

Abstract
The role of different isoforms of cyclo-oxygenase (COX) in mediating the acute (0-6 h) and late (24 h) phases of inflammation was investigated in the rat knee joint following intra-articular injection of carrageenan. The hyperaemic response was assessed transcutaneously using laser Doppler imaging (LDI). Samples were taken at corresponding time points for detection of synovial COX-1, COX-2 and inducible nitric oxide synthase (iNOS) mRNA, and measurement of urinary prostaglandin (PG) and nitric oxide metabolites (NOx). A non-selective COX inhibitor (indomethacin, 15 mg kg−1i.p.), a selective COX-2 inhibitor (SC-236, 16.8 mg kg−1i.p.) or vehicle were administered 1 h prior to carrageenan in the acute phase study. LDI scans were taken hourly for 4 h post-induction. Inflammatory hyperaemia in the vehicle group was attenuated in the indomethacin- (P < 0.001, two-way ANOVA) and SC-236-treated groups (P < 0.0001), with no difference between these treatments. At 24 h, i.v. infusion of indomethacin (0.1 mg min−1), increased vascular resistance (24 ± 7.1 %; P < 0.05) compared to vehicle infusion, whereas SC-236 (0.11 mg min−1) did not. Resistance changes to indomethacin also differed from SC-236 (P < 0.05). Knee joint diameter progressively increased over 24 h (P < 0.0001, one-way ANOVA). Urinary PG levels increased by 6 h (P < 0.05), but returned to baseline by 24 h. COX-1 mRNA was detectable at all time points; COX-2 mRNA only at 3 h. Urinary NOx levels increased progressively over 24 h (P < 0.05), paralleled by induction of iNOS in the 3 and 24 h samples. Prostaglandin production via COX-2 appears to mediate the development of acute inflammatory hyperaemia, but nitrergic mechanisms may supervene subsequently. COX-1 but not COX-2 contributes to the maintenance of basal blood flow in the hyperaemic joint at 24 h.
The production of prostaglandins (PGs), through the metabolism of arachidonic acid by cyclo-oxygenase (COX), is one of the key pathways involved in the pathogenesis of acute inflammation. Inflammatory hyperaemia forms an essential component of many rheumatological conditions including rheumatoid arthritis (Ferrell et al. 2001), and facilitates migration of leucocytes into the site of injury. This process is mediated by the release of inflammatory paracrine agents such as histamine, neuropeptides, kinins, nitric oxide and PGs, which act as vasodilators. It is now recognised that there are two COX isoforms: COX-1 and COX-2. COX-1 is constitutively expressed and performs housekeeping functions such as regulation of renal blood flow, cytoprotection of the gastric mucosa and stimulation of platelet aggregation (Vane, 1994). COX-2 is an inducible isoform, normally undetectable in most tissues. However, cytokines including tissue necrosis factor-α and various interleukins can rapidly upregulate its expression and it is considered to play an important role in prostanoid-mediated inflammation (Crofford, 1996).
Prostaglandin E2 (PGE2) and prostaglandin I2 (PGI2) are thought to be key mediators in acute inflammation, and COX inhibition is a widely used therapeutic option for the treatment of inflammatory diseases (see Davies & MacIntyre, 1992, for review). Non-selective COX inhibitors (non-steroidal anti-inflammatory drugs, NSAIDs) are used for the management of rheumatic diseases such as rheumatoid arthritis and osteoarthritis, but their long term use interferes with physiological functions of prostaglandins synthesised via COX-1, a serious adverse event being gastro-intestinal haemorrhage. Consequently, selective COX-2 inhibitors have now been developed (as reviewed by Hawkey, 1999), which offer the prospect of anti-inflammatory activity without compromising the activity of COX-1.
It is recognised that prostaglandins play a physiological role in the regulation of synovial blood flow and thereby the provision of nutrients to avascular joint structures such as cartilage. This is mediated via synovial fluid (McKibbin & Maroudas, 1979) whose production is dependent upon synovial blood flow (Levick, 1984). PGs maintain basal synovial blood flow since intravenous infusion of indomethacin leads to a decrease in basal synovial perfusion in both the rabbit (Najafipour & Ferrell, 1994) and the rat (Egan et al. 2001). This prostaglandin-dependent vasodilator tone is mediated via COX-1 since infusion of a selective COX-2 inhibitor did not alter basal perfusion in the normal joint, consistent with the lack of COX-2 mRNA detection in this tissue (Egan et al. 2001). However, the relative importance of COX-2 in mediating the development of hyperaemia in the inflamed joint has not been investigated, and forms the principal objective of the present study.
The role of COX-1 and COX-2 was investigated in the acutely inflamed knee joint of the rat using both a broad-spectrum COX inhibitor and a novel, highly selective COX-2 inhibitor, SC-236. Differential expression of COX-1 and COX-2 in this joint was characterised using reverse transcription-polymerase chain reaction (RT-PCR). Urinary PGE2 levels were used as a measure of synovial generation of PGs. Nitrergic pro-inflammatory mechanisms were also assessed, as these are known to interact with prostanoid pathways (Salvemini et al. 1993; Vane, 1994). This involved a preliminary investigation of nitrate excretion in urine and iNOS mRNA expression in synovial tissue.
METHODS
Experimental animals
Experiments were performed in male Wistar rats (300–400 g body weight; Harlan UK Ltd). Animals were housed in standard cages, had food and water available ad libitum and were maintained in a thermoneutral environment (23 ± 2 °C). All procedures were performed in accordance with Home Office regulations.
Preparatory surgery
Prior to laser Doppler imaging (LDI) experiments, routine surgical procedures were performed to allow blood pressure measurement and infusion of drugs. Animals were deeply anaesthetised by injection of urethane (Sigma, UK; 1.6 g kg−1, i.p.) and placed in dorsal recumbency. Temperature was monitored via a rectal probe and maintained at 37 ± 1 °C (mean ± s.e.m.) over the course of the experiment by a thermal pad. Tracheostomy was performed with the animals breathing spontaneously throughout the experiments. Arterial blood pressure was monitored continuously via a cannula inserted into the left common carotid artery and linked to a pressure transducer (Elcomatic EM-751, UK). Mean arterial pressure (MAP) was calculated by adding one-third of pulse pressure to the diastolic pressure. The right jugular vein was also cannulated in some experiments for infusion of drugs. At the end of the experiments all animals were killed by anaesthetic overdose (except in tissue harvesting experiments as indicated in the text).
Knee joint preparation for transcutaneous imaging
Transcutaneous LDI was performed in two groups of animals. The fur over the medial aspect of the knee joint was shaved prior to application of a proprietary depilatory cream (Boots plc, UK); 25 min later the cream was carefully removed, the knee swabbed with saline, wiped clean and dried. Vaseline petroleum jelly (Chesebrough-Pond's Ltd, UK) was thinly applied to the skin overlying the knee immediately afterwards to keep the skin moist. Cutaneous blood vessels overlying the knee were cauterised to provide an unobstructed laser Doppler image of the underlying joint vasculature. A 1 h stabilisation period was allowed prior to the beginning of the experiment.
Transcutaneous measurement of synovial perfusion by laser Doppler imaging
A detailed description of transcutaneous LDI as applied to the rat knee joint has been published previously (Abbot et al. 1996; Egan et al. 2001). Briefly, a laser Doppler perfusion imager (Lisca, Linkoping, Sweden) was used to monitor relative changes in blood flow in the underlying synovial microcirculation, using methods previously validated (Egan et al. 2001). In a darkened room, a laser beam (near infrared, 830 nm) was scanned back and forth in a raster fashion over the joint, producing a colour-coded image. These images were later analysed by dedicated software (Moor Instruments, UK) to obtain a median flux value over the knee joint region. The biological zero values were measured as described previously (Najafipour & Ferrell, 1995) and subtracted from the perfusion values. Vascular resistance was calculated by dividing MAP by the flux value. The change in flux values with the various treatments was expressed as percentage change from 0 time (immediately prior to treatment).
Induction of acute knee joint inflammation
The carrageenan-induced knee joint inflammation model has been previously described (Lam & Ferrell, 1993). Inflammation was induced in three animal groups: two for the early phase experiments (0–6 h), and one for the late phase experiments (24 h). In brief, 100 μl solution of 2 % λ-carrageenan and 4 % kaolin (w/v; Sigma) in saline (0.9 %) was injected (26-G needle) into the anterior and posterior cavities of one knee joint. Animals in the late phase experiments (n = 30) were anaesthetised initially using a mixture of 2 % halothane O2-N2O, injected with carrageenan, then allowed to recover before being taken to terminal experiment at 24 h. For this, they were re-anaesthetised using urethane. Once recovered from anaesthesia, the animals were returned to their cages. Analgesics were available but not required for any of the experimental group as adjudged by the veterinary officer. Animals in the early phase (terminal) experiments (n = 25) were anaesthetised with urethane prior to intra-articular injection of carrageenan.
Urinary nitrite/nitrate determination
In view of the known involvement of nitric oxide (NO) in joint inflammation (Grabowski et al. 1996; Stichtenoth & Frölich, 1998; reviewed by Jang & Murrell, 1998) and the cross-talk between nitrergic and prostanoid pathways, nitrate/nitrite (NOx) excretion was measured as a marker of NO generation. Urine was collected at 0, 6 and at 24 h after induction of inflammation and then frozen for later analysis. For this, 1.5 ml was centrifuged at 10 000 g for 15 min, the supernatant removed and diluted fivefold using deionised water. Nitrate standards were also reduced with vanadium to assess the percentage conversion efficiency of nitrate to nitrite. From each sample (including nitrate and nitrite standards) 50 μl was dispensed into a 96-well plate followed by 50 μl of freshly prepared vanadium chloride solution (200 mg per 25 ml 1 m HCl) and immediately followed by 50 μl of Griess reagent (2 % sulphanilamide in 1.47 m hydrochloric acid, and 0.1 % N-(1-naphthyl)ethylenediamine dihydrochloride in deionised water; Sigma; Miranda et al. 2000). After 30 min of colour development at 37 °C, absorbance was measured on a microplate spectrophotometer (Dynatech MR5000, Dynatech Laboratories Inc., VA, USA) at 570 nm with 630 nm reference filter. Each sample was assayed in triplicate. NOx values were corrected for variations in urine concentration by dividing by the corresponding urinary osmolality values.
Urinary PG determination
After 10 and 100-fold dilution of centrifuged urine samples, bicyclo PGE2, (a stable metabolite of prostaglandin E2) was quantified using competitive binding ELISA (Cayman Chemical Company, Ann Arbor, USA) according to the manufacturer's instructions. The microtitre plate was read using a spectrophotometer (Dynatech MR5000, Dynatech Laboratories Inc., VA, USA) set to 410 nm. Optical density was used to determine the percentage of labelled ligand bound. The sensitivity of the bicyclo PGE2 ELISA was 2 pg ml−1 (at 24 °C). Sample values were corrected for urine concentration.
Detection of COX-1, COX-2 and iNOS mRNA in rat synovium by RT-PCR
RT-PCR for the detection of COX isoforms has been previously described by our group (Egan et al. 2001), therefore only extensions of the technique are described here. Knee joint tissue was harvested from freshly killed rats at various time points following acute inflammation (n = 4 per group). Capsular tissue was removed (including patella and patellar ligament) and frozen immediately in liquid nitrogen, then stored at −70 °C. Total RNA was isolated according to the manufacturer's instructions (RNAzol, Biotecx Laboratories, USA). Following extraction, total RNA was re-suspended in 20 μl of RNAse-free sterile H2O and stored at −70 °C. RNA integrity and yield were assessed by UV-spectrophotometry and ethidium bromide staining. One microgram aliquots of total RNA were reverse transcribed using a reverse transcription system kit (Promega, Corp., WI, USA). Aliquots, 1 μl, of the complementary DNA (cDNA) were amplified in a 20 μl PCR mixture containing 1.2 μl MgCl2 (25 mm), 2 μl of 10 × PCR buffer, 2 μl of 10 mm dNTP cocktail, 5 U of Taq polymerase (Promega, Corp.) and 5 μm final concentration each of sense and antisense primers. Sequences for specific primers used (COX-1, COX-2, iNOS and β-actin; Operon Technologies, Inc., CA, USA) with corresponding PCR product sizes are given in Table 1. Rat β-actin primers were used to control for the efficiency of cDNA synthesis in each sample. Negative (water replacing cDNA in PCR reaction) and positive controls (cDNA for each of the targets) were also included. The amplification conditions were 94 °C for 30 s (denaturation), 55 °C for 30 s (annealing) and 72 °C for 1 min (extension) for 35 cycles. Aliquots of the PCR reactions (9 μl) were run on a 2 % (w/v) Tris-borate-EDTA (TBE) agarose gel for analysis. The identity of PCR products was confirmed by sequencing by the BigDye chain-terminator method and analysis using an ABI 3100 automated sequencer.
Table 1.
Nucleotide sequences of oligonucleotide primers used in PCR
| Gene | Oligonucleotide | Sequence (5′–3′) | PCR product (bp) |
|---|---|---|---|
| COX-1 | Sense primer | CGA GGA TGT CAT CAA GGA G | 349 |
| Antisense primer | TCA GTG AGG CTG TGT TAA CG | ||
| COX-2 | Sense primer | TCA AGA CAG ATC AGA AGC GA | 204 |
| Antisense primer | TAC CTG AGT GTC TTT GAT TG | ||
| iNOS | Sense primer | ATG GCT TGC CCT TGG AAG TTT CTC | 574 |
| Antisense primer | TCC AGG CCA TCT TGG TGG CAA AGA | ||
| β-Actin | Sense primer | GTG GGG CGC CCC AGG CAC CA | 548 |
| Antisense primer | CTC CTT AAT GTC ACG CAC GAT TTC |
Early phase response - developing inflammatory hyperaemia
Following preparatory surgery for blood pressure measurement and depilation of knees, inflammation was induced in one joint prior to transcutaneous imaging experiments. One hour prior to induction, animals were injected i.p. with either vehicle (0.5 % methylcellulose and 0.025 % Tween-20, n = 7), indomethacin (15 mg kg−1; n = 7) or SC-236 (Monsanto, 16.8 mg kg−1; n = 7). The indomethacin dose was supra-maximal for inhibition of both COX isoforms (Wallace et al. 1999). The dose of SC-236 was equimolar and three orders of magnitude below the IC50 for COX-1 inhibition (Gierse et al. 1996). A further group of four animals served as a control. LDI scans were taken before and immediately after induction of inflammation and every hour thereafter for 4 h.
Late phase response - established inflammatory hyperaemia
Twenty-four hours after induction of inflammation, animals were anaesthetised and the carotid artery and jugular vein cannulated for monitoring blood pressure and infusing drugs, respectively. Indomethacin (5 mg ml−1; n = 6), SC-236 (5.6 mg ml−1; n = 8) or vehicle (50 % DMSO; n = 6) were infused at 20 μl min−1 over 50 min and LDI scans taken over the depilated knee.
Oedema, enzyme expression and urinary NOx and PG
After intra-articular injection of carrageenan, oedema measurements were made at 0, 3, 6 and 24 h (n = 4–6 per time point). Both ipsilateral (inflamed) and contralateral knee joint diameters were measured using modified vernier callipers with a spring loaded footpad (Oditest, Kroeplin Gmbh, Germany). Measurements were compared with pre-injection diameter values. At each time point, rats were killed by stunning followed by cervical dislocation, knee diameters measured, and the synovial tissue was excised for PCR analysis and snap frozen in liquid nitrogen. Urine from the bladder was also collected and immediately frozen in liquid nitrogen for determination of PG and NO levels. Osmolality measurements (Micro-Osmometer, 13/13 DR, Roebling, Germany) were taken as a measure of urine concentration. A standard solution of 300 mosmol kg−1 was used to calibrate the system prior to urine analysis.
Statistical analysis
After image and data analysis, subsequent statistical analyses were performed using Instat software (GraphPad, USA). Comparison between group values was performed by one or two-way ANOVA as indicated in the text. Dunnett multiple comparison tests were used for post hoc comparison of time points within a group. All quoted P values are two-tailed; n values refer to the number of animals examined. Data are presented as means ± standard error of the mean (s.e.m).
RESULTS
Knee joint oedema
Following induction of inflammation, there was a progressive and highly significant (P < 0.0001, one-way ANOVA; n = 4–6) increase in knee joint diameter with a maximum measured response at 24 h (Fig. 1). The contralateral knee joint diameter also increased, but this just failed to reach significance (P = 0.054, one-way ANOVA; n = 4–6). There was a highly significant difference between the inflamed knee and its contralateral control (P < 0.0001, two-way ANOVA; n = 4–6).
Figure 1. Oedema response in carrageenan-induced joint inflammation.
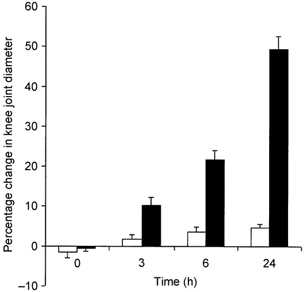
The oedema response was measured as the percentage change in knee joint diameter. ▪, ipsilateral injected knee (P < 0.0001, one-way ANOVA; n = 4–6); □, contralateral non-injected knee.
Early phase response - developing inflammatory hyperaemia
A significant (P < 0.0001, one-way ANOVA) time dependent hyperaemia (Fig. 2) was observed in the vehicle-treated group (maximum 36 ± 2 % decrease in articular vascular resistance). This hyperaemic response was significantly attenuated in the indomethacin- (P < 0.001) and SC-236-treated groups (P < 0.0001) compared to the vehicle-treated group (two-way ANOVA). However, there was no significant difference between the indomethacin- and SC-236-treated groups (P = 0.76). No significant change in resistance with time was also observed in the untreated control group (P = 0.7; one-way ANOVA), underlining the stability of the preparation. Neither the indomethacin nor the SC-236 group differed significantly from this control group (P = 0.36, P = 0.44, respectively, two-way ANOVA).
Figure 2. The role of COX in the development of acute inflammatory hyperaemia in the rat knee.
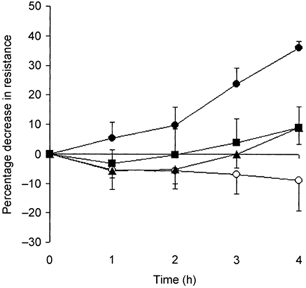
Percentage change in resistance with time (compared to pre-treatment control) is shown for vehicle (•), indomethacin (▪), SC-236 (▴) and untreated control (○) groups. A significant (P < 0.0001, one-way ANOVA) time-dependent hyperaemia was observed in the vehicle (n = 7) group. This was significantly attenuated by indomethacin (n = 7) and SC-236 (n = 7). There was no significant difference between the two COX inhibitors (P = 0.76).
Late phase response - established inflammatory hyperaemia
Knee joint diameter was maximal at the 24 h time point (Fig. 1) with basal blood flow showing a 38 % increase in the inflamed knee compared to the contralateral knee (P = 0.043, Student's paired t test, n = 6). Intravenous infusion of indomethacin (5 mg ml−1 over 50 min) produced a significant increase in articular vascular resistance (24 ± 7.1 %) compared to vehicle infusion (P = 0.026, unpaired t test, n = 6). However, no significant change in resistance was observed following infusion of an equimolar concentration of the COX-2 inhibitor, SC-236, compared to vehicle (P = 0.71, unpaired t test, n = 6). The resistance change following infusion of SC-236 did, however, differ significantly from that of the indomethacin-treated group (P < 0.05, unpaired t test, n = 6; Fig. 3).
Figure 3. The role of COX in regulating basal perfusion in the 24 h inflamed joint.
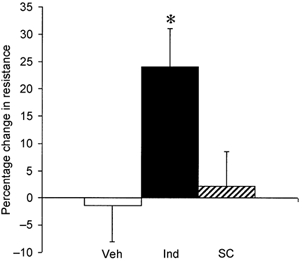
Percentage change in resistance (compared to pre-treatment control) is shown for vehicle (□), indomethacin (▪) and SC-236 ( ). Indomethacin (n = 6) produced a significant increase in articular vascular resistance (24 ± 7.1 %) compared to vehicle infusion (* P < 0.05). The change in resistance produced by infusion of an equimolar concentration of the COX-2 inhibitor, SC-236 (n = 8), differed significantly from the indomethacin-treated group (* P < 0.05), but not from the vehicle group (n = 6; P = 0.71).
). Indomethacin (n = 6) produced a significant increase in articular vascular resistance (24 ± 7.1 %) compared to vehicle infusion (* P < 0.05). The change in resistance produced by infusion of an equimolar concentration of the COX-2 inhibitor, SC-236 (n = 8), differed significantly from the indomethacin-treated group (* P < 0.05), but not from the vehicle group (n = 6; P = 0.71).
Urinary PG and NOx determination
Urinary PG levels increased significantly (P < 0.001, n = 4–6) 6 h after induction of inflammation compared to control (0 time) but decreased thereafter to baseline levels by 24 h (Fig. 4A). There was no significant difference in urinary PG levels between 24 h and control values. In contrast, urinary NOx tended to increase in a time-dependent manner (P = 0.056, one-way ANOVA, n = 4–6) reaching significance (P < 0.05) only at the 24 h time point (Fig. 4B).
Figure 4. Urinary levels at control, and 6 and 24 h after inflammation (n = 4–6), corrected for urine osmolality.
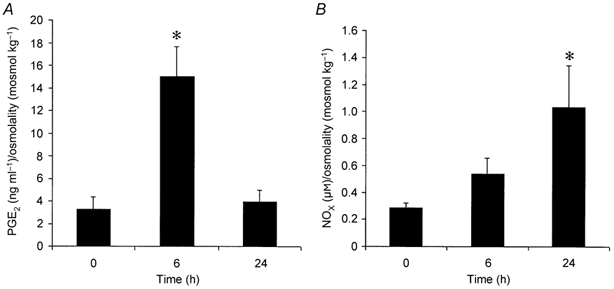
Compared to control (0 time), PG levels (A) increased significantly (* P < 0.001) only at 6 h, whereas NOx (B) tended to increase in a time-dependent manner (P = 0.056, one-way ANOVA), reaching significance (* P < 0.05) at the 24 h time point.
COX and iNOS mRNA expression
COX-1 mRNA was detectable at all time points, consistent with the constitutive nature of the enzyme. COX-2 mRNA was detectable 3 h post induction of inflammation (in three of four samples) but not in control or 24 h samples (Fig. 5). While inducible NOS mRNA was not detected in control samples, it was induced in both 3 and 24 h samples. mRNA for the housekeeping β-actin gene was detectable at all time points, demonstrating the integrity of RT-PCR.
Figure 5. COX-1, COX-2, iNOS and β-actin mRNA expression in control (0 h), and 3 and 24 h after inflammation in the joint synovium of the rat analysed by RT-PCR.
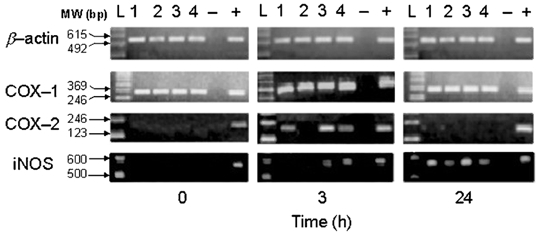
The PCR products from four rats (lanes 1–4) in each panel from the β-actin, COX-1, COX-2 and iNOS sequences generated after 35 cycles of amplification were analysed by electrophoresis in a 2 % TBE agarose gel. A 123 bp and 100 bp (iNOS) DNA ladder was used as a molecular weight standard. The sizes of the amplified DNA fragments are indicated in base pairs (see Table 1). -, negative control; +, positive control (see Methods).
DISCUSSION
This is the first study to characterise the development of hyperaemia associated with the acute inflammatory response in the joint over a 24 h period. Previous work from our group has demonstrated a physiological role for prostaglandins in maintaining joint perfusion in the rabbit (Najafipour & Ferrell, 1994) and rat knee (Egan et al. 2001). This is mediated by COX-1, not COX-2, as basal perfusion is reduced by indomethacin but not by SC-236 (Egan et al. 2001). The novel evidence, presented here, for the prostanoid regulation of the synovial vasculature under pathophysiological conditions extends these earlier observations. COX-2 is induced and mediates the early phase of inflammatory hyperaemia in the carrageenan-injected joint, since this response was virtually abolished by selective inhibition of COX-2 with SC-236. Moreover, urinary prostaglandin production substantially increased and COX-2 mRNA was detectable in synovial tissue samples, consistent with a role for prostaglandins in mediating joint inflammation. However, by 24 h the synovial vasculature is no longer regulated by COX-2, as demonstrated by the lack of vascular response to SC-236 administration. Furthermore, COX-2 mRNA was no longer detectable in the joint and urinary prostaglandin levels had returned to control values. In contrast, indomethacin significantly reduced the higher basal synovial perfusion in the 24 h inflamed joint, presumably by inhibiting PG production via COX-1, whose mRNA continues to be present in the inflamed joint. Thus, while COX-2 mediates the development of inflammatory hyperaemia in the joint, it is, as in the normal joint (Egan et al. 2001), COX-1 and not COX-2 that contributes to the maintenance of basal perfusion in the hyperaemic joint after 24 h of inflammation.
Intra-articular injection of carrageenan induces an acute arthritis characterised by local neutrophil infiltration into, and proliferation of, synovial tissue (Santer et al. 1983) and is known to be histamine, 5-hydroxytryptamine, bradykinin and prostaglandin-dependent (Di Rosa & Willoughby, 1971). In the present study, the increased urinary PGE2 excretion and detection of COX-2 mRNA in articular tissue are evidence for an increase in synovial PG production. Similar findings were reported by Salvemini et al. (1996) and Nantel et al. (1999) in the carrageenan-induced rat paw model of inflammation. This model, although not as relevant to inflammatory joint disease, does enable levels of pro-inflammatory mediators to be measured. These studies only examined the inflammatory response up to 10 h and did not assess inflammatory hyperaemia, but did find that tissue PGE2 production peaked at 3 h and fell thereafter. There is a lag phase in measuring rises of synovial prostaglandin production by urinary levels due to the time delay inherent in renal clearance. Even so, we also found that urinary PGE2 levels were elevated during the early phase of the inflammatory response, but returned to control levels by 24 h (Fig. 4A). Nantel et al. (1999) also demonstrated that COX-2 expression paralleled PG production, with mRNA levels declining after 3 h. Results from the present study show that COX-2 mRNA was present at 3 h but was no longer detectable in the joint at the 24 h time point.
COX-2 mRNA is extremely unstable with a shorter half-life than COX-1 mRNA (Dixon et al. 2000), owing to the presence of multiple instability sequences in the 3′-untranslated region, which are usually found in genes whose products are rapidly downregulated after induction (Gou et al. 1998). However, the fact that COX-2 mRNA was detected in three of four synovia studied at 3 h (Fig. 5) demonstrates that we should have been able to detect it at 24 h, had it been expressed at this later time point. That it was not detected at the later phase argues for its downregulation. Furthermore, using Western blot analysis, Tomlinson et al. (1994) detected peak COX-2 levels within the first 6 h, but only trace levels at 24 h in a carrageenan-induced model of pleurisy. This suggests that COX-2 mRNA and protein are both rapidly degraded following the early inflammatory phase.
Although the cellular location of COX-2 was not investigated in the present study, this isoform has been shown to be induced in a variety of articular cell types including vascular endothelial cells, stromal fibroblast-like cells (synoviocytes) and lymphoid aggregates which consist of monocytes and macrophages (Sano et al. 1992; Kang et al. 1996). Synoviocytes have been shown to be a major source of PGs and are located in the synovial lining and sublining layers (Crofford et al. 1994).
Despite the absence of COX-2-generated PGs at the later phase of inflammation, joint oedema reached its maximum measured levels (Fig. 1) and the hyperaemic inflammatory response that developed in the early phase still persisted, as demonstrated by significantly higher basal perfusion, at 24 h in the inflamed knee. This suggests that COX-2 is important in development but not for continued maintenance of acute inflammatory hyperaemia and oedema. Clearly, other mediators must be responsible for the sustained inflammatory response during the later phase. Certainly NO is a possible candidate, since in addition to its many physiological functions (Moncada et al. 1991; Cooke & Tsao, 1993), it is now recognised to have important pro-inflammatory roles (Gorbunov & Esposito, 1993). This supposition is supported here by the detection of iNOS mRNA by RT-PCR at 3 and 24 h (Fig. 5) and the parallel rise in urinary NOx levels (Fig. 4B). It is also supported by the similar findings of Salvemini et al. (1995) in the carrageenan-induced rat air pouch model. Moreover, we have previously reported that carrageenan-induced joint inflammation can be significantly attenuated by selective iNOS inhibitors (Lockhart & Ferrell, 2000).
Interestingly, while low levels of NO are reported to activate COX (Stadler et al. 1991; Salvemini et al. 1993), this role appears to be reversed at high levels. Lipopolysaccharide induces the release of large amounts of NO, and this inhibits both the induction and activity of COX-2 in J774 macrophages (Swierkosz et al. 1995), and the production of PGE2 in articular chondrocytes (Stadler et al. 1991). Taken together with the present study, these results suggest that the increasing levels of NO in the later phase of inflammation may inhibit the continued upregulation of COX-2 that occurs in the early phase of the carrageenan model. The mechanism may involve NO binding to the haem moiety of COX at high concentrations, and converting it to the ferrous inactive form (reviewed in Mitchell et al. 1995). Alternatively, nitrosylation of the cysteine groups on COX may undermine COX activity (Kennedy et al. 1994) and NO may also alter the availability of the transcription factors (e.g. NF-kappa B) required for COX-2 induction, thereby reducing its expression (Dela Torre et al. 1997).
Although it is generally accepted that constitutively expressed COX-1 performs housekeeping roles and that COX-2 is rapidly induced during inflammatory processes, there may be important exceptions. Evidence indicates that COX-2 is also constitutively expressed in some tissues (Harris et al. 1994; Yang et al. 1998; Ermert et al. 1998). In addition, COX-2 has been shown to have essential roles in reproduction and development (Lim et al. 1997; Davis et al. 1999; Gross et al. 2000; Komhoff et al. 2000). Together, these findings suggest that COX-2 may have physiological functions. Interestingly, some recent studies have indicated that COX-2 acts not only in the initiation of the inflammatory response but also in the resolution phase (Gilroy et al. 1999). This is supported by the findings of Wallace et al. (1998), who observed that the inflammation in the carrageenan-paw model normally resolved within 1 week, but was unmitigated over this same period in COX-2 knockout mice.
There may also be a role for COX-1-derived PGs in inflammation, as suggested by the work of Langenbach et al. (1995) and Wallace et al. (1998). In the present study, the increase in vascular resistance after infusion of indomethacin 24 h after inflammation was not significantly different (P = 0.87) from that in the normal knee (Egan et al. 2001). The fact that COX-2 mRNA was not detectable at 24 h and urinary PGE2 levels had returned to baseline levels by this time argues that the indomethacin effect at 24 h was solely attributable to COX-1. Furthermore, since the increase in vascular resistance to indomethacin was similar to that found in the normal joint, this suggests that COX-1 is not upregulated in the inflamed joint, and therefore does not contribute to joint inflammation. This could be more fully resolved by quantitative molecular biology techniques.
COX-2 inhibitors are now becoming accepted as a better therapeutic option than NSAIDs in the management of rheumatoid arthritis, as their selectivity minimises gastrointestinal side-effects. It is, however, increasingly recognised that COX inhibition does not modify disease progression (Feldmann & Maini, 1999; Fries, 1999). Nevertheless, COX-2 inhibitors are widely used for pain relief, which may be mediated via spinal nociceptive pathways by COX-2-derived prostaglandins (Malmberg & Yaksh, 1992). The present study clearly demonstrates that these inhibitors may be beneficial in reducing the hyperaemia associated with development of acute inflammatory responses, but whether they alleviate established joint hyperaemia in chronic arthritis remains to be established. Moreover, if COX-2 contributes to the resolution of inflammation, then judicial use of COX-2 inhibitors may be indicated in the management of arthritis.
Acknowledgments
This work was supported by Searle (Monsanto Corporation, USA) and the University of Paisley Research funds. We gratefully acknowledge the expert technical assistance of Lynette Dunning and Marion Drew, and Gary Jackson for sequencing the PCR products.
REFERENCES
- Abbot NC, Ferrell WR, Lockhart JC, Lowe JG. Laser Doppler imaging of skin blood flow using red and near-infrared sources. Journal of Investigative Dermatology. 1996;107:882–886. doi: 10.1111/1523-1747.ep12331185. [DOI] [PubMed] [Google Scholar]
- Cooke JP, Tsao PS. Cytoprotective effects of nitric oxide (Editorial) Circulation. 1993;88:2451–2454. doi: 10.1161/01.cir.88.5.2451. [DOI] [PubMed] [Google Scholar]
- Crofford LJ. Expression and regulation of COX-2 in synovial tissues of arthritis patients. In: Vane J, Botting J, Botting R, editors. Improved Non-steroid Anti-inflammatory Drugs: COX-2 Enzyme Inhibitors. London: Kluwer Academic Publishers and William Harvey Press; 1996. pp. 133–143. [Google Scholar]
- Crofford LJ, Wilder RL, Ristimaki AP, Remmers EF, Epps HR, Hla T. Cyclooxygenase-1 and −2 expression in rheumatoid synovial tissues: effects of interleukin-1β, phorbal ester, and corticosteroids. Journal of Clinical Investigation. 1994;93:1095–1101. doi: 10.1172/JCI117060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies P, MacIntyre DE. Prostglandins andinflammation. In: Gallin JI, Goldstein IM, Snyderman R, editors. Inflammation: Basic Principles and Clinical Correlates. New York: Raven Press Ltd; 1992. pp. 123–137. [Google Scholar]
- Davis BJ, Lennard DE, Lee CA, Tiano HF, Morham SG, Wetsel WC, Langenbach R. Anovulation in cyclooxygenase-2-deficient mice is restored by prostaglandin E2 and interleukin-1β. Endocrinology. 1999;140:2685–2695. doi: 10.1210/endo.140.6.6715. [DOI] [PubMed] [Google Scholar]
- Dela Torre A, Schroeder RA, Kuo PC. Alteration of NF-kappa B P50 binding kinetics by S-nitrosylation. Biochemical and Biophysical Research Communications. 1997;238:703–706. doi: 10.1006/bbrc.1997.7279. [DOI] [PubMed] [Google Scholar]
- Di Rosa M, Willoughby DA. Screens for anti-inflammatory drugs. Journal of Pharmacy and Pharmacology. 1971;23:297–298. doi: 10.1111/j.2042-7158.1971.tb08661.x. [DOI] [PubMed] [Google Scholar]
- Dixon DA, Kaplan CD, McIntyre TM, Zimmerman GA, Prescott SM. Post-transcriptional control of cyclooxygenase-2 gene expression. The role of the 3′-untranslated region. Journal of Biological Chemistry. 2000;275:11750–11757. doi: 10.1074/jbc.275.16.11750. [DOI] [PubMed] [Google Scholar]
- Egan CG, Lockhart JC, McLean JS, Ferrell WR. Expression of constitutive but not inducible cyclooxygenase maintains articular perfusion in the rat knee. Experimental Physiology. 2001;86:191–197. doi: 10.1113/eph8602129. [DOI] [PubMed] [Google Scholar]
- Ermert L, Ermert M, Goppelt-Struebe M, Walmrath D, Grimminger F, Steudel W, Ghofrani HA, Homberger C, Duncker H, Seeger W. Cyclooxygenase isoenzyme localization and mRNA expression in rat lungs. American Journal of Respiratory Cell and Molecular Biology. 1998;18:479–488. doi: 10.1165/ajrcmb.18.4.2939. [DOI] [PubMed] [Google Scholar]
- Feldmann M, Maini RN. The role of cytokines in the pathogenesis of rheumatoid arthritis. Rheumatology. 1999;38(suppl. 2):3–7. [PubMed] [Google Scholar]
- Ferrell WR, Balint PV, Egan CG, Lockhart JC, Sturrock RD. Metacarpophalangeal joints in rheumatoid arthritis: laser Doppler imaging - initial experience. Radiology. 2001;220:257–262. doi: 10.1148/radiology.220.1.r01jl26257. [DOI] [PubMed] [Google Scholar]
- Fries JF. How may quality of life for rheumatoid arthritis patients be enhanced by current and future treatments? Rheumatology. 1999;38(suppl. 2):35–40. [PubMed] [Google Scholar]
- Gierse JK, McDonald JJ, Hauser SD, Rangwala SH, Koboldt CM, Seibert K. A single amino acid difference between cyclooxygenase-1 (COX-1) and −2 (COX-2) reverses the selectivity of COX-2 specific inhibitors. Journal of Biological Chemistry. 1996;271:15810–15814. doi: 10.1074/jbc.271.26.15810. [DOI] [PubMed] [Google Scholar]
- Gilroy DW, Colville-Nash PR, Willis D, Chivers J, Paul-Clark MJ, Willoughby DA. Inducible cyclooxygenase may have anti-inflammatory properties. Nature Medicine. 1999;5:698–701. doi: 10.1038/9550. [DOI] [PubMed] [Google Scholar]
- Gorbunov N, Esposito E. Nitric oxide as a mediator of inflammation. International Journal of Immunopathology and Pharmacology. 1993;6:67–75. [Google Scholar]
- Gou Q, Liu CH, Ben-Av P, Hla T. Dissociation of basal turnover and cytokine-induced transcript stabilisation of the human cyclooxygenase-2 mRNA by mutagenesis of the 3′-untranslated region. Biochemical and Biophysical Research Communications. 1998;242:508–512. doi: 10.1006/bbrc.1997.7994. [DOI] [PubMed] [Google Scholar]
- Grabowski PS, England AJ, Dykhuizen R, Copland M, Benjamin N, Reid DM, Ralston SH. Elevated nitric oxide production in rheumatoid arthritis. Detection using the fasting urinary nitrate:creatinine ratio. Arthritis and Rheumatism. 1996;39:643–647. doi: 10.1002/art.1780390416. [DOI] [PubMed] [Google Scholar]
- Gross G, Imamura T, Vogt SK, Wozniak DF, Nelson DM, Sadovsky Y, Muglia LJ. Inhibition of cyclooxygenase-2 prevents inflammation-mediated preterm labor in the mouse. American Journal of Physiology — Regulatory Integrative and Comparative Physiology. 2000;278:R1415–1423. doi: 10.1152/ajpregu.2000.278.6.R1415. [DOI] [PubMed] [Google Scholar]
- Harris RC, McKanna JA, Akai Y, Jacobson HR, Dubois RN, Breyer MD. Cyclooxygenase-2 is associated with the macula densa of rat kidney and increases with salt restriction. Journal of Clinical Investigation. 1994;94:2504–2510. doi: 10.1172/JCI117620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkey CJ. COX-2 inhibitors. Lancet. 1999;353:307–314. doi: 10.1016/s0140-6736(98)12154-2. [DOI] [PubMed] [Google Scholar]
- Jang D, Murrell GA. Nitric oxide in arthritis. Free Radical Biology and Medicine. 1998;24:1511–1519. doi: 10.1016/s0891-5849(97)00459-0. [DOI] [PubMed] [Google Scholar]
- Kang RY, Freire-Moar J, Sigal E, Chu C. Expression of cyclooxygenase-2 in human and an animal model of rheumatoid arthritis. British Journal of Rheumatology. 1996;35:711–718. doi: 10.1093/rheumatology/35.8.711. [DOI] [PubMed] [Google Scholar]
- Kennedy TA, Smith CJ, Marnett LJ. Investigations of the roles of cysteines in catalysis by prostaglandin endoperoxide synthase. Journal of Biological Chemistry. 1994;269:27357–27364. [PubMed] [Google Scholar]
- Komhoff M, Wang JL, Cheng HF, Langenbach R, McKanna JA, Harris RC, Breyer MD. Cyclooxygenase-2-selective inhibitors impair glomerulogenesis and renal cortical development. Kidney International. 2000;57:414–422. doi: 10.1046/j.1523-1755.2000.00861.x. [DOI] [PubMed] [Google Scholar]
- Lam FY, Ferrell WR. Acute inflammation in the rat knee joint attenuates sympathetic vasoconstriction but enhances neuropeptide-mediated vasodilatation assessed by laser Doppler perfusion imaging. Neuroscience. 1993;52:443–449. doi: 10.1016/0306-4522(93)90170-k. [DOI] [PubMed] [Google Scholar]
- Langenbach R, Morham SG, Tiano HF, Loftin CD, Ghanayem BI, Chulada PC, Mahler JF, Lee CA, Goulding EH, Kluckman KD. Prostaglandin synthase 1 gene disruption in mice reduces arachidonic acid-induced inflammation and indomethacin-induced gastric ulceration. Cell. 1995;83:483–492. doi: 10.1016/0092-8674(95)90126-4. [DOI] [PubMed] [Google Scholar]
- Levick JR. Blood flow and mass transport in synovial joints. In: Renkin EM, Michel CC, editors. Handbook of Physiology, The Cardiovascular System, The Microcirculation. IV. Bethesda: American Physiological Society; 1984. pp. 917–947. [Google Scholar]
- Lim H, Paria BC, Das SK, Dinchuk JE, Langenbach R, Trzaskos JM, Dey SK. Multiple female reproductive failures in cyclooxygenase 2-deficient mice. Cell. 1997;91:197–208. doi: 10.1016/s0092-8674(00)80402-x. [DOI] [PubMed] [Google Scholar]
- Lockhart JC, Ferrell WR. Attenuation of joint inflammation by selective iNOS inhibitors. Nitric oxide: Biology and Chemistry. 2000;4:321. (abstract) [Google Scholar]
- McKibbin B, Maroudas A. Nutrition and metabolism. In: Freeman MAR, editor. Adult Articular Cartilage. Tunbridge Wells, UK: Pitman Medical; 1979. pp. 461–468. [Google Scholar]
- Malmberg AB, Yaksh TL. Hyperalgesia mediated by spinal glutamate or substance P receptor blocked by spinal cyclooxygenase inhibition. Science. 1992;257:1276–1279. doi: 10.1126/science.1381521. [DOI] [PubMed] [Google Scholar]
- Miranda KM, Espey MG, Wink DA. A rapid, simple spectrophotometric method for detection of nitrate and nitrite. Nitric Oxide: Biology and Chemistry. 2000;4:274. doi: 10.1006/niox.2000.0319. (abstract) [DOI] [PubMed] [Google Scholar]
- Mitchell JA, Larkin S, Williams TJ. Cyclooxygenase-2: regulation and relevance in inflammation. Biochemical Pharmacology. 1995;50:1535–1542. doi: 10.1016/0006-2952(95)00212-x. [DOI] [PubMed] [Google Scholar]
- Moncada S, Palmer RM, Higgs EA. Nitric oxide: physiology, pathophysiology, and pharmacology. Pharmacological Reviews. 1991;43:109–142. [PubMed] [Google Scholar]
- Najafipour H, Ferrell WR. Role of prostaglandins in regulation of blood flow and modulation of sympathetic vasoconstriction in normal and acutely inflamed rabbit knee joints. Experimental Physiology. 1994;79:93–103. doi: 10.1113/expphysiol.1994.sp003745. [DOI] [PubMed] [Google Scholar]
- Najafipour H, Ferrell WR. Comparison of synovial PO2 and sympathetic vasoconstrictor responses in normal and acutely inflamed rabbit knee joints. Experimental Physiology. 1995;80:209–220. doi: 10.1113/expphysiol.1995.sp003841. [DOI] [PubMed] [Google Scholar]
- Nantel F, Denis D, Gordon R, Northey A, Cirino M, Metters KM, Chan CC. Distribution and regulation of cyclooxygenase-2 in carrageenan-induced inflammation. British Journal of Pharmacology. 1999;128:853–859. doi: 10.1038/sj.bjp.0702866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvemini D, Manning PT, Zweifel BS, Seibert K, Connor J, Currie MG, Needleman P, Masferrer JL. Dual inhibition of nitric oxide and prostaglandin production contributes to the anti-inflammatory properties of nitric oxide synthase inhibitors. Journal of Clinical Investigation. 1995;96:301–308. doi: 10.1172/JCI118035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvemini D, Misko TP, Masferrer JL, Seibert K, Currie MG, Needleman P. Nitric oxide activates cyclooxygenase enzymes. Proceedings of the National Academy of Sciences of the USA. 1993;90:7240–7244. doi: 10.1073/pnas.90.15.7240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvemini D, Wang Z-Q, Wyatt PS, Bourdon DM, Marino MH, Manning PT, Currie MG. Nitric oxide: a key mediator in the early and late phase of carrageenan-induced rat paw inflammation. British Journal of Pharmacology. 1996;118:829–838. doi: 10.1111/j.1476-5381.1996.tb15475.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sano H, Hla T, Maier JAM, Crofford LJ, Case JP, Maciag T, Wilder RL. In vivo cyclooxygenase expression in synovial tissues of patients with rheumatoid arthritis and osteoarthritis and rats with adjuvant and streptococcal cell wall arthritis. Journal of Clinical Investigation. 1992;89:97–108. doi: 10.1172/JCI115591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santer V, Srirtana A, Lowther DA. Carrageenin-induced arthritis. V. A. morphological study of the development of inflammation. Seminars in Arthritis and Rheumatism. 1983;13:160–168. doi: 10.1016/0049-0172(83)90002-1. [DOI] [PubMed] [Google Scholar]
- Stadler J, Stefanovic-Racic M, Billiar TR, Curran RD, McIntyre LA, Georgescu HI, Simmons RL, Evans CH. Articular chondrocytes synthesize nitric oxide in response to cytokines and lipopolysaccharide. Journal of Immunology. 1991;147:3915–3920. [PubMed] [Google Scholar]
- Stichtenoth DO, Flichölich JC. Nitric oxide and inflammatory diseases. British Journal of Pharmacology. 1998;37:246–257. doi: 10.1093/rheumatology/37.3.246. [DOI] [PubMed] [Google Scholar]
- Swierkosz TA, Mitchell JA, Warner TD, Botting RM, Vane JR. Co-induction of nitric oxide between nitric oxide and prostanoids. British Journal of Pharmacology. 1995;114:1335–1342. doi: 10.1111/j.1476-5381.1995.tb13353.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomlinson A, Appleton I, Moore AR, Gilroy DW, Willis D, Mitchell JA, Willoughby DA. Cyclo-oxygenase and nitric oxide synthase isoforms in rat carrageenin-induced pleurisy. British Journal of Pharmacology. 1994;113:693–698. doi: 10.1111/j.1476-5381.1994.tb17048.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vane J. Towards a better aspirin. Nature. 1994;367:215–216. doi: 10.1038/367215a0. [DOI] [PubMed] [Google Scholar]
- Wallace JL, Bak A, McKnight W, Asfaha S, Sharkey KA, MacNaughton WK. Cyclooxygenase 1 contributes to inflammatory responses in rats and mice: implications for gastrointestinal toxicity [see comments] Gastroenterology. 1998;115:101–109. doi: 10.1016/s0016-5085(98)70370-1. [DOI] [PubMed] [Google Scholar]
- Wallace JL, Chapman K, McKnight W. Limited anti-inflammatory efficacy of cyclo-oxygenase-2 inhibition in carrageenan-airpouch inflammation. British Journal of Pharmacology. 1999;126:1200–1204. doi: 10.1038/sj.bjp.0702420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T, Singh I, Pham H, Sun D, Smart A, Schnermann JB, Briggs JP. Regulation of cyclooxygenase expression in the kidney by dietary salt intake. American Journal of Physiology. 1998;274:F481–489. doi: 10.1152/ajprenal.1998.274.3.F481. [DOI] [PubMed] [Google Scholar]