

Abstract
Activation of the complement system on the cell surface results in the insertion of pore forming membrane attack complexes (MAC, C5b-9). In order to protect themselves from the complement attack, the cells express several regulatory molecules, including the terminal complex regulator CD59 that inhibits assembly of the large MACs by inhibiting the insertion of additional C9 molecules into the C5b-9 complex. Using the whole cell patch clamp method, we were able to measure accumulation of homologous MACs in the membrane of CD59− human B-cells, which formed non-selective ion channels with a total conductance of 360 ± 24 pS as measured at the beginning of the steady-state phase of the inward currents. C5b-8 and small-size MAC (MAC containing only a single C9) can also form ion channels. Nevertheless, in CD59+ human B-cells in spite of small-size MAC formation, an ion current could not be detected. In addition, restoring CD59 to the membrane of the CD59− cells inhibited the serum-evoked inward current. The ion channels formed by the small-size MAC were therefore sealed, indicating that CD59 directly interfered with the pore formation of C5b-8 as well as that of small-size C5b-9. These results offer an explanation as to why CD59-expressing cells are not leaky in spite of a buildup of homologous C5b-8 and small-size MAC. Our experiments also confirmed that ion channel inhibition by CD59 is subject to homologous restriction and that CD59 cannot block the conductivity of MAC when generated by xenogenic (rabbit) serum.
One of the terminal products of the complement cascade is the membrane attack complex (MAC, C5b-9). MACs are composed of five components, C5b, C6, C7, C8 and multiple C9 molecules. When C5b binds to C6, a binding site on C6 for the C7 molecule is exposed. When C5b67 complexes are formed, they become hydrophobic and spontaneously bind to the cell membrane. The C7 component of membrane-bound C5b67 is able to bind to C8, and the resulting ring-shaped complex inserts deeply into the membrane lipid layer forming a small, leaky pore. Although C5b-8 can already function as an ion channel, it becomes more powerful when multiple C9 molecules are inserted into the ring, enlarging the MAC diameter (for a review, see Discipio, 1998).
As a means of protecting against accidental or bystander complement attack, host cells express complement regulatory proteins on the membrane (Okada et al. 1983; Okada & Tanaka, 1983; Seya et al. 1990; Liszewski & Atkinson, 1998). Decay accelerating factor (DAF, CD55) and membrane cofactor protein (MCP, CD46) control the function of C3 convertase (Liszewski & Atkinson, 1998). In addition, the 20 kDa homologous restriction factor (HRF20, CD59) inhibits assembly of complete large MACs within the membrane of a cell under attack (Davies et al. 1989; Okada et al. 1989a, b, c; Okada & Okada, 1998).
CD59 is expressed on several cell types such as leukocytes, endothelial cells and epithelial cells (Stefanova et al. 1989; Okada et al. 1989a; Nose et al. 1990; Meri et al. 1991). Erythrocytes also resist complement attack by expressing CD59 and in the case of a deficiency of this inhibitor (paroxysmal nocturnal hemoglobinuria) treatment with purified CD59 restored the resistance of the cells (Okada et al. 1990; Okada & Okada, 1998).
Although CD59 does not inhibit MAC formation of human C5b67 with rabbit C8 and C9, it inhibits when either C8 or C9 is human, indicating that CD59 interacts with both C8 and C9 (Harada et al. 1990). CD59 exerts its effect by binding tightly to the C5b-8 complex and inhibiting the incorporation of multiple C9 molecules into the complex (Morgan, 1999) thereby blocking formation of the large lytic pore. Interestingly, a single C9 molecule can still bind to the C5b-8 complex even in the presence of CD59; however, the binding of CD59 to C5b-8 hinders the necessary unfolding of C9 (Meri et al. 1990; Rollins & Sims, 1990). Partially unfolded C9 cannot expose the site required for attachment of additional C9 molecules, and consequently the large MAC ring cannot be assembled. However, it has been reported that C5b-8 complexes with an inserted single C9 molecule (small-size MAC) or even pure C5b-8 can form a transmembrane channel in an artificial membrane (Zalman & Muller-Eberhard, 1990; Discipio, 1998). If CD59 can only inhibit the build up of large MACs by preventing subsequent insertion of C9 molecules, formation of either a small-size MAC or the simple C5b-8 complex could render the attacked cells leaky, even in the presence of CD59.
Expression of CD59 has recently been demonstrated in the human central nervous system (Shen et al. 1995; Akatsu et al. 1997; Zhang et al. 1998; Singhrao et al. 1999a), and it has been found that this molecule is slightly upregulated in neurons and glial cells in neurodegenerative diseases associated with local inflammation and chronic complement activation, such as Alzheimer's and Huntington's disease (McGeer et al. 1991; Yasojima et al. 1999; Singhrao et al. 1999b). If complement activation plays a direct role in these diseases by enhancing MAC formation on neuronal cells, then CD59 upregulation would certainly be insufficient to protect these cells since neurons seem to be uniquely vulnerable to ionic imbalance caused by complement attack. This suggests that the potential [leakiness] of C5b-8 or small-size MAC could contribute to neurodegeneration.
In this study, we therefore investigated the changes in the conductivity of pores formed by C5b-8 or MAC in the presence and absence of CD59 using the whole cell patch clamp method.
METHODS
Ethics
All procedures concerning humans were carried out with written informed consent and conformed with the guidelines of the Ethical Committee of Nagoya City University Medical School and the Declaration of Helsinki.
The protocols for the animal experiments were approved by the Institutional Animal Care and Use Committee of Nagoya City University Medical School.
Cell culturing
NCU-1 is a human B-cell line established by Epstein-Barr virus transformation from a patient genetically deficient in CD59, while NCU-2 is a similar human B-cell line, isolated from a healthy volunteer, both of them of blood group B (Yamashina et al. 1990). The cells were cultured in RPMI-1640 (Nipro Company, Osaka, Japan) supplemented with 10 % fetal bovine serum (FBS) (HyClone Laboratories, Inc., Logan, UT, USA) and kept in an incubator at 37 °C under a humidified atmosphere of 95 % air-5 % CO2.
Sera
Human sera were collected from healthy volunteers. Heat inactivation of serum was carried out at 56 °C for 30 min. Rabbit sera were collected from four rabbits killed by bleeding under deep ether anaesthesia. The sera were kept in a deep freezer (-70 °C) until use.
Whole cell clamp measurements
Cells were voltage clamped at −70 mV holding potential at room temperature using a whole cell clamp configuration. The instruments used for electrophysiology were as follows: an Axopatch 200-A patch clamp amplifier, a Digidata-1200 data acquisition system and pCLAMP 6.02 software from Axon Instruments Inc. (Foster City, CA, USA). The headstage of the amplifier was fitted to an MHW-3 hydraulic manipulator produced by Narishige Inc. (Tokyo, Japan); and the cells were visualized using an Olympus IMT-2 inverted microscope. Data acquisition and analysis were performed using an IBM-compatible personal computer equipped with a 90 MHz Pentium processor. Patch electrodes (OD = 1.5 mm; thin wall; Garner Co., USA) were pulled with a PP-83 puller and polished with an MF-83 microforge (Narishige Inc.). The resistance of patch electrodes was 8–10 MΩ. The solutions used were as follows: an extracellular solution (10 mm Hepes, 140 mmNaCl, 5 mm KCl, 2 mm CaCl2, 2 mm MgCl2, 10 mm glucose, pH 7.34); an intracellular pipette solution (10 mm Hepes, 110 mm KCl, 15 mmNaCl, 0.1 mm CaCl2, 2 mm MgCl2, 1 mm EGTA, pH 7.25). Sera were applied without dilution with a puff pipette from a distance of 300–500 μm for 2 min, followed by a 3 min off period and reapplied every 5th minute until an ion-current response was observed. Recordings were carried out on several cells (n = 10 in each experiment) and they began simultaneously with the first serum application.
Average amplitudes of the inward currents were calculated using the values obtained at the beginning of the steady-state phase of the currents. Analysis of the current-voltage relation in NCU-1 cells was also carried out at this time by applying step command potentials between −90 mV and +40 mV, with 10 mV steps and width of 180 ms. Holding potential was −70 mV.
In order to restore resistance of NCU-1 cells to MAC attack, CD59 was incorporated in the cell membrane as described earlier (Okada et al. 1990). Briefly, purified CD59 (Harada et al. 1990) was added to the bath fluid of extracellular solution at a final concentration of 1 μg ml−1. The cells were then allowed to incorporate CD59 at 37 °C for 1.5 h. After washing with fresh extracellular solution thoroughly, the cells were examined with whole cell clamp electrophysiology as described above.
Antibodies and flow cytometry
Monoclonal mouse-anti-human C5b-9 antibody (Ab) (IgG; generated against a neoepitope of the C9 component of MAC) and fluorescein isothiocyanate (FITC)-labelled goat-anti-mouse IgG Ab (FITC-GAM) were purchased from DAKO Inc., Japan. The 1F5 monoclonal mouse anti-human CD59 Ab (IgG1) was produced in our laboratory, as described earlier (Okada et al. 1989a, b, c, 1990). The 1C6 monoclonal mouse-anti-human DAF primary Ab (IgG) was kindly provided by Dr T. Fujita (Fujita et al. 1987) and the monoclonal mouse-anti-human MCP Ab (IgG1) was a gift from Dr T. Seya (Seya et al. 1990). For flow cytometry measurements, Abs were diluted in Hanks' balanced salt solution (Gibco) containing 2 % fetal bovine serum (HBSS-FBS), in pre-titrated concentrations.
Following centrifugation and washing with HBSS-FBS, the cells were incubated with the diluted primary Abs on ice for 1.5 h. After washing twice with HBSS-FBS, the FITC-labelled secondary Ab was added to the cell pellet. The cells were then incubated on ice for 1.5 h. In order to determine the non-specific binding of the secondary Ab, an equal amount of FITC-GAM was also added to cells untreated with primary Abs (FITC-control). Finally, the stained cells were suspended in 0.5 ml sheath solution containing propidium iodide (PI, diluted from a stock titrated for proper concentration) and analysed by flow cytometry with FACScan and FACSCalibur (Becton-Dickinson, Mountain View, CA, USA).
In order to insert MAC into the cell membrane, alliquots of NCU-2 and NCU-1 cells were treated with undiluted human serum. Sera treatments were carried out for 1 h in an incubator (37 °C). As a control, other cells received HBSS-FBS. After 1 h the cells were labelled with the anti-human C5b-9 Ab and then FITC-GAM or with the FITC-GAM only (FITC-control) and examined by flow cytometry, as described above.
Statistical analysis
Statistical analyses were performed with the Statistica software package (StatSoft, Inc., Tulsa, OK, USA). Averaged statistical data are presented as means ± s.e.m.
RESULTS
Expression of complement regulatory molecules on NCU cell lines
In order to detect the presence or absence of DAF, MCP and CD59 in membranes of NCU-1 and NCU-2 cells, the cells were stained with anti-DAF, anti-MCP and anti-CD59 Abs by indirect immunofluorescence assay. Using flow cytometry, we demonstrated that NCU-2 cells expressed all of these molecules (Fig. 1A). However, NCU-1 cells expressed DAF and MCP only, while CD59 was completely absent (Fig. 1B), confirming the earlier diagnosis of CD59 deficiency in the patient from whom the NCU-1 cell line was isolated. The expression of DAF was almost identical in both cell lines. Nevertheless, expression of MCP in NCU-1 cells was approximately 30 % higher than that found in NCU-2 cells.
Figure 1. Flow cytometry of NCU-2 and NCU-1 cells after incubation with anti-CD59 (dashed line), anti-MCP (dashed-dotted line) and anti-DAF (dotted line) antibodies.
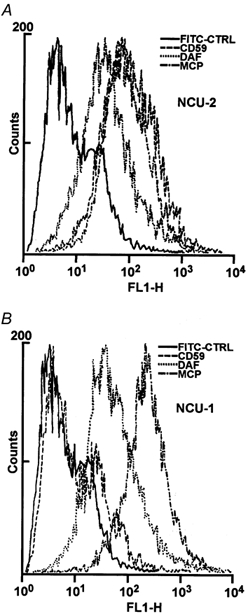
FACS analysis revealed the presence of complement regulatory molecules in the membrane of NCU-2 cells (A) whereas NCU-1 cells expressed DAF and MCP but not CD59 (B). The continuous line represents the FITC control.
MAC formation in the cell membrane
Both NCU-2 and NCU-1 cells were treated with homologous human serum (obtained from a blood group-A donor). Exposure to serum resulted in the deposition of complement as demonstrated by an accumulation of MAC in the cell membrane which we detected using indirect immunofluorescence and flow cytometry by targeting the neoantigen on the C5b-9 complex. Both NCU-1 and NCU-2 cells stained, as compared to the respective FITC controls and to the staining of the cells which were not treated with serum (Fig. 2A and B), demonstrating the presence of MAC in serum-treated cells. Nevertheless, intensity of the fluorescescence was approximately two times stronger in NCU-1 than in NCU-2 cells, showing more intensive MAC formation.
Figure 2. Flow cytometry of NCU-2 (A) and NCU-1 (B) cells after human serum treatment and incubation with anti-MAC antibody.
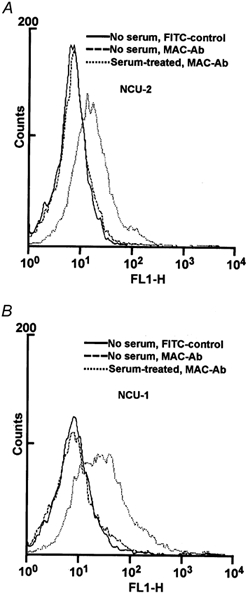
Both cell types harbour MAC in their membranes (dotted line). The fluorescence signal of cells incubated with anti-MAC antibody without serum treatment (dashed line) does not differ significantly from the signal of the FITC control (continuous line).
Whole cell clamp measurements
For whole cell clamp measurements, cells were treated with blood group-A serum administered through a puff pipette while the recording of ion current response was carried out. The recorded inward current indicated intensive pore formation, corresponding to an ongoing formation of MACs. The average amplitude of the current using NCU-1 cells was 115 ± 20.6 pA (Fig. 3A). The reversal potential during the initial, activation phase of the current in the first 20–30 s was −38 ± 5.6 mV; however it rapidly dropped to −13.0 ± 5.4 mV in 2–3 min and later decreased further to its final value (-3.6 ± 2.4 mV). The current appeared as early as 10 ± 5.5 min after opening the puff pipette. The current weakened when the puff pipette was closed.
Figure 3. Whole cell clamp electrophysiology of NCU-1 and NCU-2 cells during homologous (human, A-J) or heterologous (rabbit, K-L) serum treatment.
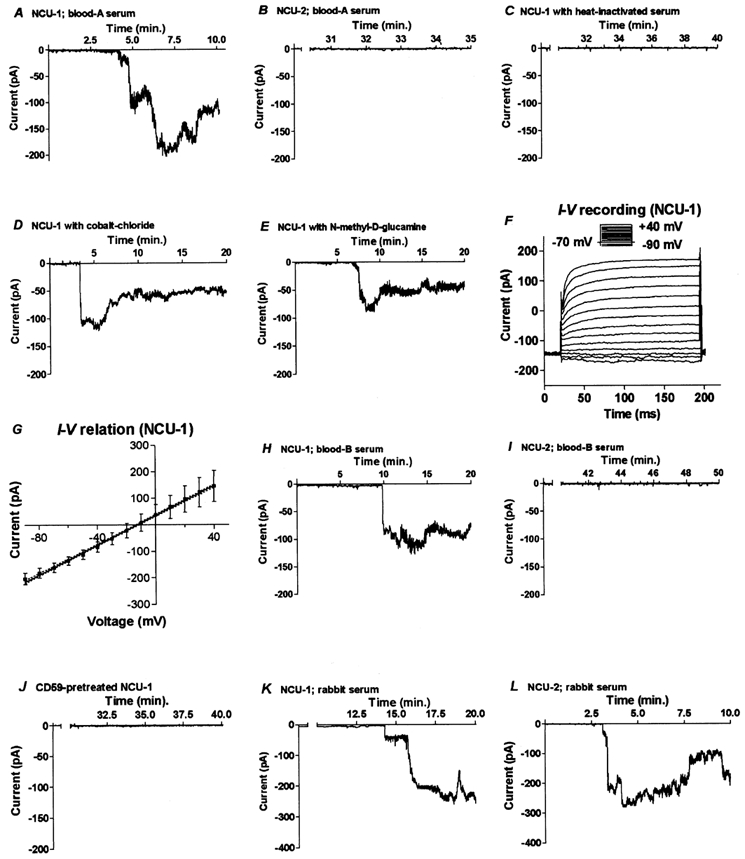
Inward current was evoked in NCU-1 cells as a result of extracellular serum administration (A). However, similar serum application could not trigger current pulses in NCU-2 cells expressing CD59 (B). A background current was observed only when heat-inactivated serum was applied to the NCU-1 cells (C), showing that the inward current evoked by normal human serum was due to complement activation. The calcium channel blocker cobalt chloride diminished but did not abolish the amplitude of the current in the NCU-1 cells (D). When NaCl was replaced with N-methyl-d-glucamine in the extracellular solution, the ion current with decreased amplitude was still recorded (E) suggesting the presence of non-selective channels in the membrane of NCU-1 cells, probably MACs. This was further supported by the linear current-voltage relationship demonstrated both in individual NCU-1 cells (F) and on averaged amplitudes of currents followed by linear regression (G). Human serum prepared from B-type blood also evoked current pulses in NCU-1 cells (H), but not in NCU-2 cells (I). Involvement of CD59 in blocking the serum-evoked current was further confirmed when CD59 was incorporated in the membrane of NCU-1 cells (J). Heterologous (rabbit) serum triggered current pulses in cells which were unrelated to the expression (NCU-2 cells; L) or absence of expression (NCU-1 cells; K) of CD59 on the membrane. Except G, each graph shows representative recording of 10 measurements.
In order to verify whether the currents measured in NCU-1 cells were caused by complement activation triggered by interaction between the serum and the cell membrane, heat-inactivated serum was applied to the NCU-1 cells. As the recording data indicate, the heat-inactivated serum failed to trigger any current (Fig. 3C), demonstrating that the current seen in Fig. 3A was a result of complement activation. Extracellularly applied Ca2+ blocker (0.2 mm CoCl2) reduced (89 ± 15.3 pA), but did not eliminate completely, the serum-induced current in NCU-1 cells (Fig. 3D). The currents with decreased amplitude (54 ± 13.2 pA) were recorded again, when the extracellular NaCl had been exchanged for N-methyl-d-glucamine (Fig. 3E). Statistical analysis (one-way ANOVA) of the control, cobalt- and glucamine-treated cells demonstrated that the average amplitudes of the recorded currents were significantly different (P < 0.05).
In contrast to NCU-1 cells, only a background current with an average amplitude of 10 ± 2.6 pA was recorded when NCU-2 cells were treated with human serum, indicating that conductive ion channels failed to form in the presence of CD59 during the entire length of the experiments (Fig. 3B).
Current-voltage analysis showed that the relationship between these parameters was linear as demonstrated by the recorded traces of the currents (Fig. 3F). The small inset above the recorded currents represents the protocol of voltage commands. Calculation of linear regression was carried out after averaging of the currents for several cells (n = 10; Fig. 3G). The high value of goodness of fit (r2 = 0.998) and that the [slope deviation from zero] is significant (P < 0.001) verified that the relation is strongly linear and the slope conductance (360 ± 24 pS) is significantly different from zero.
Measurements were repeated using type-B serum. With this serum, the average amplitude of the inward current of the NCU-1 cells was 107 ± 19.2 pA (Fig. 3H). Similar to results obtained previously, no current was triggered in NCU-2 cells using this serum (Fig. 3I).
When the NCU-1 cells were previously allowed to incorporate purified CD59, human serum failed to evoke the inward current (Fig. 3J), showing involvement of CD59 in the examined phenomenon.
Heterologous (rabbit) serum was also administered to the cells through the puff pipette. Soon after beginning serum administration to the NCU-1 cells, an inward current with an amplitude of 218 ± 32.4 pA was recorded (Fig. 3K). The electrophysiological changes during heterologous complement attack on NCU-2 cells (which resisted the homologous serum attack) were indistinguishable (196 ± 35.2 pA; Fig. 3L) from those seen with NCU-1 cells.
DISCUSSION
Using flow cytometry and antibodies, we confirmed that NCU-2 cells express CD59 whereas NCU-1 cells do not. It may be interesting that antibody to another complement regulatory molecule, MCP, stained CD59-deficient NCU-1 cells more strongly than NCU-2 cells. Similar inverse expression of complement regulatory molecules has been reported in different organs and cells (Bora et al. 1993; Guc et al. 2000). However, our results also demonstrate that MCP and DAF cannot completely block the complement cascade both on NCU-1 and on NCU-2 cells in the experimental condition used, underlining the importance of the terminal complex regulator CD59 (Morgan & Gasque, 1996). Measuring actual pore formation using electrophysiological techniques confirmed an unequivocally beneficial role for CD59 in the membrane of NCU-2 cells by protecting these cells from complement attack. In addition to preventing the development of large lytic pores of MAC, no inward current or current pulses were recorded in the NCU-2 cells and in the NCU-1 cells after restoring CD59, i.e. there was no leaky pore formation.
Lysis by MAC involves formation of non-selective ion channels in the membrane of the attacked cell through which ions and other small molecules may pass, causing osmotic lysis (Morgan, 1999). Our whole cell clamp experiments confirmed that the formed channel was not ion selective: elimination of Na+ from the extracellular solution or application of the Ca2+ channel blocker cobalt chloride significantly decreased but did not abolish the currents being recorded. This conclusion was further supported by the linear regression calculation of the current-voltage relationship. The presence of homoreactive antibodies, which are found in most sera in the form of naturally occurring anti-cholesterol and anti-phospholid antibodies (Visvanathan & McNeil, 1999), may be required for the potent complement activation we observed, although blood group antibodies were not essential, as sera from A and B blood groups were equally efficient in inducing MAC formation.
It is known that metabolically active cells can recover from MAC attack by removing freshly formed MACs from the membrane, either by shedding, vesiculation or endocytosis (Morgan et al. 1987; Scolding et al. 1989; Hansch & Shin, 1998). This removal takes some time, and therefore it is essential to determine how effective the available CD59 would be in decreasing the lytic effect of MAC during this critical period. Both homeostasis and the overall metabolism of the cell could be strongly affected to the point of irreversible damage, which would reduce the time available for MAC removal and the chance of recovery. Alternatively, strong physiological responses could also be triggered by sublethal doses of MAC, as in the case of MAC-mediated pro-inflammatory responses, which in turn could elicit pathophysiological changes during autoimmune reactions (Morgan, 1989, 1994; Kilgore et al. 1997). The idea that a functional channel may be assembled, even in the presence of bound CD59, is supported by results of size analysis of the pore complexes. Sucrose passage experiments, liposome swelling assays and measuring kinetics of release of different molecules demonstrated that the estimated diameter of C5b-8 and the C5b-9, which incorporates a single C9 molecule, can be as small as 0.9 and 1.5 nm, respectively (Zalman & Muller-Eberhard, 1990; Discipio, 1998). This is much smaller than the diameter of the fully developed large-MAC containing multiple C9 molecules, which can be as large as 5–10 nm (Morgan, 1999). However, the sizes of small-MAC and C5b-8 complexes closely match the average size of other known ion channels: the pore diameter of ion-selective channels such as that of the Na+ channel is 0.74 nm and that of the K+ channel is 0.81 nm whereas the diameter of non-selective channels is larger, for example 1.3 nm (nAChR) and 1.1 nm (GABAA) (Hille, 1994). In contrast to these earlier data suggesting that the C5b-9 complex could function as a channel since the insertion of one C9 molecule can occur even in the presence of CD59 (Meri et al. 1990; Rollins & Sims, 1990; Zalman & Muller-Eberhard, 1990; Rollins et al. 1991; Discipio, 1998), our whole cell clamp experiments demonstrated that CD59 can not only block incorporation of subsequent C9 molecules, but can inhibit the passage of ions and small substances through MAC in the form of C5b-8 or C5b-9 with a single C9.
At present, the mechanism by which CD59 blocks leaky channel formation of the C5b-8 and small-size C5b-9 is unknown. However, the most obvious way of blocking a pore is to plug it (Hille, 1994). Analysis of the 3-dimensional structure of CD59 predicts a rather flat, disc-shaped molecule, with a surface area of 32 nm2, which is closely attached to the membrane via a PI anchor (Fletcher et al. 1994; Kieffer et al. 1994). However, lack of a transmembrane region and the absence of an intracellular portion preclude the possibility that the CD59 molecule could plug the intracellular end of the pore. In addition, lack of a flexible hinge-like region within CD59 indicates that the molecule has a rather rigid, flat structure, which renders extracellular [plug] formation rather improbable.
Although the active site of CD59 has been identified (Yu et al. 1997) and peptide domains for recognition of human C8 and C9 binding to CD59 have already been reported (Chang et al. 1994; Lockert et al. 1995), the 3-dimensional structure of the CD59-C5b-8/9 complex is not yet available. Nevertheless, it is well established that the binding of CD59 to the C5b-8/9 complex inhibits the necessary partial unfolding of C9, which would expose a novel oligomerization site for additional C9 insertion. Interaction with CD59 interferes either with this conformational change, which is probably necessary for maintaining the integrity of the pore and the function of the channel, or the small C5b-8 complex as well (Morgan, 1999). Strict requirements for the specificity of the stereochemical reaction are underlined by the finding that in cases where a heterologous complement source (rabbit) was used, CD59 did not block either C9 insertion or channel formation.
In summary, CD59 can function not only as an inhibitor of the formation of large MACs, but can allow cells to eliminate newly formed MACs by blocking the early, functional channel formation of the C5b-8 and C5b-9 complexes. Therefore, even in the presence of weak chronic complement activation, the burden of leaky pore formation will not be high enough to cause cell lysis or to induce a strong imbalance in the homeostasis of the cell. We cannot, however, exclude the importance of other effects of MAC formation, which can result in pathological changes (Morgan, 1989, 1994; Kilgore et al. 1997). Non-lytic dose of MAC can activate the cell, for example by triggering cell cycle induction mediated by activation of Ras, Raf-1 and extracellular signal-related kinase (ERK1) in association with increased DNA synthesis (Rus et al. 1996; Niculescu et al. 1997). In addition, it is also necessary to keep in mind the beneficial effects of MAC, for example that sublytic MAC can inhibit apoptosis of cells by inducing enhanced synthesis of the anti-apoptotic Bcl-2 and by inhibition of caspase-3 activation (Soane et al. 1999) and that non-lethal amount of MAC can protect the cells even from lytic doses of the complement attack (Morgan, 1988; Reiter et al. 1992).
Acknowledgments
The authors would like to thank Catherine Campbell and Dr William Campbell for English editing of the manuscript, Dr Imre Ocsovszky and the Hungarian branch of Becton Dickinson Inc., for their contribution to converting the flow cytometry data, and to Mihaly Dezso for his help with the artwork. This research was conducted with the support of the fellowship program of the Japanese Society for the Promotion of Science (JSPS) and by a research grant from the Organization for Pharmaceutical Safety and Research.
REFERENCES
- Akatsu H, Yamada T, Okada N, Yamamoto T, Yamashina M, Okada H. Unique expression of HRF20 (CD59) in human nervous tissues. Microbiology and Immunology. 1997;41:321–329. doi: 10.1111/j.1348-0421.1997.tb01208.x. [DOI] [PubMed] [Google Scholar]
- Bora NS, Gobleman CL, Atkinson JP, Pepose JS, Kapla H. Differential expression of the complement regulatory proteins in the human eye. Investigation in Ophthalmology and Visual Sciences. 1993;34:3579–3584. [PubMed] [Google Scholar]
- Chang C, Husler T, Zhao J, Wiedmer T, Sims PJ. Identity of a peptide domain of human C9 that is bound by the cell-surface complement inhibitor, CD59. Journal of Biological Chemistry. 1994;269:26424–26430. [PubMed] [Google Scholar]
- Davies A, Simmons DL, Hale G, Harrison RA, Tighe H, Lachmann PJ, Waldmann H. CD59, an Ly-6-like protein expressed in human lymphoid cells, regulates the action of the complement membrane attack complex on homologous cells. Journal of Experimental Medicine. 1989;170:637–654. doi: 10.1084/jem.170.3.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Discipio RG. Late components. In: Rother K, Till GO, Hansch GM, editors. The Complement System. 2. Berlin, Heidelberg: Springer Verlag; 1998. pp. 50–68. [Google Scholar]
- Fletcher CM, Harrison RA, Lachmann PJ, Neuhaus D. Structure of a soluble, glycosylated form of the human complement regulatory protein CD59. Structure. 1994;185:185–199. doi: 10.1016/s0969-2126(00)00020-4. [DOI] [PubMed] [Google Scholar]
- Fujita T, Inoue T, Ogawa K, Iida K, Tamura N. The mechanism of action of decay-accelerating factor (DAF). DAF inhibits the assembly of C3 convertases by dissociating C2a and Bb. Journal of Experimental Medicine. 1987;155:1221–1228. doi: 10.1084/jem.166.5.1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guc D, Canpinar H, Kucukaksu C, Kansu E. Expression of complement regulatory proteins CR1, DAF, MCP and CD59 in haematological malignancies. European Journal of Haematology. 2000;64:3–9. doi: 10.1034/j.1600-0609.2000.80097.x. [DOI] [PubMed] [Google Scholar]
- Hansch GM, Shin ML. Complement attack phase. In: Rother K, Till GO, Hansch GM, editors. The Complement System. 2. Berlin, Heidelberg: Springer Verlag; 1998. pp. 115–146. [Google Scholar]
- Harada R, Okada N, Fujita T, Okada H. Purification of 1F5 antigen that prevents complement attack on homologous cell membranes. Journal of Immunology. 1990;144:1823–1828. [PubMed] [Google Scholar]
- Hille B. Ionic Channels of Excitable Membranes. 2. Sunderland, MA, USA: Sinauer Associates; 1994. [Google Scholar]
- Kieffer B, Driscoll PC, Campbell ID, Willis AC, van der Merwe PA, Davis SJ. Three-dimensional solution structure of the extracellular region of the complement regulatory protein CD59, a new cell-surface protein domain related to snake venom neurotoxins. Biochemistry. 1994;33:4471–4482. [PubMed] [Google Scholar]
- Kilgore KS, Schmid E, Shanley TP, Flory CM, Maheswari V, Tramontini NL, Cohen H, Ward PA, Friedl HP, Warren JS. Sublytic concentration of the membrane attack complex of complement induce endothelial interleukin-8 and monocyte chemoattractant protein-1 through nuclear factor-kappaB activation. American Journal of Pathology. 1997;150:2019–2031. [PMC free article] [PubMed] [Google Scholar]
- Liszewski MK, Atkinson JP. Membrane cofactor protein (CD46) and decay accelerating factor (CD55) In: Rother K, Till GO, Hansch GM, editors. The Complement System. 2. Berlin, Heidelberg: Springer Verlag; 1998. pp. 146–163. [Google Scholar]
- Lockert DH, Kaufman KM, Chang C, Husler T, Sodetz JM, Sims PJ. Identity of the segment of human complement C8 recognized by complement regulatory protein CD59. Journal of Biological Chemistry. 1995;270:19723–19728. doi: 10.1074/jbc.270.34.19723. [DOI] [PubMed] [Google Scholar]
- McGeer PL, Walker DG, Akiyama H, Kawamata T, Guan AL, Parker CJ. Detection of the membrane inhibitor of reactive lysis (CD59) in diseased neurons of Alzheimer brain. Brain Research. 1991;544:315–319. doi: 10.1016/0006-8993(91)90071-3. [DOI] [PubMed] [Google Scholar]
- Meri S, Morgan BP, Davies A, Daniels RH, Olavessen MG, Waldmann H, Lachmann PJ. Human protectin (CD59), an 18,000–20,000 MW complement lysis restricting factor, inhibits C5b-8 catalysed insertion of C9 into lipid bilayers. Immunology. 1990;71:1–9. [PMC free article] [PubMed] [Google Scholar]
- Meri S, Waldmann H, Lachmann PJ. Distribution of protectin (CD59), a complement membrane attack inhibitor, in normal human tissues. Laboratory Investigation. 1991;65:532–537. [PubMed] [Google Scholar]
- Morgan BP. Non-lethal complement membrane-attack on human neutrophils: transient cell swelling and metabolic depletion. Immunology. 1988;63:71–77. [PMC free article] [PubMed] [Google Scholar]
- Morgan BP. Mechanism of tissue damage by the membrane attack complex of complement. Comparative Inflammation. 1989;6:104–111. doi: 10.1159/000463082. [DOI] [PubMed] [Google Scholar]
- Morgan BP. Clinical complementology: recent progress and future trends. European Journal of Clinical Investigation. 1994;24:219–228. doi: 10.1111/j.1365-2362.1994.tb01078.x. [DOI] [PubMed] [Google Scholar]
- Morgan BP. Regulation of the complement membrane attack pathway. Critical Reviews in Immunology. 1999;19:173–198. [PubMed] [Google Scholar]
- Morgan BP, Dankert JR, Esser AF. Recovery of human neutrophils from complement attack: removal of the membrane attack complex by endocytosis and exocytosis. Journal of Immunology. 1987;138:246–253. [PubMed] [Google Scholar]
- Morgan BP, Gasque P. Expression of complement in the brain: role in health and disease. Immunology Today. 1996;17:461–466. doi: 10.1016/0167-5699(96)20028-f. [DOI] [PubMed] [Google Scholar]
- Niculescu F, Rus H, Van Biesen T, Shin ML. Activation of Ras and mitogen-activated protein kinase pathway by terminal complement complexes is G-protein dependent. Journal of Immunology. 1997;158:4405–4412. [PubMed] [Google Scholar]
- Nose M, Katoh M, Okada N, Kyogoku M, Okada H. Tissue distribution of HRF20, a novel factor preventing the membrane attack of homologous complement, and its predominant expression on endothelial cells in vivo. Immunology. 1990;70:145–149. [PMC free article] [PubMed] [Google Scholar]
- Okada N, Harada R, Fujita T, Okada H. A novel membrane glycoprotein capable of inhibiting membrane attack by homologous complement. International Immunology. 1989a;1:205–208. doi: 10.1093/intimm/1.2.205. [DOI] [PubMed] [Google Scholar]
- Okada N, Harada R, Fujita T, Okada H. Monoclonal antibodies capable of causing hemolysis of neuraminidase-treated human erythrocytes by homologous complement. Journal of Immunology. 1989b;143:2262–2266. [PubMed] [Google Scholar]
- Okada N, Harada R, Okada H. Erythrocytes of patients with paroxysmal nocturnal haemoglobinuria acquire resistance to complement attack by purified 20-kD homologous restriction factor. Clinical and Experimental Immunology. 1990;80:109–113. doi: 10.1111/j.1365-2249.1990.tb06449.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada H, Nagami Y, Takahashi K, Okada N, Hideshima T, Takizawa H, Kondo J. 20 kDa homologous restriction factor of complement resembles T cell activating proteins. Biochemical and Biophysical Research Communications. 1989c;162:1553–1559. doi: 10.1016/0006-291x(89)90852-8. [DOI] [PubMed] [Google Scholar]
- Okada H, Okada N. Membrane-bound inhibitors of C5b-9. In: Rother K, Till GO, Hansch GM, editors. The Complement System. 2. Berlin, Heidelberg: Springer Verlag; 1998. pp. 174–178. [Google Scholar]
- Okada H, Tanaka H. Species-specific inhibition by glycophorins of complement activation via the alternative pathway. Molecular Immunology. 1983;20:1233–1236. doi: 10.1016/0161-5890(83)90148-7. [DOI] [PubMed] [Google Scholar]
- Okada H, Tanaka H, Okada N. Prevention of complement activation on the homologous cell membrane of nucleated cells as well as erythrocytes. European Journal of Immunology. 1983;13:340–344. doi: 10.1002/eji.1830130413. [DOI] [PubMed] [Google Scholar]
- Reiter Y, Ciobotaru A, Fishelson Z. Sublytic complement attack protects tumor cells from lytic doses of antibody and complement. European Journal of Immunology. 1992;22:1207–1213. doi: 10.1002/eji.1830220515. [DOI] [PubMed] [Google Scholar]
- Rollins SA, Sims PJ. The complement-inhibitory activity of CD59 resides in its capacity to block incorporation of C9 into membrane C5b-9. Journal of Immunology. 1990;144:3478–3483. [PubMed] [Google Scholar]
- Rollins SA, Zhao J, Ninomiya H, Sims PJ. Inhibition of homologous complement by CD59 is mediated by a species-selective recognition conferred through binding to C8 within C5b-8 or C9 within C5b-9. Journal of Immunology. 1991;146:2345–2351. [PubMed] [Google Scholar]
- Rus H, Niculescu F, Shin ML. Sublytic complement attack induces cell cycle in oligodendrocytes. Journal of Immunology. 1996;156:4892–4900. [PubMed] [Google Scholar]
- Scolding NJ, Morgan BP, Houston WAJ, Linington C, Campbell AK, Compston DAS. Vesicular removal by oligodendrocytes of membrane attack complexes formed by activated complement. Nature. 1989;339:620–622. doi: 10.1038/339620a0. [DOI] [PubMed] [Google Scholar]
- Seya T, Hara T, Matsumoto M, Akedo H. Quantitative analysis of membrane cofactor protein (MCP) of complement. High expression of MCP on human leukemia cell lines, which is down-regulated during cell differentiation. Journal of Immunology. 1990;145:238–245. [PubMed] [Google Scholar]
- Shen Y, Halperin JA, Lee C. Complement-mediated neurotoxicity is regulated by homologous restriction. Brain Research. 1995;671:282–292. doi: 10.1016/0006-8993(94)01264-i. [DOI] [PubMed] [Google Scholar]
- Singhrao SK, Neal JW, Morgan BP, Gasque P. Increased complement biosynthesis by microglia and complement activation on neurons in Huntington's disease. Experimental Neurology. 1999b;159:362–376. doi: 10.1006/exnr.1999.7170. [DOI] [PubMed] [Google Scholar]
- Singhrao SK, Neal JW, Rushmere NK, Morgan BP, Gasque P. Differential expression of individual complement regulators in the brain and choroid plexus. Laboratory Investigation. 1999a;79:1247–1259. [PubMed] [Google Scholar]
- Soane L, Rus H, Niculescu F, Shin ML. Inhibition of oligodendrocyte apoptosis by sublytic C5b-9 is associated with enhanced synthesis of Bcl-2 and mediated by inhibition of caspase-3 activation. Journal of Immunology. 1999;163:6132–6138. [PubMed] [Google Scholar]
- Stefanova I, Hilgert I, Kristofova H, Brown R, Low MG, Horejsi V. Characterization of a broadly expressed human leukocyte surface antigen MEM-43 anchored in membrane through phosphatidylinositol. Molecular Immunology. 1989;26:153–161. doi: 10.1016/0161-5890(89)90097-7. [DOI] [PubMed] [Google Scholar]
- Visvanathan S, McNeil HP. Cellular immunity to the beta 2-glycoprotein-1 in patients with the antiphospholipid syndrome. Journal of Immunology. 1999;162:6919–6925. [PubMed] [Google Scholar]
- Yamashina M, Ueda E, Kinoshita T, Takami T, Ojima A, Ono H, Tanaka H, Kondo N, Orii T, Okada N, Okada H, Inoue K, Kitani T. Inherited complete deficiency of 20-kilodalton homologous restriction factor (CD59) as a cause of paroxysmal nocturnal hemoglobinuria. New England Journal of Medicine. 1990;323:1184–1189. doi: 10.1056/NEJM199010253231707. [DOI] [PubMed] [Google Scholar]
- Yasojima K, McGeer EG, McGeer PL. Complement regulators C1 inhibitor and CD59 do not significantly inhibit complement activation in Alzheimer disease. Brain Research. 1999;833:297–301. doi: 10.1016/s0006-8993(99)01514-0. [DOI] [PubMed] [Google Scholar]
- Yu J, Abagyan R, Dong S, Gilbert A, Nussenzweig V, Tomlinson S. Mapping the active site of CD59. Journal of Experimental Medicine. 1997;185:745–753. doi: 10.1084/jem.185.4.745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zalman LS, Muller-Eberhard HJ. Comparison of channels formed by poly C9, C5b-8 and the membrane attack complex of complement. Molecular Immunology. 1990;27:533–537. doi: 10.1016/0161-5890(90)90072-8. [DOI] [PubMed] [Google Scholar]
- Zhang K, Junikkala S, Erlander MG, Guo H, Westberg JA, Meri S, Andersson LC. Up-regulated expression of decay-accelerating factor (CD55) confers increased complement resistance to sprouting neural cells. European Journal of Immunology. 1998;28:1189–1196. doi: 10.1002/(SICI)1521-4141(199804)28:04<1189::AID-IMMU1189>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]