

Abstract
In vascular endothelial cells, elevation of cytosolic free calcium concentration ([Ca2+]i) causes activation of nitric oxide synthase (NOS) and release of nitric oxide (NO). The goal of the study was to characterize the interplay between [Ca2+]i and NO production in this cell type. Simultaneous measurements of [Ca2+]i and intracellular NO concentration ([NO]i) in cultured bovine vascular endothelial cells (CPAE cell line) with the fluorescent indicators fura-2 and DAF-2, respectively, revealed that Ca2+ influx following agonist-induced intracellular Ca2+ store depletion (capacitative Ca2+ entry, CCE) represents the preferential Ca2+ source for the activation of the Ca2+-calmodulin-dependent endothelial NOS (eNOS). Exposure to the NO donor sodium nitroprusside (SNP) showed that high NO levels suppressed CCE and had an inhibitory effect on Ca2+ extrusion by the plasmalemmal Ca2+-ATPase. This inhibitory effect on CCE was mimicked by the membrane-permeant cGMP analogue 8-bromo-cGMP, but was reversed by the NO scavenger haemoglobin and prevented by the inhibitor of the NO-sensitive guanylate cyclase ODQ. Brief exposure to SNP reduced the peak of ATP-induced Ca2+ release from the endoplasmic reticulum (ER) and accelerated Ca2+ reuptake into the ER. Prolonged incubation with SNP resulted in enhanced Ca2+ loading of the ER, as revealed by direct measurements of store content with the ER-entrapped low-affinity Ca2+ indicator mag-fura-2. The results suggest that in vascular endothelial cells, NO synthesis is under autoregulatory control that involves NO-dependent [Ca2+]i regulation. Via cGMP-dependent inhibition of CCE and acceleration of Ca2+ sequestration into the ER, NO can lower [Ca2+]i and therefore exert an autoregulatory negative feedback on its own Ca2+-dependent synthesis.
The vascular endothelium forms a selective permeability barrier between the bloodstream and the vessel wall, which plays a key role in blood pressure regulation and cardiovascular homeostasis (e.g. Moncada et al. 1991). The endothelium responds to neurohumoral and physical stimuli by producing nitric oxide (NO), which relaxes vascular smooth muscle cells (VSMCs) (Carrier et al. 1997), inhibits platelet aggregation (Radomski et al. 1987), modulates leukocyte adhesion (Kubes et al. 1991), and prevents VSMC proliferation (Garg & Hassid, 1989). NO is generated from the amino acid l-arginine by nitric oxide synthase (NOS). Three isoforms of NOS have been identified. The neuronal-type (nNOS or NOS I) and the endothelial-type (eNOS or NOS III) are constitutive and their activation is Ca2+-calmodulin-dependent. In contrast, the third isoform (the macrophage-type NOS II or iNOS) is inducible and Ca2+ independent (Moncada et al. 1991; Forstermann et al. 1994). Agonists that raise intra-cellular Ca2+ concentration ([Ca2+]i) activate constitutively expressed NOS transiently, generating NO in submicromolar concentrations (Forstermann et al. 1994; Blatter et al. 1995; Nakatsubo et al. 1998). In contrast, inducible NOS requires immunological activation of cells by cytokines or endotoxins. Typically, the enzyme is active for prolonged periods of time, producing NO in micromolar concentrations (Forstermann et al. 1994; Brorson et al. 1999). Therefore, inducible NOS appears to be more important in pathophysiological conditions, such as hypertension, atherosclerosis, diabetes, cerebral ischaemia and septic shock (Hobbs et al. 1999; Li & Forstermann, 2000). In target cells, most of the actions of NO are mediated by the activation of the soluble guanylate cyclase and formation of cyclic GMP (cGMP) (Hobbs et al. 1999).
Vascular endothelial cells respond to stimulation by vasoactive agonists with a transient elevation of [Ca2+]i. Agonist-induced elevations of [Ca2+]i result from release of Ca2+ from intracellular stores (endoplasmic reticulum, ER) through inositol 1,4,5-trisphosphate (IP3)-sensitive release channels, followed by activation of capacitative Ca2+ entry (CCE) through plasma membrane Ca2+ influx channels (Holda et al. 1998). Removal of Ca2+ from the cytosol occurs via extrusion by the plasma membrane Ca2+-ATPase pump (PMCA) (Klishin et al. 1998; Sedova & Blatter, 1999) and the plasmalemmal Na+–Ca2+ exchanger (Goto et al. 1996; Sedova & Blatter, 1999) as well as sequestration into intracellular stores by the sarcoplasmic- endoplasmic reticulum Ca2+-ATPase (SERCA) (Morgan & Jacob, 1998).
NO and cGMP-activated kinases have been shown to modulate virtually all Ca2+ transporting systems in a variety of non-endothelial target cells including VSMCs (Blatter & Wier, 1994; Clementi, 1998). In contrast, the paracrine and autocrine effects of NO on Ca2+ homeostasis in endothelial cells themselves have seldom been subject of investigations, leaving a largely incomplete picture. What is known is scant, and the few findings about [Ca2+]i regulation in endothelial cells by NO are contradictory. To shed some light onto the confusing picture regarding autocrine and paracrine effects of NO on Ca2+ homeostasis in vascular endothelial cells, we set out to systematically investigate the effect of NO on release and uptake of Ca2+ from the ER as well as on Ca2+ entry and extrusion in vascular endothelial cells. Our results demonstrate that in pulmonary artery endothelial cells, NO inhibits capacitative Ca2+ entry, reduces the peak of ATP-induced ER Ca2+ release and PMCA activity, and enhances filling and accumulation of Ca2+ into intracellular stores. The results suggest that autocrine and paracrine modulation of [Ca2+]i by NO exerts an important autoregulatory feedback on NO synthesis in vascular endothelial cells.
Part of this work has been presented in abstract form (Dedkova & Blatter, 2001).
METHODS
Cell culture and solutions
Experiments were performed on calf pulmonary artery endothelial (CPAE) cells in non-confluent cultures. The CPAE cell line was obtained as passage 15 from American Type Culture Collection (ATCC CCL−209; Manassas, VA, USA). The cells were cultured in Eagle's minimum essential medium, supplemented with 10% fetal bovine serum (GIBCO, Grand Island, NY, USA) and 2 mm l-glutamine, and kept at 37 °C in an atmosphere of 5% CO2 and 95% air. Once per week, the cells were dispersed using a Ca2+-free (0.1% EDTA) 0.25% trypsin solution, and subcultured onto glass coverslips for later experimentation. Cells were passaged up to six times after they were obtained from ATCC. Cells from all these six passages were used for experimentation. For each series of experiments, cells from typically two to nine different passages were used.
All experiments were carried out at room temperature (20–22 °C) on single cells in non-confluent cultures within 1 week after plating. During the experiments, cells were superfused continuously with a physiological salt solution (standard Tyrode solution) containing (mm): 135 NaCl, 5 KCl, 1 MgCl2, 2 CaCl2, 10 glucose and 10 Hepes; titrated to pH 7.3 with NaOH. In nominally Ca2+-free solution, CaCl2 was omitted. In the experiments measuring cellular [NO]i, 100 μm l-arginine was added to the standard Tyrode solution. In experiments where NOS was inhibited with Nω-nitro-l-arginine (l-NNA; Sigma Chemical Co., St Louis, MO, USA), l-arginine was omitted. 1−[1,2,4]Oxadiazolo[4,3-a]quinoxalin-1-one (ODQ) was obtained from Tocris Cookson Inc. (Ballwin, MO, USA).
Fluorescence measurements
[Ca2+]i measurements
Spatially averaged [Ca2+]i measurements from single endothelial cells were performed using the fluorescent ratiometric Ca2+ indicator fur−2. Cells were loaded with the dye by exposure to 1 ml standard Tyrode solution containing 5 μm fur−2 acetoxymethyl ester (fura-2 AM; Molecular Probes, Eugene, OR, USA) and 5 μl of a Pluronic F-127 stock solution (0.2 g ml−1 Pluronic F-127 dissolved in DMSO) for 20 min at room temperature. Fura-2 was excited by alternately illuminating the cells at 360 nm (F360) and 380 nm (F380) through a rotating filter wheel. Emitted cellular fluorescence was recorded at 540 nm. Single-cell fluorescence signals were recorded with a photomultiplier tube (model R2693; Hamamatsu Corp.) by masking off individual cells with an iris positioned in the emission path. Changes in [Ca2+]i were expressed as changes of the ratio R = F360/F380.
[NO]i measurements
For direct intracellular NO ([NO]i) measurements, cells were incubated with the fluorescent NO-sensitive dye 4,5-diaminofluorescein (DAF-2; Kojima et al. 1998; Nakatsubo et al. 1998). Cells were exposed to the membrane-permeant DAF-2 diacetate ([DAF-2 DA] = 5 μm; Calbiochem, San Diego, CA, USA) for 30 min at room temperature in 1 ml standard Tyrode solution containing 100 μm l-arginine. Cells were subsequently washed for 10 min. DAF-2 fluorescence was excited at 480 nm (F480). Emitted cellular fluorescence was recorded at 540 nm. Changes in cellular DAF-2 fluorescence intensities (F) in each experiment were normalized to the level of fluorescence recorded prior to stimulation (F0). Changes in [NO]i are expressed as F/F0, thus representing percentage increases above basal levels. Endogenous NO production or NO accumulation after exposure to NO donors was quantified by calculating the rate of increase from a linear fit of the DAF-2 F/F0 signal (d(F/F0)/dt) under various experimental conditions.
For simultaneous measurement of [Ca2+]i and [NO]i, CPAE cells were incubated simultaneously with 5 μm fura-2 AM and 5 μm DAF-2 DA for 30 min at room temperature and washed for 10 min to allow sufficient time for de-esterification of the indicators. Fluorescence was excited by alternately illuminating the cells at 360 nm (F360) and 380 nm (F380) for fura-2 measurements and 480 nm (F480) for DAF-2 measurements.
Intra-ER [Ca2+] measurements
For measurements of the ER [Ca2+], we used an approach based on the ability of the low-affinity fluorescent Ca2+ dye mag-fura-2 AM to compartmentalize into cell organelles (Hofer & Machen, 1993). Cells were loaded with mag-fura-2 by exposure to 1 ml standard Tyrode solution containing 2 μm mag-fura-2 acetoxymethyl ester (mag-fura-2 AM; Molecular Probes) and 5 μl of Pluronic F-127 stock solution (see above) for 60 min at 37 °C. During the last 20 min of dye loading, 10 μm BAPTA AM was added to the loading solution. Control experiments (not shown) confirmed that the presence of the Ca2+ buffer BAPTA in the cytosol eliminated contamination of the mag-fura-2 signal by changes in cytosolic [Ca2+]. Emitted mag-fura-2 fluorescence from single cells was measured at 540 nm in response to alternating illumination with light of 340 nm (F340) and 380 nm (F380). Under the described experimental conditions, changes of the ratio R = F340/F380 reflect changes of [Ca2+] in the intracellular Ca2+ stores (Hofer & Machen, 1993).
Measurement of CCE activity
Mn2+ was used as a substitute for Ca2+ ions to characterize unidirectional ion flux through the CCE pathway. As established previously (for details see Sedova et al. 2000), the rate of Mn2+ entry ([Mn2+]o = 100 μm), and therefore CCE activity, was inferred from the rate of quenching of fura-2 fluorescence excited at the Ca2+-insensitive (isosbestic) wavelength of 360 nm (F360). Because small cell-to-cell differences of the isosbestic excitation wavelength were observed, we used a linear combination of F360 and 5–10% of F380 to generate a truly Ca2+-insensitive fura-2 signal. This was verified by applying a brief ATP pulse to elicit a [Ca2+]i transient at the beginning of the experiment. After correction, F360 remained unaffected by a rise of [Ca2+]i. At the end of each Mn2+ quench experiment, cells were permeabilized with digitonin (10 μm). The remaining fluorescence signal was subtracted from the original signal and the F360 traces were normalized to the fluorescence level encountered before the addition of Mn2+.
Statistical analysis
Statistical differences among the data were determined with the Student's t test for unpaired data and considered significant at P < 0.05. Results are reported as means ± s.e.m. for the indicated number (n) of cells. Each experiment was conducted on a separate cell culture.
RESULTS
Direct measurements of [NO] in CPAE cells
For the direct measurement of endogenous NO production by single endothelial cells, we used the recently developed fluorescent NO-sensitive indicator DAF-2, which has been employed successfully to estimate NO levels in endothelial cells produced by constitutive eNOS (Nakatsubo et al. 1998; Berkels et al. 2000a). Stimulation of CPAE cells with the purinergic agonist ATP (5 μm) caused a gradual increase in DAF-2 fluorescence, which plateaued at about 25% above baseline after about 10 min (Fig. 1A; n = 16 cells). The initial rate of increase of F/F0 was (0.36 ± 0.05) × 10−3 s−1. After 5–10 min, the rise of F/F0 slowed down considerably (remaining rate of increase (0.09 ± 0.02) × 10−3 s−1), reflecting a decrease in endogenous NO production. In the presence of the NOS inhibitor l-NNA, ATP stimulation failed to elevate [NO]i, suggesting that the elevation of NO level was due to endogenous NO production through activation of eNOS (d(F/F0)/dt in the presence of l-NNA was (0.02 ± 0.01) × 10−3 s−1; n = 8; P < 0.001). The control trace indicates basal NO production in the absence of ATP (n = 5). To confirm that the intracellular DAF-2 indeed sensed NO, cells were treated with the exogenous NO donor sodium nitroprusside (SNP, 100 μm; n = 25), which led to a robust increase in DAF-2 fluorescence (Fig. 1B). The rate of increase of F/F0 was (3.07 ± 0.44) × 10−3 s−1, approximately ninefold faster than observed upon stimulation with ATP (Fig. 1A). The SNP-induced rise of DAF-2 fluorescence was not affected by l-NNA (rate of increase of F/F0 = (2.58 ± 0.94) × 10−3 s−1; not statistically significant), as would be expected from an exogenous source of NO (n = 6).
Figure 1. Effect of NOS inhibition on the ATP- and SNP-induced [NO]i elevations.
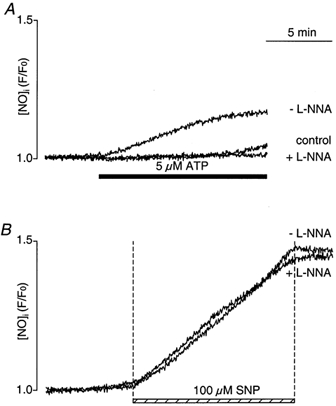
A, ATP (5 μm) induced an increase of [NO]i that was completely abolished when cells were preincubated with the NOS inhibitor l-NNA (100 μm). The control trace represents basal [NO]i levels in the absence of ATP. B, effect of exposure to SNP on [NO]i in the presence and absence of l-NNA. Cells were pretreated with l-NNA for > 40 min before exposure to SNP. DAF-2 fluorescence intensities in each experiment were normalized to the level of fluorescence recorded prior to stimulation.
Simultaneous measurements of [Ca2+]i and [NO]i: Effect of NO on ATP-induced increase of [Ca2+]i
We simultaneously measured changes in [Ca2+]i and NO production in single CPAE cells in response to ATP stimulation (5 μm) before and after cells were exposed to SNP (100 μm). Stimulation of CPAE cells with ATP induced a typical biphasic [Ca2+]i response (Fig. 2A) consisting of a rapid transient increase due to Ca2+ release from IP3-sensitive internal Ca2+ stores, followed by a maintained plateau resulting from Ca2+ influx from extracellular space via CCE (Holda et al. 1998). The rise of DAF-2 fluorescence lagged behind the peak of the [Ca2+]i transient and was predominantly observed during the plateau phase of the [Ca2+]i transient, suggesting that Ca2+ entering via CCE may be the preferential source for activation of eNOS (see also Discussion). The rate of rise of the DAF-2 F/F0 signal was (0.38 ± 0.06) × 10−3 s−1 (n = 13). In the same experiment, we also attempted to evaluate the effect of high levels of NO on [Ca2+]i. When cells were exposed to SNP later during the plateau phase of the ATP-induced [Ca2+]i transient, there was a robust increase of DAF-2 fluorescence as well as a decrease of [Ca2+]i (Fig. 2A). After exposure to SNP, the rate of rise of DAF-2 F/F0 increased significantly (approximately 10-fold) from, on the average, (0.38 ± 0.06) × 10−3 s−1 to (3.74 ± 0.62) × 10−3 s−1 (n = 13; P < 0.0001).
Figure 2. Simultaneous recordings of [Ca2+]i and [NO]i in CPAE cells.
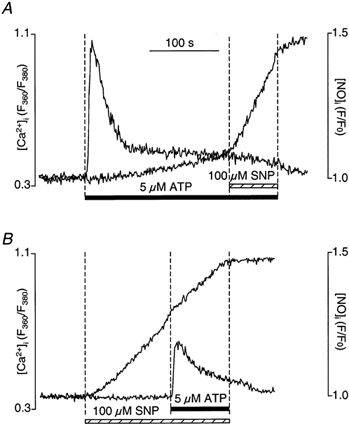
A, ATP stimulation (5 μm) induced a biphasic [Ca2+]i increase and NO production that became more prominent during the plateau phase of the [Ca2+]i transient. Subsequent SNP (100 μm) exposure significantly increased [NO]i level and suppressed the sustained [Ca2+]i plateau phase. B, high NO levels, achieved by SNP application 120 s before ATP stimulation, significantly decreased the ATP-induced [Ca2+]i transient.
When cells were exposed to SNP prior to stimulation with ATP (Fig. 2B), the DAF-2 F/F0 signal rose at a typical rate (d(F/F0)/dt = (3.31 ± 0.06) × 10−3 s−1; n = 6). The ATP-mediated rise of [Ca2+]i, however, was reduced significantly. On average (n = 6), SNP reduced the peak and the plateau phase by 59 ± 8% (P < 0.005) and 53 ± 6% (P < 0.001), respectively.
The initial results demonstrated directly that not only is the endogenous NO production Ca2+ dependent, but [Ca2+]i itself is also affected by NO in endothelial cells. We therefore set out to identify the Ca2+ source for eNOS activation and the Ca2+ regulatory pathways and mechanisms that are affected by NO in this cell type.
Ca2+ source for activation of endogenous NO production
The results shown in Fig. 2A suggested that Ca2+ entering the cell might be more proficient in activating eNOS than Ca2+ released from intracellular stores. The protocol applied in this experiment, however, did not allow a conclusive answer regarding the preferential Ca2+ source, because ATP stimulation in the presence of extracellular Ca2+ results in Ca2+ release and entry that overlap in time. (In separate experiments (n = 7; data not shown), we determined that in the presence of extracellular Ca2+, about 61% of the amplitude of the ATP-induced [Ca2+]i transient was due to store release, whereas the remainder resulted from Ca2+ entry.) Because of this overlap between release and entry, we applied an experimental protocol that separated Ca2+ release from entry in time, but also resulted in [Ca2+]i elevations of similar amplitude (Fig. 3). For that purpose, we exposed cells to the SERCA blocker thapsigargin (Thastrup et al. 1990) in the absence of extracellular Ca2+. Exposure to thapsigargin led to a transient increase of [Ca2+]i due to Ca2+ leakage from the ER and depletion of the stores due to inhibition of Ca2+ reuptake. Subsequent exposure to 2 mm extracellular Ca2+ resulted in an increase of [Ca2+]i, which under these conditions reflects Ca2+ influx through the CCE pathway (e.g. Holda et al. 1998) and will be referred to here as CCE transient. Simultaneous measurement of [NO]i showed that [NO]i increased mainly during activation of CCE (d(F/F0)/dt = (0.31 ± 0.04) × 10−3 s−1) and not during the thapsigargin-induced elevation of [Ca2+]i (d(F/F0)/dt = (0.05 ± 0.02) × 10−3 s−1; n = 6; P < 0.001), even though both [Ca2+]i signals reached similar amplitudes. This result demonstrates that Ca2+ entering the cell seems to be more efficient at activating eNOS than Ca2+ liberated from intracellular stores (see also Fig. 4).
Figure 3. Ca2+ source for eNOS activation.
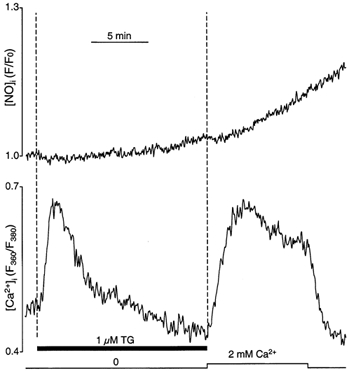
Simultaneous recordings of [Ca2+]i (bottom trace) and [NO]i (top trace) from CPAE cells loaded with fura-2 and DAF-2. Intracellular Ca2+ stores were depleted by exposure to thapsigargin (TG, 1 μm) in the absence of extracellular Ca2+. CCE was elicited by increasing [Ca2+]o to 2 mm.
Figure 4. Inhibition of ATP-induced CCE by NO.
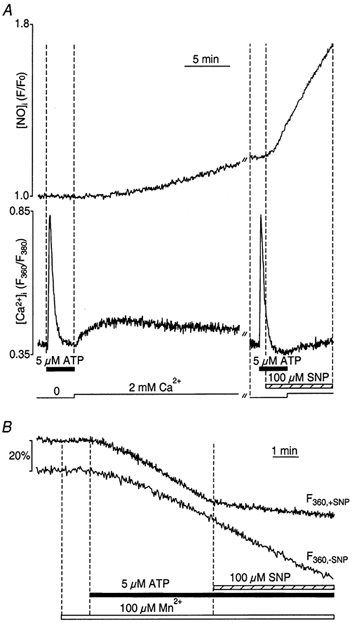
A, simultaneous recordings of [Ca2+]i (bottom trace) and [NO]i (top trace) from CPAE cells. Cells were exposed to ATP (5 μm) in the absence of extracellular Ca2+ to deplete intracellular Ca2+ stores. After recovery of the ATP-induced [Ca2+]i transient, 2 mm Ca2+ was added to induce CCE. SNP, added 120 s before Ca2+ application, significantly inhibited ATP-induced CCE. B, the rate of Mn2+ entry ([Mn2+]o= 100 μm; [Ca2+]o= 0) was monitored as the rate of quenching of fura-2 fluorescence excited at the Ca2+-insensitive (isosbestic) wavelength of 360 nm (F360; normalized). The fast Mn2+ influx triggered by ATP stimulation was significantly suppressed by SNP (100 μm). The Mn2+ quench F360 trace labelled ‘-SNP’ was obtained from a cell stimulated with ATP but not subsequently exposed to SNP.
Effects of NO on capacitative Ca2+ entry
In a next series of experiments, we directly measured the effect of NO, through the use of the NO-donor SNP, on capacitative Ca2+ entry. To activate CCE, we depleted intracellular Ca2+ stores either by stimulation with ATP or exposure to the SERCA blocker thapsigargin.
CCE activation and NO production after ATP-induced store depletion
To deplete ER Ca2+ stores, single CPAE cells were stimulated with ATP (5 μm) in Ca2+-free medium. Changes in [Ca2+]i and NO production were measured simultaneously in cells loaded with fura-2 and DAF-2 (Fig. 4A). CCE was characterized by analysis of the changes of [Ca2+]i that occurred upon re-addition of 2 mm extracellular Ca2+ following store depletion. Single endothelial cells were stimulated twice with ATP in the absence of extracellular Ca2+ (Fig. 4A). Application of ATP caused a reproducible transient increase of [Ca2+]i reaching a peak after 10–20 s and returning to the basal level within 1–2 min. No measurable change in [NO]i was observed during the ATP-induced [Ca2+]i transient. The subsequent addition of 2 mm extracellular Ca2+ led to an increase of [Ca2+]i, which under these conditions resulted from Ca2+ influx through the CCE pathway. During the CCE transient, NO production was initiated, leading to a gradual increase of the cellular DAF-2 signal (the rate of rise of the DAF-2 F/F0 signal was (0.31 ± 0.07) × 10−3 s−1; n = 5). Consistent with the data shown in Fig. 2A and Fig. 3, these experiments also confirmed that Ca2+ entering the cell is more effective for the activation of NO synthesis than Ca2+ released from internal stores.
To determine the effect of NO on CCE itself, SNP (100 μm) was applied 2 min before Ca2+ re-addition after the second stimulation with ATP of the same cell. Besides the expected robust increase of the DAF-2 fluorescence (d(F/F0)/dt = (2.48 ± 0.67) × 10−3 s−1) signal, the application of SNP significantly decreased both the rate and the amplitude of the CCE transient. In the presence of SNP, on average (n = 5), the rate and the amplitude decreased to 22 ± 4% (P < 0.001) and 23 ± 4% (P < 0.001), respectively, compared with control (see also Fig. 6).
Figure 6. Effect of NO on CCE parameters after store depletion with thapsigargin and ATP.
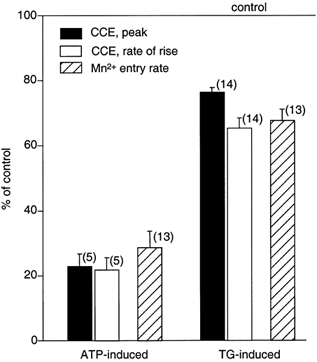
Summary of the effects of SNP on CCE parameters after store depletion with ATP stimulation (left) and thapsigargin (TG) exposure (right). The columns represent the amplitude of the CCE transient (filled column; measured 1 min after addition of Ca2+), the rate of rise of [Ca2+]i during the CCE transient (open column) and the rate Mn2+ entry measured as the rate of quenching of fura-2 fluorescence (hatched column). The data are presented as average percentages ±s.e.m. of control, i.e. the same parameters measured in the absence of SNP. Numbers in parentheses indicate the number of individual cells. The rate of rise of [Ca2+]i and the rate of Mn2+ entry were measured as the slope of the linear portion of the signals presented in Figs 4 and 5.
We also used the Mn2+ quench technique to assess the effect of NO on CCE in the CPAE cells. As established previously (Sedova et al. 2000), Mn2+ can act as a surrogate for Ca2+ ions to study ion permeation through the CCE pathway. The use of Mn2+ has the following advantages: Mn2+ permeates the CCE channel similarly to Ca2+, is only a poor substrate for Ca2+-ATPases, and can be used to estimate CCE activity from the rate of quenching of the fluorescence from cytosolic Ca2+ indicators such as fura-2 when measured at a Ca2+-independent wavelength. Exposure of CPAE cells to 100 μm Mn2+ had little effect on the fura-2 signal measured at the 360 nm excitation wavelength (Fig. 4B). Application of ATP (5 μm) significantly increased the decline of F360 as a result of Mn2+ entry and quenching of fura-2. Subsequent exposure to SNP in the continued presence of ATP, however, induced a marked decrease in quenching rate indicative of a NO-induced inhibition of CCE. On average (n = 13), in the presence of SNP, the rate of Mn2+ entry was only 29 ± 5% of that observed under control conditions (P < 0.001). The second trace in panel B shows that, at the point in time when SNP was applied, quenching of fura-2 continued at an unchanged rate in the absence of SNP. Thus, the change in quenching rate of the top F360 trace reflects an effect caused by SNP, and was not the due to the fact that fura-2 quenching might be nearing completion. The data presented in Fig. 4 suggest that Ca2+/Mn2+ permeable plasma membrane channels and CCE are potential targets for an inhibitory effect of NO.
CCE activation by thapsigargin-induced store depletion
We applied a second protocol to evaluate the role of NO for CCE regulation. Intracellular Ca2+ stores were depleted of Ca2+ by preincubating CPAE cells for 15 min in Ca2+-free solution containing 1 μm thapsigargin. Re-application of 2 mm external Ca2+ evoked the typical biphasic CCE transient (Fig. 5A). The CCE transient was characterized by an initial rise of [Ca2+]i that rapidly reached a peak, followed by a slower decline of [Ca2+]i to a sustained plateau of elevated [Ca2+]i. Upon removal of extracellular Ca2+, [Ca2+]i decreased quickly to the resting level. As established previously, under these conditions this decay reflects solely Ca2+ extrusion that occurs predominantly by the PMCA (Klishin et al. 1998; Sedova & Blatter, 1999). As shown in Fig. 5A, exposure to extracellular Ca2+ in the presence of 100 μm SNP also caused an increase in [Ca2+]i, but the rate of Ca2+ entry was slower (only 64 ± 2% of control; n = 14; P < 0.001), and the amplitude was lower than in the absence of the NO donor (75 ± 2% compared with control; n = 14; P < 0.001).
Figure 5. Effect of NO on CCE triggered by thapsigargin-induced store depletion.
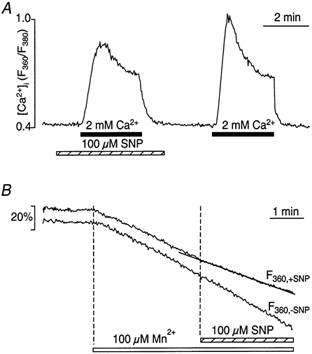
A, CCE was triggered by exposure of CPAE cells to 2 mm Ca2+ (filled bar). Intracellular Ca2+ stores were depleted previously (not shown) by exposure to thapsigargin (TG, 1 μm) for 15 min in the absence of extracellular Ca2+. Activation of CCE in the presence of SNP inhibited CCE, resulting in a slower rate of rise of [Ca2+]i, a lower amplitude and slower decline of the CCE transient. B, addition of Mn2+ (100 μm) to thapsigargin-pretreated cells stimulated a fast Mn2+ influx, which was partially suppressed by SNP ([Ca2+]o= 0). The control Mn2+ quench F360 trace labelled ‘-SNP’ was obtained from a thapsigargin-treated cell not subsequently exposed to SNP.
We also examined the effect of SNP on Mn2+ influx after previous store depletion with thapsigargin. The F360 signal of a representative experiment is shown in Fig. 5B. The rate of quenching of the fura-2 signal increased after exposure to Mn2+ because of Mn2+ entry through store-operated cation influx channels. Application of 100 μm SNP resulted in a slowing of the fura-2 quenching rate, suggesting inhibition of CCE, while in the absence of SNP, the quenching rate remained constant. In the presence of SNP, the rate of Mn2+ entry after store depletion was only 68 ± 5% (n = 13; P < 0.001) of the quenching rate in the absence of SNP.
Figure 6 summarizes the results regarding inhibition of CCE by NO. When CCE was activated by thapsigargin depletion of internal stores, several parameters of CCE activity, i.e. the amplitude of the CCE transient (filled column), the rate of rise of the CCE transient (open column), as well as the rate of Mn2+ entry (hatched column), were reduced by approximately 25–35% in the presence of SNP. The effect of NO was much more pronounced when CCE was activated by ATP-dependent store depletion. Under these circumstances, all three parameters decreased by 75–80% in the presence of NO. Under the premise that both protocols led to store depletion and CCE activation, this discrepancy in the ability of NO to inhibit CCE suggested that other Ca2+ regulatory mechanisms are involved and affected by NO. This, as well as the mechanism through which NO acted on CCE, was further investigated in the following series of experiments.
Inhibition of CCE by SNP is due to NO
To confirm that the observed effects of SNP on CCE were indeed the result of the action of NO itself, we performed two types of control experiments. In a set of experiments (n = 4), we established that a different NO donor (S-nitroso-N-acetyl-penicillamine or SNAP, 100 μm) had the same inhibitory effect on CCE as SNP (data not shown), suggesting that the effects of SNP were probably due to its ability to produce NO and not due to some non-specific action of this substance.
To further establish this point, we treated cells with SNP in the presence of the NO scavenger haemoglobin (10 μm), which is known to effectively bind NO (Moncada et al. 1991; Blatter & Wier, 1994). As shown in Fig. 7A, the effects of SNP on amplitude (104 ± 2% of control) and kinetics (rate of entry 98 ± 2% and rate of decay 102 ± 3% of control) of the CCE transient were reversed in the presence of haemoglobin (n = 4; all parameters not significantly different from control). The effect of haemoglobin indicated that inhibition of CCE by SNP was mediated by NO.
Figure 7. Inhibition of CCE by SNP is mediated by NO and cGMP.
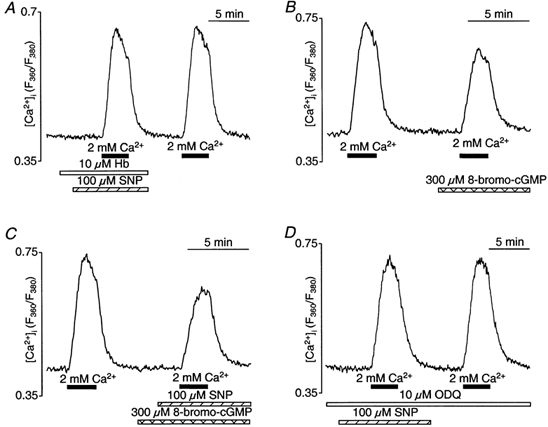
CCE transients elicited with 2 mm extracellular Ca2+ after store depletion with thapsigargin (1 μm). Changes of [Ca2+]i evoked by thapsigargin itself are not shown. A, pretreament of CPAE cells with the NO scavenger haemoglobin (Hb) abolished the inhibitory effect of SNP on CCE. Hb (10 μm) was added 100 s before SNP application. B, preincubation with the membrane-permeant cGMP analogue 8-bromo-cGMP (300 μm) before addition of 2 mm Ca2+ inhibited CCE. C, CCE transient in the absence and presence of SNP (100 μm) and 8-bromo-cGMP (300 μm). D, effect of SNP on CCE in the presence of the NO-sensitive guanylate cyclase inhibitor ODQ (10 μm). Cells were preincubated for 40 min.
Inhibition of CCE by NO is mediated by cGMP
There is ample evidence that many effects of NO in various target cells are mediated by activation of soluble guanylate cyclase and elevation of cGMP levels (e.g. Xu et al. 1994; Kwan et al. 2000). To test whether the observed SNP effects on CCE in CPAE cells might also be mediated by increased cGMP levels, we used the exogenous cell-permeant cGMP analogue 8-bromo-cGMP (300 μm). For these experiments, we applied the thapsigargin protocol to deplete intracellular stores and to activate CCE. Figure 7B shows CCE transients recorded in the absence and presence of 8-bromo-cGMP treatment. Similar to SNP (Fig. 5A), 8-bromo-cGMP altered the shape of the CCE transient by decreasing the rate of Ca2+ entry (65 ± 3% of control; n = 6; P < 0.001), its amplitude (72 ± 3% of control; n = 6; P < 0.001) and decay kinetics after removal of extracellular Ca2+ (72 ± 4% of control; n = 6; P < 0.001). When 8-bromo-cGMP and SNP were applied simultaneously (Fig. 7C), the effect on amplitude (69 ± 4% of control; n = 3), rate of rise (57 ± 6%) and decay (76 ± 5%) of the CCE transient was very similar and not significantly different from the effect of cGMP alone. Thus, maximally active concentrations of cGMP and NO did not have additive effects on the kinetics and amplitude of the CCE transient.
Further evidence for the involvement of the NO-cGMP signalling pathway was established by the use of 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ), a rather selective inhibitor of the NO-sensitive guanylate cyclase (Fig. 7D). In the presence of 10 μm ODQ (n = 5), the effect of SNP was completely abolished. With ODQ, the amplitude as well as the rise and decay kinetics of the CCE transient were not significantly different with and without SNP or compared with control.
The results presented in Fig. 7 strongly suggested that store-operated Ca2+ entry in endothelial cells is modulated via an NO-mediated increase in cGMP levels.
Inhibition of PMCA by NO
We have established previously a protocol that can be used to investigate the kinetics of PMCA activity and its regulation in CPAE cells (Klishin et al. 1998). To do so, we use the thapsigargin depletion protocol and subsequent re-exposure to 2 mm extracellular Ca2+ to elicit a CCE transient. We have demonstrated that under these circumstances, [Ca2+]i in CPAE cells is determined solely by Ca2+ entry via CCE and extrusion across the cell membrane. As shown previously (Sedova & Blatter, 1999), Ca2+ extrusion is carried by the PMCA, and Ca2+ removal via Na+-Ca2+ exchange becomes relevant only when the PMCA is blocked. Consequently, under control conditions, changes of [Ca2+]i during the CCE transient that are observed after removal of extracellular Ca2+ reflect Ca2+ removal by the PMCA.
Figure 8A shows two CCE transients that were elicited in thapsigargin-depleted cells. In contrast to Fig. 7, in this experiment, cells were exposed to 2 mm extracellular Ca2+ only for a brief period of time (< 1 min) and Ca2+ was removed again during the rising phase of the CCE transient. The experiment clearly demonstrates that SNP not only slowed CCE, resulting in a significantly lower amplitude of the CCE transient, but it also slowed the decay of the CCE transient after removal of Ca2+. The slow decline of the CCE transient resulted from NO-dependent inhibition of the PMCA. This result is similar to the CCE transients presented in Figs 5A, 7B and 7C, which also revealed a significant slowing of the decay kinetics upon removal of extracellular Ca2+ in the presence of SNP (Fig. 5A), 8-bromo-cGMP (Fig. 7B) or both compounds (Fig. 7C). Figure 8B shows the decay kinetics of normalized CCE transients after removal of extracellular Ca2+ in the presence and absence of SNP (left) and 8-bromo-cGMP (right). In both cases, the kinetics of the decay of the CCE transient, expressed as the time constant, τ, of a monoexponential fit to the [Ca2+]i signal, were significantly slowed by SNP and 8-bromo-cGMP, respectively. In fact, on average (Fig. 8E), τ of the decline of the CCE transient was only 66 ± 4% (n = 7; P < 0.001) in the presence of SNP, and only 72 ± 4% (n = 6; P < 0.001) of control with 8-bromo-cGMP present.
Figure 8. Inhibition of the PMCA by NO.
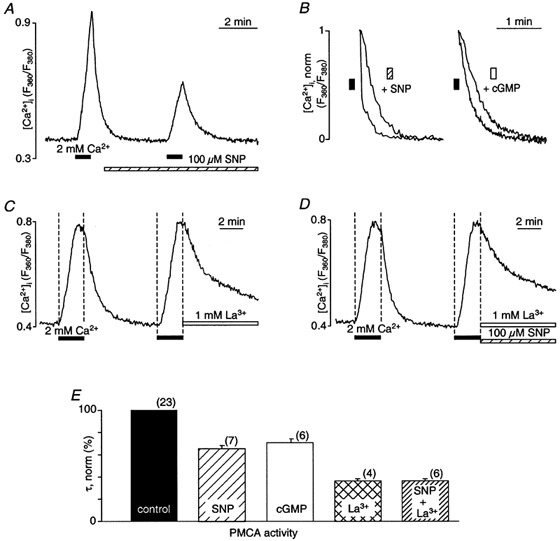
A, CCE transients elicited by brief (< 50 s) exposure to 2 mm extracellular Ca2+ in the absence and presence of SNP (100 μm). Extracellular Ca2+ was removed again during the rising phase of the CCE transient. Intracellular Ca2+ stores were previously depleted by thapsigargin treatment. B, normalized decaying phases of the CCE transients (taken from Figs 5A and 7B) after removal of extracellular Ca2+ under control conditions and in the presence of SNP and 8-bromo-cGMP (cGMP), respectively. The [Ca2+]i signals were normalized to the level of [Ca2+]i encountered at the time of removal of extracellular Ca2+. C, inhibition of the PMCA by lanthanum ([La3+]= 1 mm). La3+ was applied when Ca2+ was removed from the extracellular solution. D, inhibition of the PMCA by La3+ in the presence of SNP. E, average normalized time constants (τ) of the decay phase of the CCE transients under control conditions (100 %) and in the presence of SNP, 8-bromo-cGMP (cGMP), La3+ and La3++ SNP. τ was determined by a monoexponential fit to the declining phase of the CCE transient. The numbers in parentheses indicate the number of cells for each group.
When the PMCA was blocked with lanthanum ([La3+] = 1 mm), the decay of the CCE transient after removal of extracellular Ca2+ was slowed significantly (37 ± 1% compared with control; n = 4; P < 0.001) and remained incomplete (Fig. 8C and E); however, SNP had no additional inhibitory effect (37 ± 2% of control; n = 6; P < 0.001; Fig. 8D and E). These data suggested that the observed effects of SNP and cGMP on the decay phase of the CCE transient were indeed due to inhibition of the PMCA.
In summary, these results indicate that NO inhibits the PMCA in CPAE cells and that this inhibition is mediated by a signalling mechanism involving cGMP.
NO enhances Ca2+ uptake into intracellular Ca2+ stores
As demonstrated so far (summarized in Fig. 6), NO inhibited CCE, independently of whether CCE was activated by ATP- or thapsigargin-induced store depletion; however, the effect of SNP on the thapsigargin-induced cation influx was significantly smaller. Bearing in mind that the difference between the two experimental protocols was the fact that the SERCA pump was blocked (by thapsigargin) or not, we focused on the possibility that NO may influence refilling of intracellular Ca2+ stores.
In a first set of experiments, we explored to what extent NO affects the filling status of Ca2+ stores from which Ca2+ could be released by ATP stimulation. We used the Ca2+ ionophore ionomycin to estimate releasable Ca2+ store content. Exposure of unstimulated CPAE cells to ionomycin (1 μm) in the absence of extracellular Ca2+ led to a rapid and substantial increase of [Ca2+]i (Fig. 9A), giving an estimate of releasable Ca2+ from intact filled Ca2+ stores. The transient nature of the ionomycin-induced rise of [Ca2+]i was due to subsequent removal of Ca2+ from the cell. When cells were stimulated with ATP (5 μm) in Ca2+-free medium to release Ca2+, subsequent ionomycin exposure released approximately three times less Ca2+ from the internal stores, suggesting a substantial depletion by the preceding ATP stimulation (Fig. 9B). The amount of releasable Ca2+ by ionomycin under these experimental conditions could be due to resequestration of Ca2+ after ATP stimulation and/or liberation of Ca2+ from an ionomycin-sensitive store that is not depleted by ATP. To investigate whether NO affects the filling of Ca2+ stores in ATP-activated cells, SNP (100 μm) was applied before ATP (Fig. 9C). Pretreatment with SNP resulted in the following differences: SNP slightly attenuated the amplitude of the ATP-induced [Ca2+]i response (compared with panel B); it accelerated the decay of the ATP-induced [Ca2+]i transient; and most notably, it increased the remaining amount of Ca2+ in the stores that could be released by ionomycin (Fig. 9C). On average, the amplitude of the ionomycin-induced [Ca2+]i signal in the presence of SNP was 54 ± 13% higher (P < 0.005; n = 8) than in the absence of SNP (Fig. 9B). Similar results were obtained when SNP was added not prior to but at the peak of the ATP-activated [Ca2+]i response. In this case, the ionomycin-induced release was 44 ± 8 % (n = 3) higher than in the absence of SNP (not shown).
Figure 9. Effect of SNP on the amount of releasable Ca2+ from ionomycin-sensitive Ca2+ stores.
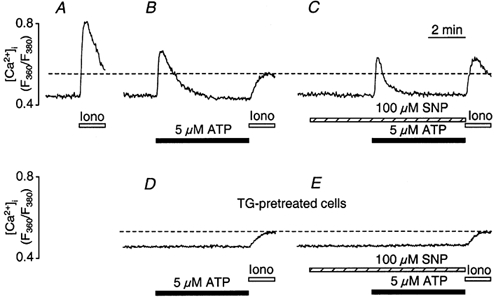
Ionomycin (Iono, 1 μm) was applied in the absence of extracellular Ca2+ at the end of each experiment, to estimate the amount of releasable Ca2+ from intracellular stores. A, control [Ca2+]i transient due to ionomycin-induced Ca2+ release. B, stimulation with ATP (5 μm) prior to ionomycin exposure decreased the releasable amount of Ca2+. C, ionomycin treatment following ATP stimulation in the presence of SNP (100 μm, applied 200 s prior to ATP stimulation). The presence of SNP reduced the amplitude of the ATP-induced [Ca2+]i transient, accelerated its recovery and augmented the amount of store Ca2+ releasable by ionomycin exposure. D, ionomycin-releasable [Ca2+]i in a CPAE cell with disabled ER. ER Ca2+ release was abolished by preceding exposure to thapsigargin (TG, 1 μm for 15 min) and verified by the lack of a response to ATP stimulation. E, ionomycin-induced [Ca2+]i signal in a thapsigargin-treated cell in the presence of SNP. Panels A to E are from different cells.
When the ER Ca2+ store was eliminated (Fig. 9D) by prolonged pretreatment with thapsigargin (substantiated by the lack of a response to ATP stimulation), exposure to ionomycin resulted in a small elevation of [Ca2+]i (n = 4). This small (< 20 % of control) increase of [Ca2+]i was consistent with the liberation of Ca2+ from intracellular organelles other than the ER that are capable of sequestering Ca2+ (e.g. mitochondria). As shown in Fig. 9E, however, preincubation with SNP had no effect on the ionomycin-induced response in thapsigargin-treated cells (n = 5), suggesting that NO produced by 100 μm SNP did not affect sequestration of Ca2+ into non-ER organelles (such as mitochondria) in a relevant fashion.
To further distinguish whether this effect of SNP was due to an activation of SERCA or an inhibition of Ca2+ release from the stores, the following experiments were performed. Cells were stimulated twice with 5 μm ATP in the absence of extracellular Ca2+. Between the two ATP stimulations, 2 mm extracellular Ca2+ was re-introduced to replenish the Ca2+ stores. As shown in Fig. 10A, two subsequent exposures to 5 μm ATP resulted in [Ca2+]i transients with virtually identical amplitudes and time courses. Exposure to SNP 1 min prior to the second stimulation with ATP led to a small decrease in the amplitude of the ATP-induced [Ca2+]i transient (Fig. 10B), possibly due to some inhibitory effect of NO on Ca2+ release. In contrast, when cells (n = 5) were incubated in SNP during the entire period of store reloading (> 20 min; Fig. 10C), the ATP-induced [Ca2+]i transient was significantly larger in amplitude (190 ± 39 % of control; P < 0.05) and the rate of decay was accelerated significantly (227 ± 63 % of control; P < 0.05). These results suggested that prolonged exposure to SNP resulted in an NO-mediated enhanced filling of intracellular Ca2+ stores.
Figure 10. Effect of SNP on Ca2+ reuptake into the stores.
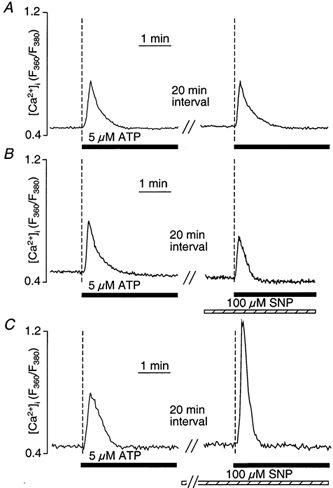
A, fura-2-loaded CPAE cells were stimulated twice with ATP (5 μm) in the absence of extracellular Ca2+. Between ATP stimulations, Ca2+ stores were allowed to refill in the presence of 2 mm extracellular Ca2+ for 20 min. B, ATP stimulation in the presence of SNP. SNP was introduced for 1 min prior to the second ATP stimulation; 20 min was allowed for refilling of the stores between ATP stimulations. C, ATP stimulation after store loading in the presence of SNP. SNP was present during the entire 20 min reloading period between the two ATP stimulations.
This hypothesis was confirmed by direct measurements of Ca2+ store content. For this purpose, we used the membrane-permeant form of the low-affinity Ca2+ indicator mag-fura-2, which was used to monitor the changes in free [Ca2+] in the internal Ca2+ stores without cell permeabilization (Hofer & Machen, 1993). Figure 11 shows typical recordings of free luminal [Ca2+] in control experiments (Fig. 11A) and in the presence of SNP (Fig. 11B). Single CPAE cells were stimulated with 5 μm ATP for 100 s in a Ca2+-containing medium. As shown in Fig. 11A, exposure to ATP caused a fast decrease of the mag-fura-2 signal as a result of Ca2+ release from the stores. After removal of ATP, the mag-fura-2 signal increased to previous levels due to reuptake of Ca2+. Twenty minutes were allowed for sufficient refilling of the stores prior to the second stimulation with ATP. In contrast, when Ca2+ stores refilled in the presence of SNP (Fig. 11B), the magnitude of Ca2+ mobilization from the stores by ATP stimulation was higher (147 ± 13 %; n = 5; P < 0.01), and the rate of Ca2+ reuptake into the stores was significantly faster (246 ± 27 %; n = 5; P < 0.001). Moreover, the free [Ca2+] in the ER was higher after prolonged incubation with SNP. Finally, even though the magnitude of Ca2+ release was larger in the presence of SNP, the absolute level of store Ca2+ was still higher during ATP stimulation compared with control conditions.
Figure 11. NO-dependent enhancement of Ca2+ loading of the ER revealed by direct measurements of store content with the low-affinity indicator mag-fura-2.
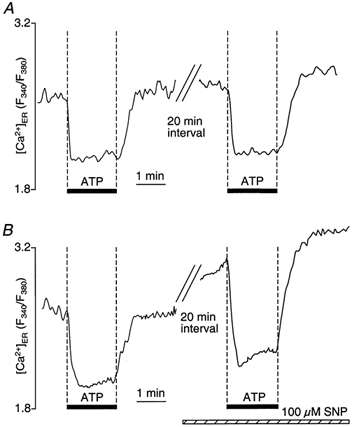
CPAE cells were loaded with mag-fura-2 AM and BAPTA AM. Changes in store [Ca2+] are expressed as changes of the fluorescence ratio F340/F380, reflecting Ca2+ movements in and out of intracellular Ca2+ stores. A, stimulation of cells with ATP (5 μm) in Ca2+-containing medium evoked release of Ca2+ from the stores, resulting in a rapid decline of the mag-fura-2 ratio. Removal of ATP initiated reuptake of Ca2+ into stores. After a 20 min refilling period, accumulated Ca2+ was released by a second stimulation with ATP. B, SNP (100 μm) application during the 20 min refilling period after the first stimulation with ATP resulted in enhanced store loading. A subsequent second challenge with ATP resulted in enhanced Ca2+ release and acceleration of Ca2+ reuptake.
We conclude from these experiments that increasing NO levels accelerates reuptake of Ca2+ into the intracellular stores. This allows more rapid refilling of the stores, and, as a consequence results in a de-activation of CCE into CPAE cells.
DISCUSSION
Ca2+ dependence of NO synthesis in vascular endothelial cells
Although it is well established that eNOS is regulated in a Ca2+-calmodulin-dependent manner, recent findings provided interesting evidence that the source of Ca2+ appears to be relevant for the efficient activation of NOS. Several studies have shown that Ca2+ entering via CCE is the preferential source for eNOS activation (Wang et al. 1996; Lin et al. 2000). Our simultaneous measurements of [Ca2+]i and [NO]i provided direct evidence for this idea (Figs 2, 3 and 4). The rise of [NO]i clearly lagged behind the peak [Ca2+]i increase when CPAE cells were stimulated with ATP. [NO]i started to rise during the plateau phase of the ATP-induced [Ca2+]i transient (Fig. 2A), during which cytosolic [Ca2+] remains elevated due to Ca2+ entry via CCE, and not because of release from internal stores. A similar temporal dissociation between peak [Ca2+]i and maximal NO production was observed previously in bradykinin-stimulated vascular endothelial cells (Blatter et al. 1995). To clearly distinguish between the efficacy of released Ca2+versus Ca2+ entering the cell to activate NO production, and to further exclude the possibility that the ATP-induced [Ca2+]i transient in the absence of extracellular Ca2+ (Fig. 4A) was just too short to activate eNOS, we also measured [NO]i during exposure to thapsigargin (Fig. 3). The transient elevations of [Ca2+]i during thapsigargin exposure are the result of liberation of Ca2+ from the stores, and resemble the CCE transients in amplitude and time course (Holda et al. 1998; Klishin et al. 1998). Under these conditions as well, NO production was observed predominantly during activation of CCE and not during release of Ca2+ from the stores. Thus, it appears that in endothelial cells, Ca2+ entering across the surface membrane is more efficient in activating NOS. This is consistent with previous studies showing that eNOS is mainly associated with the plasma membrane in endothelial cells from numerous vascular beds (Hecker et al. 1994; Garcia-Cardena et al. 1996), from where it can be translocated to various intracellular targets upon receptor stimulation (Prabhakar et al. 1998). In addition, recent evidence showed that eNOS is associated with caveolae, plasma membrane structures that have been implicated in important cellular signalling pathways (Garcia-Cardena et al. 1996; Igarashi et al. 1999).
Autoregulation of NO synthesis in endothelial cells
NO levels in stimulated endothelial cells typically do not exceed micromolar levels (Blatter et al. 1995; Nakatsubo et al. 1998), suggesting regulatory mechanisms that limit and terminate NO synthesis under physiological conditions. This is particularly important because excessive NO levels are detrimental to cells and have been implicated in various pathological conditions of the cardiovascular system. For example, high NO levels have been suggested to play a role in the hyporesponsiveness to vasoconstrictors observed in animals with septic shock (Julou-Schaeffer et al. 1990). An increased production of nitrogen oxides was also observed in vascular endothelium during early stages of hypercholesterolaemia concomitant with an upregulation of eNOS (Kanazawa et al. 1996).
Ca2+ plays a key role in the activation of eNOS and affects Ca2+ homeostasis in target cells of endothelial NO release (such as VSMCs). This raises the question of whether endothelial NO also influences [Ca2+]i regulation in endothelial cells themselves and thereby exerts autocrine regulation of its own synthesis. Few data are available on the effect of NO on endothelial [Ca2+]i regulation and the results are inconclusive. For example, it was shown that NO (applied in the form of SNP) could stimulate Ca2+ influx in porcine aortic endothelial cells (Berkels et al. 2000b). In contrast, NO dose-dependently decreased endothelin 1-stimulated mobilization of Ca2+ in microvascular endothelium from human brain (Chen et al. 1999), a response that was mimicked by cGMP and abrogated by inhibitors of guanylate cyclase or cGMP-dependent protein kinase. Furthermore, neither elevation of NO levels (by exposure to the NO donor SNP) and concomitant increase of cGMP nor a reduction of endogenous [NO] by treatment with the NOS inhibitor l-NNA had any effect on the histamine-induced [Ca2+]i increases in human umbilical vein endothelial cells (Bolz & Pohl, 1997), whereas the NOS inhibitor Nω-nitro-l-arginine methyl ester (l-NAME) enhanced the histamine-induced [Ca2+]i elevations in primary cultures of human cerebral microvascular endothelial cells (Li et al. 1999). In addition, high doses of l-arginine, the substrate for NOS, slightly reduced [Ca2+]i in endothelial cells and markedly decreased the [Ca2+]i in VSMCs in co-culture, demonstrating both autocrine and paracrine effects of NO (Wang et al. 1996). There is also evidence that in vascular endothelial cells, endogenous NO is capable of enhancing CCE, but possibly also of activating the PMCA (Chen et al. 2000). Because NO is capable of influencing Ca2+ homeostasis in endothelial cells, it may also affect the Ca2+-dependent NO synthesis in NO-producing cells. There is indeed evidence to indicate that NO may have an autoregulatory role via a negative feedback inhibition of NOS activity in rat cerebellum (Rogers & Ignarro, 1992), murine macrophages (Assreuy et al. 1993) and vascular endothelial cells (Buga et al. 1993; Ma et al. 1996).
Our own results revealed that Ca2+ entering via CCE activates NOS, but higher levels of NO in turn inhibit CCE. Inhibition of CCE occurs either by direct blockade of the entry pathway or by enhancing and accelerating reuptake of Ca2+ into depleted intracellular stores by a cGMP-dependent mechanism. Our results lend support for both mechanisms acting in parallel. This conclusion is drawn from the following observations. (1) Exogenous NO from the NO donor SNP significantly suppressed the ATP-induced capacitative Ca2+ entry and Mn2+ influx (Fig. 4). (2) The inhibitory effect of SNP on CCE and Mn2+ entry was smaller when CCE was activated by inhibition of SERCA with thapsigargin rather than with ATP stimulation (Fig. 6). This observation is not unprecedented. Loss of the inhibitory effect of NO on capacitative Ca2+ entry upon SERCA blockade was shown previously in platelets (Okamoto et al. 1995; Trepakova et al. 1999), human embryonic kidney (HEK 293) cells (Bischof et al. 1997), rat megakaryocytes (Uneyama et al. 1998) and VSMCs (Cohen et al. 1999). This is consistent with the idea that with inhibition of SERCA, refilling of the stores is prevented completely, and therefore the signal to activate CCE is likely to remain strong and thereby counteracts the inhibitory effect of NO on CCE. Furthermore, acceleration of Ca2+ uptake and store refilling by NO inhibits CCE through inactivation of this pathway. (3) In thapsigargin-pretreated cells, SNP not only failed to activate, but slightly inhibited, the PMCA (Fig. 5A and Fig. 8). This argues against the possibility that the acceleration of the decline of the ATP-induced [Ca2+]i transient in the presence of NO (Fig. 9 and Fig. 10) was the result of enhanced Ca2+ extrusion from cells, because the PMCA appears to be inhibited by NO (Fig. 8). Measurement of Mn2+ influx revealed directly that NO inhibited Ca2+ entry and might act on the Ca2+-permeable plasma membrane channels, an observation made previously for voltage-gated Ca2+ channels in VSMCs (Blatter & Wier, 1994). It is conceivable, however, that NO could affect other membrane ion channels, possibly leading to membrane depolarization. Depolarization would result in a diminished driving force for Ca2+ during CCE and therefore reduce the magnitude of the CCE transient. This alternative explanation is unlikely because SNP did not affect the membrane potential significantly in CPAE cells as measured with the voltage-sensitive dye Di-8-ANEPPS (data not shown). (4) We have clearly shown that NO affected Ca2+ store content. Estimating store content by exposure to ionomycin in the presence and absence of SNP revealed a higher degree of store filling in the presence of NO. Stimulation with ATP after brief preincubation with SNP caused [Ca2+]i transients with slightly smaller amplitudes, suggesting that NO also affected release. This would be in line with data demonstrating inhibition of IP3-induced Ca2+ release by NO in VSMCs (Ji et al. 1998) and platelets (Cavallini et al. 1996). Our data, however, provide strong evidence that the predominant action of NO on ER Ca2+ handling in endothelial cells is an enhancement of Ca2+ uptake. Our notion was based on the experimental data showing that SNP, when applied during the 20 min recovery period after the first addition of ATP, evoked a Ca2+ release with a significantly larger amplitude during the second application of ATP in the same cell (Fig. 10C). The significantly higher Ca2+ content of the ER and the accelerated rate of refilling were directly demonstrated in the experiments with mag-fura-2-loaded cells. As shown in Fig. 11, prolonged exposure to NO led to a larger degree of filling and an enhanced release upon stimulation, followed by acceleration of subsequent refilling of the stores.
cGMP is involved in the autocrine action of NO in endothelial cells
The effect of NO appears to be mediated via increased cGMP levels and the subsequent activation of cytosolic G kinase. In our study, the effect of NO was abolished in the presence of the NO-sensitive guanylate cyclase inhibitor ODQ (Fig. 7D), and the membrane-permeant analogue of cGMP, 8-bromo-cGMP, mimicked the effect of SNP on CCE. The involvement of the cGMP-G kinase signalling cascade in mediating NO effects has been shown in a variety of tissues including VSMCs (Cornwell & Lincoln, 1989), platelets (Geiger et al. 1992), PC12 cells and fibroblasts (Clementi et al. 1995). For example, it was shown that cGMP increases Ca2+ sequestration into VSMC Ca2+ stores (Twort & van Breemen, 1988) due to phosphorylation of phospholamban (Cornwell et al. 1991) and by isolated ER vesicles from the bovine main pulmonary artery (Raeymaekers et al. 1990).
Conclusion
The effect of NO on Ca2+ regulation in endothelial cells is controversial. For example, in contrast to our data, it has been shown that endogenous NO can lower basal [Ca2+]i levels due to activation of Ca2+ extrusion by the PMCA in transfected endothelial cells with blocked IP3-dependent Ca2+ release and potentiate CCE in intact cells (Chen et al. 2000). This discrepancy may be explained by the concentration dependence of the NO effect on the Ca2+ homeostasis in vascular endothelial cells, where low NO concentrations may have a stimulatory effect on CCE, whereas at higher concentrations, NO becomes inhibitory. Similar concentration-dependent effects have been demonstrated for pancreatic acinar cells (Gukovskaya & Pandol, 1994; Xu et al. 1994) and colonic epithelial cells (Bischof et al. 1995).
In conclusion, there is converging evidence that Ca2+ entering via the CCE pathway is the preferential source for activation of the endothelial Ca2+-calmodulin-dependent NOS to elevate NO levels. As [NO]i levels increase, NO in turn is capable of lowering [Ca2+]i by inhibition of CCE and acceleration of reuptake of Ca2+ into intracellular stores. Through this mechanism, cellular NO levels exert an autoregulatory negative feedback by regulating [Ca2+]i itself and therefore the Ca2+-dependent eNOS. This inhibition occurs at different levels: (1) through direct inhibition of CCE; (2) through clearance of cytosolic Ca2+ via enhanced reuptake of Ca2+ into intracellular stores; and (3) through de-activation of CCE as a result of enhanced refilling of the stores. Thus, this mechanism has a dual function: it serves as a protection against excessive [Ca2+]i levels and detrimentally high NO production in the vascular endothelium.
Acknowledgments
We thank Drs J. Kockskämper and S. L. Lipsius as well as K. A. Sheehan for critically reading the manuscript. Holly R. Gray provided expert technical assistance. Financial support was provided by grants from the National Institutes of Health (HL-51941, HL-62231) and the American Heart Association National Center (Established Investigator Award 95002520).
REFERENCES
- Assreuy J, Cunha FQ, Liew FY, Moncada S. Feedback inhibition of nitric oxide synthase activity by nitric oxide. British Journal of Pharmacology. 1993;108:833–837. doi: 10.1111/j.1476-5381.1993.tb12886.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkels R, Dachs C, Roesen R, Klaus W. Simultaneous measurement of intracellular Ca2+ and nitric oxide: A new method. Cell Calcium. 2000a;25:281–286. doi: 10.1054/ceca.2000.0119. [DOI] [PubMed] [Google Scholar]
- Berkels R, Suerhoff S, Roesen R, Klaus W. Nitric oxide causes a cGMP-independent intracellular calcium rise in porcine endothelial cells - a paradox? Microvascular Research. 2000b;59:38–44. doi: 10.1006/mvre.1999.2191. [DOI] [PubMed] [Google Scholar]
- Bischof G, Serwold TS, Machen TE. Does nitric oxide regulate capacitative Ca2+ influx in HEK 293 cells? Cell Calcium. 1997;21:135–142. doi: 10.1016/s0143-4160(97)90037-3. [DOI] [PubMed] [Google Scholar]
- Blatter LA, Wier WG. Nitric oxide decreases [Ca2+]i in vascular smooth muscle by inhibition of the calcium current. Cell Calcium. 1994;15:122–131. doi: 10.1016/0143-4160(94)90051-5. [DOI] [PubMed] [Google Scholar]
- Bolz SS, Pohl U. Indomethacin enhances endothelial NO release - evidence for a role of PGI2 in the autocrine control of calcium-dependent autacoid production. Cardiovascular Research. 1997;36:437–444. doi: 10.1016/s0008-6363(97)00197-1. [DOI] [PubMed] [Google Scholar]
- Brorson JR, Schumacker PT, Zhang H. Nitric oxide inhibits neuronal energy production. Journal of Neuroscience. 1999;19:147–158. doi: 10.1523/JNEUROSCI.19-01-00147.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buga GM, Griscavage JM, Rogers NE, Ignarro LJ. Negative feedback regulation of endothelial cell function by nitric oxide. Circulation Research. 1993;73:808–812. doi: 10.1161/01.res.73.5.808. [DOI] [PubMed] [Google Scholar]
- Carrier GO, Fuchs LC, Winecoff AP, Giulumiam AD, White RE. Nitrovasodilators relax mesenteric microvessels by cGMP-induced stimulation of Ca2+-activated K channels. American Journal of Physiology. 1997;273:H76–84. doi: 10.1152/ajpheart.1997.273.1.H76. [DOI] [PubMed] [Google Scholar]
- Cavallini L, Coassin M, Borean A, Alexandre A. Prostacyclin and sodium nitroprusside inhibit the activity of the platelet inositol 1,4,5-trisphosphate receptor and promote its phosphorylation. Journal of Biological Chemistry. 1996;271:5545–5551. doi: 10.1074/jbc.271.10.5545. [DOI] [PubMed] [Google Scholar]
- Chen J, Wang Y, Wang Y, Nakajima T, Iwasawa K, Hikiji H, Sunamoto M, Choi D-K, Yoshida Y, Sakaki Y, Toyo-oka T. Autocrine action and its underlying mechanism of nitric oxide on intracellular Ca2+ homeostasis in vascular endothelial cells. Journal of Biological Chemistry. 2000;275:28739–28749. doi: 10.1074/jbc.M000910200. [DOI] [PubMed] [Google Scholar]
- Chen Y, McCarron RM, Bembry J, Ruetzler C, Azzam N, Lenz FA, Spatz M. Nitric oxide modulates endothelin 1-induced Ca2+ mobilization and cytoskeletal F-actin filaments in human cerebromicrovascular endothelial cells. Journal of Cerebral Blood Flow and Metabolism. 1999;19:133–138. doi: 10.1097/00004647-199902000-00003. [DOI] [PubMed] [Google Scholar]
- Clementi E. Role of nitric oxide and its intracellular signalling pathways in the control of Ca2+ homeostasis. Biochemical Pharmacology. 1998;55:713–718. doi: 10.1016/s0006-2952(97)00375-4. [DOI] [PubMed] [Google Scholar]
- Clementi E, Vecchio I, Sciorati C, Nistro G. Nitric oxide modulation of agonist evoked intracellular Ca2+ release in neurosecretory PC-12 cells: Inhibition of phospholipase C activity via cyclic GMP-dependent protein kinase I. Molecular Pharmacology. 1995;47:517–524. [PubMed] [Google Scholar]
- Cohen RA, Weisbrod RM, Gericke M, Yaghoubi M, Bierl C, Bolotina VM. Mechanism of nitric oxide-induced vasodilatation: Refilling of intracellular stores by sarcoplasmic reticulum Ca2+ ATPase and inhibition of store-operated Ca2+ influx. Circulation Research. 1999;84:210–219. doi: 10.1161/01.res.84.2.210. [DOI] [PubMed] [Google Scholar]
- Cornwell TL, Lincoln TM. Regulation of intracellular Ca2+ levels in cultured vascular smooth muscle cells. Reduction of Ca2+ by atriopeptin and 8-bromo-cyclic GMP is mediated by cyclic GMP-dependent protein kinase. Journal of Biological Chemistry. 1989;264:1146–1155. [PubMed] [Google Scholar]
- Cornwell TL, Pryzwansky KB, Wyatt TA, Lincoln TM. Regulation of sarcoplasmic reticulum protein phosphorylation by localized cyclic GMP-dependent protein kinase in vascular smooth muscle cells. Molecular Pharmacology. 1991;40:923–931. [PubMed] [Google Scholar]
- Dedkova EN, Blatter LA. Nitric oxide inhibits capacitative Ca2+ entry in vascular endothelial cells. Biophysical Journal. 2001;80:617a. doi: 10.1113/jphysiol.2001.013258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forstermann U, Closs EI, Pollock JS, Nakane M, Schwarz P, Gath I, Kleinert H. Nitric oxide isozymes. Characterization, purification, molecular cloning, and functions. Hypertension. 1994;23:1121–1131. doi: 10.1161/01.hyp.23.6.1121. [DOI] [PubMed] [Google Scholar]
- Garcia-Cardena G, Oh P, Liu J, Schnitzer JE, Sessa WC. Targeting of nitric oxide synthase to endothelial cell caveolae via palmitoylation: Implications for nitric oxide signaling. Proceedings of the National Academy of Sciences of the USA. 1996;93:6448–6453. doi: 10.1073/pnas.93.13.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg UC, Hassid A. Nitric oxide-generating vasodilators and 8-bromo-cyclic guanosine monophosphate inhibit mitogenesis and proliferation of cultured rat vascular smooth muscle cells. Journal of Clinical Investigation. 1989;83:1174–1777. doi: 10.1172/JCI114081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiger J, Nolte C, Butt E, Sage SO, Walter U. Role of cGMP and cGMP-dependent protein kinase in nitrovasodilator inhibition of agonist-evoked calcium elevation in human platelets. Proceedings of the National Academy of Sciences of the USA. 1992;89:1031–1035. doi: 10.1073/pnas.89.3.1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto Y, Miura M, Iijima T. Extrusion mechanisms of intracellular Ca2+ in human aortic endothelial cells. European Journal of Pharmacology. 1996;314:185–192. doi: 10.1016/s0014-2999(96)00532-8. [DOI] [PubMed] [Google Scholar]
- Gukovskaya A, Pandol SJ. Nitric oxide production regulates cGMP formation and calcium influx in pancreatic acinar cells. American Journal of Physiology. 1994;11:477–486. doi: 10.1152/ajpgi.1994.266.3.G350. [DOI] [PubMed] [Google Scholar]
- Hecker M, Mulsch A, Bassenge E, Forstermann E, Busse R. Subcellular localization and characterization of nitric oxide synthase(s) in endothelial cells: Physiological implications. Biochemical Journal. 1994;299:247–252. doi: 10.1042/bj2990247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobbs AJ, Higgs A, Moncada S. Inhibition of nitric oxide synthase as a potential therapeutic target. Annual Reviews in Pharmacology and Toxicology. 1999;39:191–220. doi: 10.1146/annurev.pharmtox.39.1.191. [DOI] [PubMed] [Google Scholar]
- Hofer AM, Machen TE. Technique for in situ measurement of calcium in IP3-sensitive stores using the fluorescent indicator mag-fura-2. Proceedings of the National Academy of Sciences of the USA. 1993;90:2598–2602. doi: 10.1073/pnas.90.7.2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holda JR, Klishin A, Sedova M, Hüser J, Blatter LA. Capacitative calcium entry. News in Physiological Sciences. 1998;13:157–163. doi: 10.1152/physiologyonline.1998.13.4.157. [DOI] [PubMed] [Google Scholar]
- Igarashi J, Thatte HS, Prabhakar P, Golan DE, Michel T. Calcium-independent activation of endothelial nitric oxide synthase by ceramide. Proceedings of the National Academy of Sciences of the USA. 1999;96:12583–12588. doi: 10.1073/pnas.96.22.12583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji J, Benishin CG, Pang PT. Nitric oxide selectively inhibits intracellular Ca2+ release elicited by inositol trisphosphate but not caffeine in rat vascular smooth muscle. Journal of Pharmacology and Experimental Therapeutics. 1998;285:16–21. [PubMed] [Google Scholar]
- Julou-Schaeffer G, Gray GA, Fleming I, Schott C, Parratt JR, Stoclet JC. Loss of vascular responsiveness induced by endotoxin involves l-arginine pathway. American Journal of Physiology. 1990;259:H1038–1043. doi: 10.1152/ajpheart.1990.259.4.H1038. [DOI] [PubMed] [Google Scholar]
- Kanazawa K, Kawashima S, Mikami S, Miwa Y, Hirata K, Suematsu M, Hayashi Y, Itoh H, Yokoyama M. Endothelial constitutive nitric oxide synthase protein and mRNA increased in rabbit atherosclerotic aorta despite impaired endothelium-dependent vascular relaxation. American Journal of Pathology. 1996;148:1949–1956. [PMC free article] [PubMed] [Google Scholar]
- Klishin A, Sedova M, Blatter LA. Time-dependent modulation of capacitative Ca2+ entry signals by plasma membrane Ca2+ pump in endothelium. American Journal of Physiology. 1998;274:C1117–1128. doi: 10.1152/ajpcell.1998.274.4.C1117. [DOI] [PubMed] [Google Scholar]
- Kojima H, Nakatsubo N, Kikuchi K, Kawahara S, Kirino Y, Nagoshi H, Hirata Y, Nagano T. Detection and imaging of nitric oxide with novel fluorescent indicators: Diaminofluoresceins. Analytical Chemistry. 1998;70:2446–2453. doi: 10.1021/ac9801723. [DOI] [PubMed] [Google Scholar]
- Kubes P, Suzuki M, Grander DN. Nitric oxide: An endogenous modulator of leukocyte adhesion. Proceedings of the National Academy of Sciences of the USA. 1991;88:4651–4655. doi: 10.1073/pnas.88.11.4651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwan HY, Huang Y, Yao X. Store-operated calcium entry in vascular endothelial cells is inhibited by cGMP via a protein kinase G-dependent mechanism. Journal of Biological Chemistry. 2000;275:6758–6763. doi: 10.1074/jbc.275.10.6758. [DOI] [PubMed] [Google Scholar]
- Li H, Forstermann U. Nitric oxide in the pathogenesis of vascular disease. Journal of Pathology. 2000;190:244–254. doi: 10.1002/(SICI)1096-9896(200002)190:3<244::AID-PATH575>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Li L, Bressler B, Prameya R, Dorovini-Zis K, van Breemen C. Agonist-stimulated calcium entry in primary cultures of human cerebral microvascular endothelial cells. Microvascular Research. 1999;57:211–226. doi: 10.1006/mvre.1998.2131. [DOI] [PubMed] [Google Scholar]
- Lin S, Fagan KA, Li KX, Shaul PW, Cooper DMF, Rodman DM. Sustained endothelial nitric-oxide synthase activation requires capacitative Ca2+ entry. Journal of Biological Chemistry. 2000;275:17979–17985. doi: 10.1074/jbc.275.24.17979. [DOI] [PubMed] [Google Scholar]
- Ma XL, Lopez BL, Christopher TA, Birenbaum DS, Vinten-Johansen J. Exogenous NO inhibits basal NO release from vascular endothelium in vitro and in vivo. American Journal of Physiology. 1996;271:H2045–2051. doi: 10.1152/ajpheart.1996.271.5.H2045. [DOI] [PubMed] [Google Scholar]
- Moncada S, Palmer RM, Higgs EA. Nitric oxide: Physiology, pathophysiology, and pharmacology. Pharmacological Reviews. 1991;43:109–142. [PubMed] [Google Scholar]
- Morgan AJ, Jacob R. Differential modulation of the phases of a Ca2+ spike by the store Ca2+-ATPase in human umbilical vein endothelial cells. Journal of Physiology. 1998;513:83–101. doi: 10.1111/j.1469-7793.1998.083by.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatsubo N, Kojima H, Kikuchi K, Nagoshi H, Hirata Y, Maeda D, Imai Y, Irimura T, Nagano T. Direct evidence of nitric oxide production from bovine aortic endothelial cells using new fluorescence indicators: Diaminofluoresceins. FEBS Letters. 1998;427:263–266. doi: 10.1016/s0014-5793(98)00440-2. [DOI] [PubMed] [Google Scholar]
- Okamoto Y, Ninomiya H, Miwa S, Masaki T. Capacitative Ca2+ entry in human platelets is resistant to nitric oxide. Biochemical and Biophysical Research Communications. 1995;212:90–96. doi: 10.1006/bbrc.1995.1940. [DOI] [PubMed] [Google Scholar]
- Prabhakar P, Thatte HS, Goetz RM, Cho MR, Golan DE, Michel T. Receptor-regulated translocation of endothelial nitric-oxide synthase. Journal of Biological Chemistry. 1998;273:27383–27388. doi: 10.1074/jbc.273.42.27383. [DOI] [PubMed] [Google Scholar]
- Radomski MW, Palmer RM, Moncada S. The anti-aggregating properties of vascular endothelium: Interactions between prostacyclin and nitric oxide. British Journal of Pharmacology. 1987;92:639–646. doi: 10.1111/j.1476-5381.1987.tb11367.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raeymaekers L, Eggermont JA, Wuytack F, Casteels R. Effects of cyclic nucleotide dependent protein kinases on the endoplasmic reticulum Ca2+ pump of bovine pulmonary artery. Cell Calcium. 1990;11:261–268. doi: 10.1016/0143-4160(90)90002-c. [DOI] [PubMed] [Google Scholar]
- Rogers NE, Ignarro LJ. Constitutive nitric oxide synthase from cerebellum is reversibly inhibited by nitric oxide formed from l-arginine. Biochemical and Biophysical Research Communications. 1992;189:242–249. doi: 10.1016/0006-291x(92)91550-a. [DOI] [PubMed] [Google Scholar]
- Sedova M, Blatter LA. Dynamic regulation of [Ca2+]i by plasma membrane Ca2+-ATPase and Na+-Ca2+ exchange during capacitative Ca2+ entry in bovine vascular endothelial cells. Cell Calcium. 1999;25:333–343. doi: 10.1054/ceca.1999.0036. [DOI] [PubMed] [Google Scholar]
- Sedova M, Klishin A, Hüser J, Blatter LA. Capacitative Ca2+ entry is graded with degree of intracellular Ca2+ store depletion in bovine vascular endothelial cells. Journal of Physiology. 2000;523:549–559. doi: 10.1111/j.1469-7793.2000.t01-3-00549.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thastrup O, Cullen PJ, Drobak BK, Hanley MR, Dawson AP. Thapsigargin, a tumor promoter, discharges intracellular Ca2+ stores by specific inhibition of the endoplasmic reticulum Ca2+-ATPase. Proceedings of the National Academy of Sciences of the USA. 1990;87:2466–2470. doi: 10.1073/pnas.87.7.2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trepakova ES, Cohen RA, Bolotina VM. Nitric oxide inhibits capacitative cation influx in human platelets by promoting sarcoplasmic/endoplasmic reticulum Ca2+-ATPase-dependent refilling of Ca2+ stores. Circulation Research. 1999;84:201–209. doi: 10.1161/01.res.84.2.201. [DOI] [PubMed] [Google Scholar]
- Twort CH, van Breemen C. Cyclic guanosine monophosphate-enhanced sequestration of Ca2+ by sarcoplasmic reticulum in vascular smooth muscle. Circulation Research. 1988;62:961–964. doi: 10.1161/01.res.62.5.961. [DOI] [PubMed] [Google Scholar]
- Uneyama C, Uneyama H, Akaike N, Takahashi M. Cyclic GMP inhibits cytoplasmic Ca2+ oscillation by increasing Ca2+-ATPase activity in rat megakaryocytes. European Journal of Pharmacology. 1998;347:355–361. doi: 10.1016/s0014-2999(98)00123-x. [DOI] [PubMed] [Google Scholar]
- Wang Y, Shin WS, Kawaguchi H, Inukai M, Kato M, Sakamoto A, Uehara Y, Miyamoto M, Shimamoto N, Korenaga R, Ando J, Toyo-oka T. Contribution of sustained Ca2+ elevation for nitric oxide production in endothelial cells and subsequent modulation of Ca2+ transient in vascular smooth muscle cells in coculture. Journal of Biological Chemistry. 1996;271:5647–5655. doi: 10.1074/jbc.271.10.5647. [DOI] [PubMed] [Google Scholar]
- Xu X, Star RA, Tortorici G, Muallem S. Depletion of intracellular Ca2+ stores activates nitric-oxide synthase to generate cGMP and regulate Ca2+ influx. Journal of Biological Chemistry. 1994;269:12645–12653. [PubMed] [Google Scholar]