (e.g. a TTX-insensitive background Na+ leak channel or electroneutral Na+−K+-2Cl− cotransport; Drewnowska & Baumgarten, 1991). We argued above that NCX is thermodynamically closer to equilibrium in resting rat myocytes, and therefore it may be surprising that Ni2+ reduced resting Na+ influx by almost 40 % in rat. However, sarcoplasmic reticulum Ca2+ leak occurs as local Ca2+ sparks, which are more frequent in resting rat than rabbit cells (Satoh et al. 1997). The resulting high local [Ca2+]i may drive local Ca2+ extrusion and Na+ influx via NCX. Lack of selectivity of Ni2+ could partly explain this effect. The fact that combined application of TTX, HOE 642 and Ni2+ has a smaller effect on Na+ influx than would be predicted from the individual effect of each inhibitor supports this hypothesis. On the other hand, if this Ni2+-sensitive component is all NCX, this would correspond to diastolic Na+ influx of ∼5 μm s−1, which would extrude ∼2 μm Ca2+ s−1. This is a plausible number for resting Ca2+ efflux, and must also match Ca2+ influx in the steady state (Choi et al. 2000).
In summary, the results of this study indicate that SBFI can provide accurate values for resting [Na+]i in cardiac myocytes. We confirmed that the resting [Na+]i is higher in rat than in rabbit ventricle and we showed that this difference is due to a greater Na+ influx in rat (rather than lower Na+ extrusion). Multiple pathways, including Na+ channels, Na+-H+ exchanger and Na+-Ca2+ exchanger contribute to the enhanced Na+ influx in rat. There is little difference in Na+ transport rate by the Na+−K+ pump for a given [Na+]i for values below 10 mm, but Vmax is about twice as high in rat. Moreover, the higher Vmax for the Na+ pump in rat may be an adaptation to a chronically higher [Na+]i which is caused by higher diastolic Na+ influx.
Acknowledgments
We thank Jorge Acevedo for preparation of myocytes. This work was supported by National Institutes of Health grants HL-30077, HL-64724 (D.M.B.) and HL-46929 (S.M.P.).
REFERENCES
- Baartscheer A, Schumacher CA, Fiolet JWT. Small changes in cytosolic sodium in rat ventricular myocytes measured with SBFI in emission ratio mode. Journal of Molecular and Cellular Cardiology. 1997;29:3375–3383. doi: 10.1006/jmcc.1997.0567. [DOI] [PubMed] [Google Scholar]
- Barmashenko G, Kockskamper J, Glitsch HG. Depolarization increases the apparent affinity of the Na+-K+ pump to cytoplasmic Na+ in isolated guinea-pig ventricular myocytes. Journal of Physiology. 1999;517:691–698. doi: 10.1111/j.1469-7793.1999.0691s.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassani JWM, Bassani RA, Bers DM. Relaxation in rabbit and rat cardiac cells: species-dependent differences in cellular mechanisms. Journal of Physiology. 1994;476:279–293. doi: 10.1113/jphysiol.1994.sp020130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bers DM. Excitation-Contraction Coupling and Cardiac Contractile Force. The Netherlands: Kluwer Academic Publishers; 2001. [Google Scholar]
- Borzak S, Reers M, Arruda J, Sharma VK, Sheu SS, Smith TW, Marsh JD. Na+ efflux mechanisms in ventricular myocytes: measurement of [Na+]i with Na+-binding benzofuran isophthalate. American Journal of Physiology. 1992;263:H866–874. doi: 10.1152/ajpheart.1992.263.3.H866. [DOI] [PubMed] [Google Scholar]
- Choi HS, Trafford AW, Eisner DA. Measurement of calcium entry and exit in quiescent rat ventricular myocytes. Pflügers Archiv. 2000;440:600–608. doi: 10.1007/s004240000295. [DOI] [PubMed] [Google Scholar]
- Desilets M, Baumgarten CM. K+, Na+, Cl− activities in ventricular myocytes isolated from rabbit heart. American Journal of Physiology. 1986;251:C197–208. doi: 10.1152/ajpcell.1986.251.2.C197. [DOI] [PubMed] [Google Scholar]
- Despa S, Steels P, Ameloot M. Fluorescence lifetime microscopy of the sodium indicator sodium-binding benzofuran isophthalate in HeLa cells. Analytical Biochemistry. 2000;280:227–241. doi: 10.1006/abio.2000.4505. [DOI] [PubMed] [Google Scholar]
- Donoso P, Mill JG, O'Neil SC, Eisner DA. Fluorescence measurements of cytoplasmic and mitochondrial sodium concentration in rat ventricular myocytes. Journal of Physiology. 1992;448:493–509. doi: 10.1113/jphysiol.1992.sp019053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drewnowska K, Baumgarten CM. Regulation of cellular volume in rabbit ventricular myocytes: bumetanide, chlorothiazide, and ouabain. American Journal of Physiology. 1991;260:C122–131. doi: 10.1152/ajpcell.1991.260.1.C122. [DOI] [PubMed] [Google Scholar]
- Frampton JE, Harrison SM, Boyett MR, Orchard CH. Ca2+ and Na+ in rat myocytes showing different force-frequency relationships. American Journal of Physiology. 1991;261:C739–750. doi: 10.1152/ajpcell.1991.261.5.C739. [DOI] [PubMed] [Google Scholar]
- Gao J, Mathias RT, Cohen IS, Baldo GJ. Two functionally different Na/K pumps in cardiac ventricular myocytes. Journal of General Physiology. 1995;106:995–1030. doi: 10.1085/jgp.106.5.995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glitsch HG, Krahn T, Pusch H. The dependence of sodium pump on internal Na concentration and membrane potential in cardioballs from sheep Purkinje fibres. Pflügers Archiv. 1989;414:52–58. doi: 10.1007/BF00585626. [DOI] [PubMed] [Google Scholar]
- Harrison SM, Frampton JE, McCall E, Boyet MR, Orchard CH. Contraction and intracellular Na+, Ca2+ and H+ during acidosis in rat ventricular myocytes. American Journal of Physiology. 1992a;262:C348–357. doi: 10.1152/ajpcell.1992.262.2.C348. [DOI] [PubMed] [Google Scholar]
- Harrison SM, McCall E, Boyet MR. The relationship between contraction and intracellular sodium in rat and guinea-pig ventricular myocytes. Journal of Physiology. 1992b;449:517–550. doi: 10.1113/jphysiol.1992.sp019100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung DW, Apel LM, Brierley GP. Transmembrane gradients of free Na+ in isolated heart mitochondria estimated using a fluorescent probe. American Journal of Physiology. 1992;262:C1047–1055. doi: 10.1152/ajpcell.1992.262.4.C1047. [DOI] [PubMed] [Google Scholar]
- Levi AJ, Lee CO, Brooksby P. Properties of the fluorescent sodium indicator SBFI in rat and rabbit cardiac myocytes. Journal of Cardiovascular Electrophysiology. 1994;5:241–257. doi: 10.1111/j.1540-8167.1994.tb01161.x. [DOI] [PubMed] [Google Scholar]
- Liu X, Songu-Mize E. Effect of Na on Na+,K+-ATPase α-subunit expression and Na pump activity. European Journal of Pharmacology. 1998;351:113–119. doi: 10.1016/s0014-2999(98)00278-7. [DOI] [PubMed] [Google Scholar]
- Mubagwa K, Lin W, Sipido K, Bosteels S, Flameng W. Monensin-induced reversal of positive force-frequency relationship in cardiac muscle: role of intracellular sodium in rest-dependent potentiation of contraction. Journal of Molecular and Cellular Cardiology. 1997;29:977–989. doi: 10.1006/jmcc.1996.0342. [DOI] [PubMed] [Google Scholar]
- Nakao M, Gadsby DC. [Na] and [K] dependence of the Na/K pump current-voltage relationship in guinea pig ventricular myocytes. Journal of General Physiology. 1989;94:539–565. doi: 10.1085/jgp.94.3.539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negulescu PA, Machen TE. Intracellular ion activities and membrane transport in parietal cells measured with fluorescent dyes. Methods in Enzymology. 1990;192:38–81. doi: 10.1016/0076-6879(90)92062-i. [DOI] [PubMed] [Google Scholar]
- Push M, Neher E. Rates and diffusional exchange between small cells and a measuring pipette. Pflügers Archiv. 1988;411:204–211. doi: 10.1007/BF00582316. [DOI] [PubMed] [Google Scholar]
- Satoh H, Blatter LA, Bers DM. Effects of [Ca]i, Ca2+ load and rest on Ca2+ spark frequency in ventricular myocytes. American Journal of Physiology. 1997;272:H657–668. doi: 10.1152/ajpheart.1997.272.2.H657. [DOI] [PubMed] [Google Scholar]
- Satoh H, Delbridge LM, Blatter LA, Bers DM. Surface:volume relationship in cardiac myocytes studied with confocal microscopy and membrane capacitance measurements: species-dependence and developmental effects. Biophysical Journal. 1996;70:1494–1504. doi: 10.1016/S0006-3495(96)79711-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh H, Hayashi H, Noda N, Terada H, Kobayashi A, Hirano M, Yamashita Y, Yamazaki N. Regulation of [Na+]i and [Ca2+]i in guinea pig myocytes: dual loading of fluorescent indicators SBFI and fluo 3. American Journal of Physiology. 1994;266:H568–576. doi: 10.1152/ajpheart.1994.266.2.H568. [DOI] [PubMed] [Google Scholar]
- Semb SO, Sejersted OM. Calcium-induced contracture stimulates Na,K pump rate in isolated sheep cardiac Purkinje fibers. Journal of Molecular and Cellular Cardiology. 1997;29:2197–2212. doi: 10.1006/jmcc.1997.0455. [DOI] [PubMed] [Google Scholar]
- Shattock MJ, Bers DM. Rat vs. rabbit ventricle: Ca flux and intracellular Na assessed by ion-selective microelectrodes. American Journal of Physiology. 1989;256:C813–822. doi: 10.1152/ajpcell.1989.256.4.C813. [DOI] [PubMed] [Google Scholar]
- Shattock MJ, Matsuura H, Ward JPT. Sodium pump current measured in cardiac ventricular myocytes isolated from control and potassium depleted rabbits. Cardiovascular Research. 1994;28:1854–1862. doi: 10.1093/cvr/28.12.1854. [DOI] [PubMed] [Google Scholar]
- Shivkumar K, Deutsch NA, Lamp ST, Khuu K, Goldhaber JI, Weiss JN. Mechanism of hypoxic K loss in rabbit ventricle. Journal of Clinical Investigation. 1997;100:1782–1788. doi: 10.1172/JCI119705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutko JL, Bers DM, Reeves JP. Postrest inotropy in rabbit ventricle: Na+-Ca2+ exchange determines sarcoplasmic reticulum Ca2+ content. American Journal of Physiology. 1986;250:H654–661. doi: 10.1152/ajpheart.1986.250.4.H654. [DOI] [PubMed] [Google Scholar]
- Trafford AW, Diaz ME, O'Neill SC, Eisner DA. Comparison of subsarcolemmal and bulk calcium concentration during spontaneous calcium release in rat ventricular myocytes. Journal of Physiology. 1995;488:577–586. doi: 10.1113/jphysiol.1995.sp020991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber CR, Ginsburg KS, Philipson KD, Shannon TR, Bers DM. Allosteric regulation of Na+-Ca2+ exchange current by cytosolic Ca in intact cardiac myocytes. Journal of General Physiology. 2001;117:119–131. doi: 10.1085/jgp.117.2.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao A, Su Z, Nonaka A, Zubair I, Spitzer KW, Bridge JH, Muelheims G, Ross J, Barry WH. Abnormal myocyte Ca2+ homeostasis in rabbits with pacing-induced heart failure. American Journal of Physiology. 1998;275:H1441–1448. doi: 10.1152/ajpheart.1998.275.4.H1441. [DOI] [PubMed] [Google Scholar]