

Abstract
Prostaglandins are important mediators of pain and inflammation. We have examined the effects of prostanoids on voltage-activated calcium currents (ICa) in acutely isolated mouse trigeminal sensory neurons, using standard whole cell voltage clamp techniques. Trigeminal neurons were divided into two populations based on the presence (Type 2) or absence (Type 1) of low voltage-activated T-type ICa. The absence of T-type ICa is highly correlated with sensitivity to μ-opioid agonists and the VR1 agonist capsaicin. In both populations of cells, high voltage-activated ICa was inhibited by PGE2 with an EC50 of about 35 nm, to a maximum of 30 %. T-type ICa was not inhibited by PGE2. Pertussis toxin pre-treatment abolished the effects of PGE2 in Type 2 cells, but not in Type 1 cells, whereas treatment with cholera toxin prevented the effects of PGE2 in Type 1 cells, but not in Type 2 cells. Inhibition of ICa by PGE2 was associated with slowing of current activation and could be relieved with a large positive pre-pulse, consistent with inhibition of ICa by G protein βγ subunits. Reverse transcription-polymerase chain reaction of mRNA from trigeminal ganglia indicated that all four EP prostanoid receptors were present. However, in both Type 1 and Type 2 cells the effects of PGE2 were only mimicked by the selective EP3 receptor agonist ONO-AE-248, and not by selective agonists for EP1 (ONO-DI-004), EP2 (ONO-AE1–259) and EP4 (ONO-AE1–329) receptors. These data indicate that two populations of neurons in trigeminal ganglia differing in their calcium channel expression, sensitivity to μ-opioids and capsaicin also have divergent mechanisms of PGE2-mediated inhibition of calcium channels, with Gi/Go type G proteins involved in one population, and Gs type G proteins in the other.
The pain produced in response to many forms of noxious stimulation can be facilitated by prostaglandins (Ferreira, 1972). Prostanoids, including prostaglandin E2 (PGE2; Willis, 1969; Greaves et al. 1971), are generated in response to injury or inflammation and can increase the excitability of sensory nerves (Handwerker, 1976; Chahl & Iggo, 1977; Mense, 1981) in vivo and in vitro (Baccaglini & Hogan, 1983; Pitchford & Levine, 1991). The heightened responsiveness to other noxious stimuli produced by PGE2 is known as sensitization and is reflected at a cellular level by modulation of a number of different ion channels (see below) and the facilitation of neuropeptide release (Nicol et al. 1992; Vasko et al. 1994; Cui & Nicol, 1995). In sensory neurons from various species, PGE2 shifts the activation of tetrodotoxin-resistant sodium currents (TTX-R INa) to more negative potentials (Gold et al. 1996; England et al. 1996), potentiates capsaicin-induced currents (Pitchford & Levine, 1991; Lopshire & Nicol, 1998) and increases the activation of the hyperpolarization-activated cation current (Ih; Ingram & Williams, 1994, 1996), all of which will increase the excitability of sensory neurons. PGE2 also attenuates voltage-dependent (Nicol et al. 1997) and calcium-activated potassium currents (Fowler et al. 1985). The effects of PGE2 on sensory neuron excitability (Ferreira & Nakamura, 1979) and neuropeptide release (Cui & Nicol, 1995; Hingtgen et al. 1995) have largely been attributed to elevation of cAMP. Cyclic AMP acts both directly (Ingram & Williams, 1994, 1996) and via protein kinase A to mediate its cellular sensitizing effects (Pitchford & Levine, 1991; England et al. 1996; Lopshire & Nicol, 1998; Gold et al. 1998; Evans et al. 1999). These effects are consistent with the predominant coupling of prostanoid receptors to Gs proteins (Narumiya et al. 1999).
High voltage-activated ICa (HVA ICa) and low voltage-activated T-type ICa contribute to the excitability of sensory neurons (e.g. Yoshida et al. 1978; White et al. 1989; Abdulla & Smith, 1997) and HVA ICa contributes directly to action potential-dependent neurotransmitter release from primary afferent terminals. The inhibition of sensory neuron voltage-dependent calcium channels at the primary afferent synapse is thought to be important in mediating the spinal analgesic effects of opioid agonists (Hori et al. 1992) and invertebrate neurotoxin analgesics (Xiao & Bennett, 1995). Consistent with its role as a pro-nociceptive mediator, PGE2 has been reported to increase HVA ICa in embryonic avian sensory neurons (Nicol et al. 1992), although the mechanism was not determined. In contrast, in rat sympathetic neurons (Ikeda 1992; Ito et al. 2000), bovine adrenal chromaffin cells (Currie & Fox, 2000) and rat melanotrophs (Tanaka et al. 1998), PGE2 inhibits calcium channels. In the light of these contrasting findings, we examined the effects of PGE2 on HVA ICa in mammalian sensory neurons. We found that PGE2 inhibited HVA ICa in almost all acutely isolated mouse trigeminal sensory neurons, and that PGE2 receptors are differentially coupled to Gi/o- and Gs-type G proteins in separate pharmacologically and electrophysiologically defined populations of sensory neurons.
METHODS
Isolation of trigeminal ganglion neurons
129SvEv/C57BL/6 mice (4–6 weeks old) of either sex were used for this study. All procedures were approved by the Animal Ethics Committee of the University of Sydney, Australia. Mice were anaesthetized with halothane (4 %) and decapitated, and the trigeminal ganglia were removed and placed in cold (4 °C) physiological saline containing (mm): NaCl 126; KCl 2.5; CaCl2 2.5; MgCl2 1.2; NaH2PO4 1.2; NaHCO3 24; and glucose 10, gassed with 95 % O2-5 % CO2. Using a simplified version of the methods outlined in Eckert et al. (1997), ganglia were cut up with iridectomy scissors and incubated at 32–34 °C for 30 min in physiological saline. The ganglia pieces were then transferred to oxygenated Hepes buffered saline (HBS) containing 20 units ml−1 papain and incubated at 32–34 °C for 15 min. In a few experiments the ganglia were incubated with 3 mg ml−1 collagenase Type IV (Worthington) before incubation with papain. This procedure reduced the amount of trituration needed to release the cells. HBS contained (mm): NaCl 154; KCl 2.5; CaCl2 2.5; MgCl2 1.5; Hepes 10; glucose 10; pH 7.2 (NaOH), 330 ± 5 mosmol l−1. The papain digestion was terminated with addition of HBS containing 1 mg ml−1 bovine serum albumin (BSA) and 1 mg ml−1 trypsin inhibitor. Minced ganglia were washed free of enzyme and enzyme inhibitors with room temperature HBS. Cells were released by gentle trituration through decreasing bore, silanized Pasteur pipettes with fire-polished tips. The cells were plated onto plastic culture dishes and kept at room temperature in HBS. Cells remained viable for up to 10 h after dissociation and could be cultured overnight.
Pertussis toxin and cholera toxin treatments
Trigeminal ganglion cells were isolated as above. Neurons in HBS were left to attach to plastic culture dishes before changing medium to L15 Lebovitz (Sigma) containing 5 % fetal bovine serum (Gibco) and penicillin-streptomycin (50 u, 5 μg ml−1), and incubated at 25 °C in a humidified incubator. Pertussis toxin (PTX; 100 ng ml−1) or cholera toxin (CTX; 250 ng ml−1) was added to the culture medium prior to overnight incubation.
Electrophysiological recordings
Ionic currents from mouse trigeminal neurons were recorded in the whole-cell configuration of the patch-clamp method (Hamill et al. 1981) at room temperature (22–24 °C). Dishes were continually perfused with HBS. For isolating calcium currents, an extracellular solution was used containing (mm): TEACl 140; CsCl 2.5; CaCl2 2.5; Hepes 10; MgCl2 1; glucose 10; pH 7.2 (CsOH), 330 ± 5 mosmol l−1. The intracellular solution contained (mm): CsCl 130; Hepes 10; EGTA 10; CaCl2 2; MgATP 5; NaGTP 0.2; NaCl 5; pH 7.3 (CsOH), 285 ± 3 mosmol l−1.
Recordings were made using an EPC-9 patch-clamp amplifier and corresponding Pulse software from Heka Electroniks (Lambrecht, Germany) or an Axopatch-1D amplifier (Axon Instruments, Union City, CA) using pCLAMP acquisition software (Axon Instruments). Currents were sampled at 20–50 kHz and recorded on hard disk for later analysis. The recordings were filtered at 2.83 kHz (Heka) or 2 kHz (Axopatch). Patch pipettes were pulled from borosilicate glass (AM Systems, Everett, WA, USA). The pipette input resistance ranged between 0.7 and 1.5 MΩ. The capacitance of individual cells ranged between 4 and 40 pF with a series resistance between 1 and 5 MΩ. A series resistance compensation of at least 80 % was used in all experiments. Capacitance transients were compensated automatically using a built-in procedure of the Heka amplifier or by using the manual compensation on the Axopatch-1D. Leak current was subtracted on line using a P/8 protocol by the Pulse software (Heka Electroniks) unless otherwise noted (see Fig. 1A). The average current settling time (from 12 randomly selected cells) was 0.6 ± 0.05 ms. A liquid junction potential of approximately −8 mV has not been corrected for.
Figure 1. PGE2 inhibits HVA ICa in Type 1 mouse trigeminal neurons.
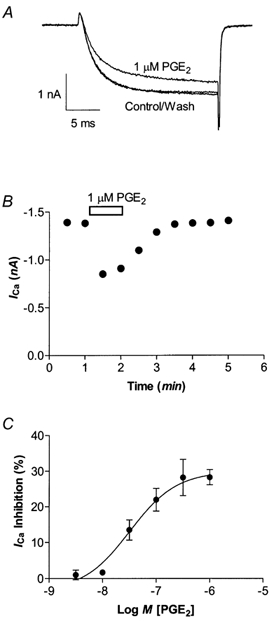
A, HVA ICa elicited by stepping the membrane potential from −80 to 0 mV was inhibited by 1 μm PGE2 in a Type 1 neuron. B, a representative time plot illustrating that inhibition of HVA ICa by 1 μm PGE2 completely reversed upon washout of the drug. C, Type-1 neurons were superfused with various concentrations of PGE2 for 1 min and then washed until the current returned to pre-drug treatment amplitude. Each point represents the mean and s.e.m. of 6–12 cells.
Peak HVA ICa in each cell was determined by stepping the membrane potential from a holding potential of −80 mV to between −60 and +60 mV, for 30 ms, in 10 mV increments. Following this procedure the test current was evoked every 30s and monitored for current stability before drugs were applied. Cells were rejected if the current increased or decreased by more than 2 % in the first 90 s after running the I-V protocol. Cells were exposed to drugs via a series of flow pipes positioned above the cells. The inhibition by drugs was quantified by measuring the current isochronically from the peak of the control current in the presence and absence of the drug.
RNA isolation and reverse transcription-polymerase chain reaction
Total RNA was isolated from whole trigeminal ganglia of mice using the RNeasy kit (Qiagen, Germany). Total RNA was treated with RQ1 RNase-free DNase (Promega, Madison, WI, USA) at 37 °C for 30 min to remove any contaminating genomic DNA. The enzyme was removed by a phenol-chloroform extraction. Optical density readings were performed to estimate the amount of total RNA before it was used in reverse transcription- polymerase chain reaction (RT-PCR).
A total of 1 μg of RNA and 1 μg of oligodT primers (Promega) were incubated at 70 °C for 5 min, and then cooled on ice. The RNA was reverse transcribed at 37 °C in a 15 μl reaction mixture containing (mm): Tris-HCl 50 (pH 8.5); KCl 75; MgCl2 3; dithiothreitol 10; and 400 μm each of deoxyadenosinetriphosphate (dATP), deoxythymidinetriphosphate (dTTP), deoxycytidinetriphosphate (dCTP), and deoxyguanosinetriphosphate (dGTP); 20 U of SUPERase•In RNase inhibitor (Ambion Inc., Austin, TX, USA) and 200 U of M-MLV reverse transcriptase (Promega). The RNA from each animal was incubated both with and without M-MLV reverse transcriptase to confirm the absence of contaminating genomic DNA.
PCR was performed in a 20 μl reaction volume containing (mm): Tris-HCl 10 (pH 9); KCl 50; MgCl2 1.5; 0.1 % Triton X-100; and 200 μm each of dATP, dTTP, dCTP and dGTP; 500 μm of each primer; 1 U of Taq DNA polymerase (Promega) and 5 μl of the appropriate RT product. Amplifications were performed in a Perkin-Elmer 2400 thermal cycler with an initial denaturation step at 94 °C for 1 min followed by 35 cycles. Each cycle consisted of a denaturation step at 94 °C for 30 s, an annealing step at 55 °C for 30 s, and an elongation step at 72 °C for 30 s. This was followed by a final elongation step at 72 °C for 5 min. The PCR products were separated on a 2 % agarose gel stained with ethidium bromide.
Data analysis
All data are expressed as means ± s.e.m. unless otherwise indicated. Concentration response data were pooled for each group and fitted to the Hill equation using the software package GraphPad Prism v. 3. Where noted, significant differences between means were tested, using Student's paired or unpaired, two tailed t test.
Drugs and chemicals
PGE2 was from Cayman Chemical Co. (Ann Arbor, MI, USA). ONO-DI-004, ONO-AE1–329, ONO-AE-248 and ONO-AE1–259 were kindly supplied by Ono Pharmaceuticals Co. Ltd (Osaka, Japan). Baclofen was from Research Biochemicals International (Natick, MA, USA). Buffer salts were from BDH Australia or Sigma Australia. Papain and collagenase were from Worthington Biochemical Corp. (Freehold, NJ, USA). BSA and trypsin inhibitor (chicken egg white ovomucoid, Type II-O), pertussis toxin and cholera toxin were from Sigma Australia. Methionine-enkephalin was from Auspep (Melbourne, Australia).
PGE2 was dissolved in ethanol and stored at −20 °C. More dilute aqueous solutions were made daily and kept on ice; ethanol alone (up to 0.01 %) did not affect ICa. Other compounds were dissolved in dimethylsulfoxide (DMSO); DMSO alone (up to 0.01 %) did not affect ICa. At higher concentrations (up to 0.1 %) of DMSO, the osmotic effects of the solvent were minimized by diluting the recording buffer before addition of the drugs (ONO-AE-248 and ONO-AE1–259) or the DMSO alone. For these higher concentrations of DMSO, the control was always defined as the current in the appropriate concentration of DMSO.
RESULTS
Acute application of prostaglandin E2 reversibly inhibited HVA ICa in almost all trigeminal neurons. We have previously characterized two populations of trigeminal sensory neurons based on sensitivity to opioid receptor agonists and capsaicin (Type 1 neurons) or the presence of a prominent T-type calcium current and insensitivity to opioid agonists and capsaicin (Type 2 cells, Borgland et al. 2001). In Type 1 neurons PGE2 inhibited HVA ICa in 234 of 262 cells examined, with an EC50 of 41 nm (logEC50 −7.4 ± 0.1) to a maximum of 28 ± 2 % (Fig. 1C). In Type 2 neurons PGE2 inhibited HVA ICa in 203 of 218 cells examined, with an EC50 of 34 nm (logEC50 −7.5 ± 0.1) and inhibited HVA ICa to a maximum of 32 ± 1 % (Fig. 2C). In Type 2 neurons that were sensitive to PGE2, the T-type ICa itself was not inhibited (Fig. 2A; n = 10).
Figure 2. Calcium currents in Type 2 trigeminal neurons are inhibited by PGE2.
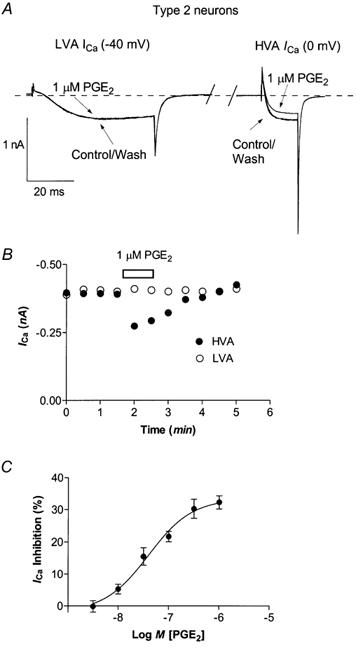
A, to elicit the low voltage-activated T-type ICa, neurons were stepped from −80 mV to −40 mV. Neurons recovered for 80 ms (approximately 65 ms has been removed for clarity) before stepping the membrane potential to 0 mV to elicit HVA ICa. T-type ICa was not inhibited by 1 μm PGE2. This trace was not leak subtracted and zero current is denoted by the dashed line.B, time course of inhibition of HVA ICa by 1 μm PGE2 in the same cell. C, Type 2 neurons were superfused with various concentrations of PGE2 for 1 min and then washed until the current returned to pre-drug treatment amplitude. Each point represents the mean and s.e.m. of 6–12 cells.
When current-voltage (I-V) relationships for ICa were determined in trigeminal neurons, the peak amplitude ICa of cells with (Fig. 3B) or without (Fig. 3A) T-type ICa occurred at either 0 or +10 mV (except in 6 cells with T-type ICa where the peak was at −40 mV). Inhibition of ICa by PGE2 occurred over a range of potentials in both types of cell. The inhibition of ICa was not due indirectly to a PGE2-induced increase in an outward current, because PGE2 did not modulate the current evoked in the presence of 30 μm Cd2+ when cells were stepped from −80 mV to either 0 mV (Fig. 3C, n = 9; 3D, n = 8) or +60 mV (n = 13, data not shown).
Figure 3. Characteristics of PGE2 modulation of HVA ICa.
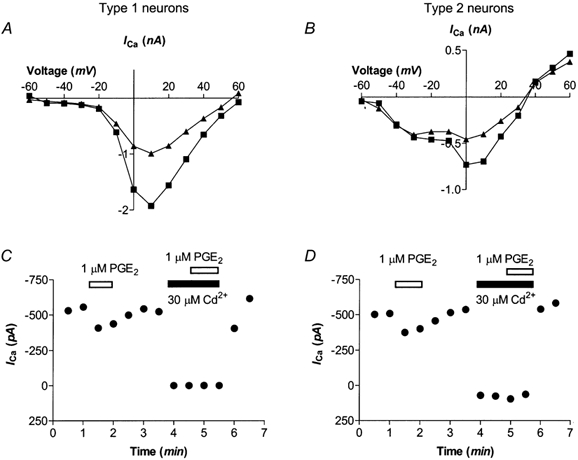
A and B, HVA ICa was elicited by stepping the membrane potential from −80 mV to potentials between −60 mV and +60 mV in 10 mV increments (Control, black squares). PGE2 (1 μm; triangles) inhibited ICa over a range of membrane potentials. A, a representative example of a current-voltage relationship of a Type 1 cell. B, current-voltage relationship for a Type 2 cell. In Type 1 cells (C) and Type 2 cells (D) 1 μm PGE2 did not alter Cd2+-insensitive current when HVA ICa was elicited by stepping the cells from the holding potential to 0 mV.
PGE2 inhibits ICa in two subpopulations of trigeminal sensory neurons by distinct G protein mechanisms
To examine the possible role of G proteins in the PGE2 inhibition of ICa, trigeminal neurons were treated overnight with either pertussis toxin (PTX, 100 ng ml−1), an irreversible inhibitor of Gi/o proteins, or cholera toxin (CTX, 250 ng ml−1), an activator of Gs proteins which uncouples Gs α-subunits from their receptors (Fig. 4). PTX treatment significantly attenuated the inhibition of ICa caused by 1 μm PGE2 in Type 2 cells (Fig. 4A, P < 0.001, n = 8), but not in Type 1 cells (Fig. 4A, n = 9). In contrast, CTX did not affect PGE2-mediated inhibition of ICa in Type 2 cells (n = 10), but it did abolish PGE2 inhibition of ICa in Type 1 cells (Fig. 4A, n = 10). CTX treatment alone did not alter the activation kinetics of the peak ICa (0–50 % rise times for ICa: Type 1 cells, 1.6 ± 0.2 ms vs. 1.6 ± 0.2 ms after CTX; Type 2 cells, 1.3 ± 0.1 ms vs. 1.3 ± 0.1 ms after CTX). In Type 1 cells, PTX treatment abolished inhibition of ICa by methionine-enkephalin (ME) (Fig. 4B, n = 6–8 each, and in Type 2 cells PTX treatment abolished the effects of baclofen (30 μm; Fig. 4C, n = 6–8 each). CTX treatment had no effect on the responses to ME in Type 1 cells or to baclofen in Type 2 cells. (Fig. 4C, n = 6–10 cells for each).
Figure 4. Inhibition of HVA ICa by PGE2 is mediated via multiple G proteins.
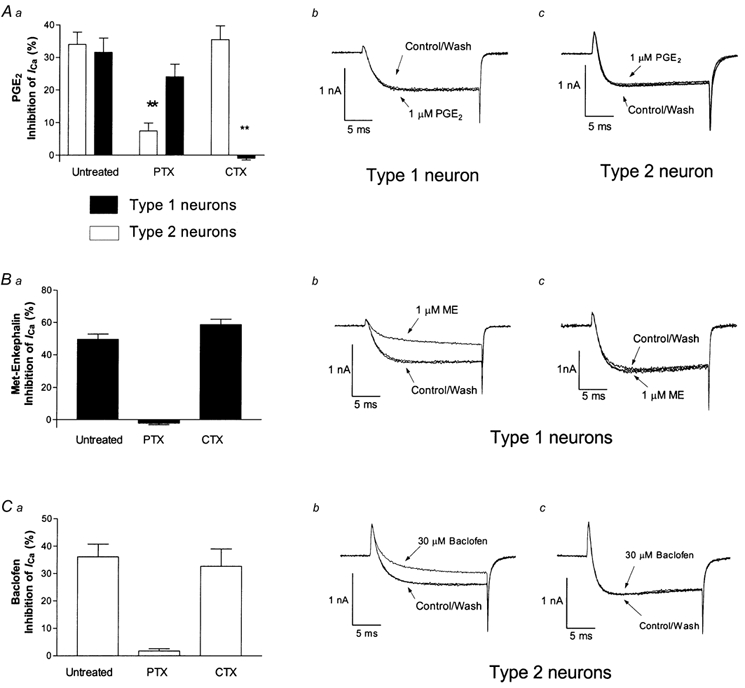
Neurons were treated overnight with either 100 ng ml−1 PTX or 250 ng ml−1 CTX. Aa, CTX selectively abolished PGE2-mediated inhibition of HVA ICa in Type 1 neurons, while PTX treatment selectively reduced effects of PGE2 in Type 2 neurons. Type 2 neurons pre-treated with PTX had significantly less inhibition of HVA ICa than controls (P < 0.001). Ab, a representative trace of a CTX treated Type 1 neuron in the presence and absence of 1 μm PGE2. Ac, a representative trace of a PTX treated Type 2 neuron in the presence and absence of 1 μm PGE2. B, in Type 1 neurons, inhibition of ICa caused by 1 μm ME was abolished by PTX but not CTX. Representative traces of a CTX treated neuron (Bb) and a PTX treated neuron (Bc) in the presence and absence of 1 μm ME. C, ICa inhibition caused by 30 μm baclofen was abolished by PTX, but not CTX in Type 2 cells. Representative traces of a CTX treated Type 2 neuron (Cb) and a PTX treated Type 2 neuron (Cc) in the presence and absence of 30 μm baclofen. Records illustrated are superimposed currents recorded before, during and after drug application.
The inhibition of ICa by PGE2 was associated with a significant slowing of the rise time of the evoked current, consistent with direct inhibition of ICa by G protein βγ subunits (see Table 1). To address the involvement of G protein βγ in the PGE2-mediated inhibition of ICa, a depolarizing conditioning step designed to reduce affinity of G protein βγ subunits for the calcium channel was used to test the voltage dependence of inhibition. In these experiments we required different voltage protocols for Type 1 and Type 2 cells to achieve relief of PGE2-mediated ICa inhibition. For both cell types ICa was evoked by two test steps to 0 mV, separated by a conditioning step applied 100 ms after the first test step (S1). A conditioning step to +100 mV for 50 ms was applied to Type 2 neurons and a step to +130 mV for 70 ms was applied to Type 1 neurons (Fig. 5). In Type 2 cells, PGE2 (1 μm) inhibition of ICa after the conditioning step was reduced by 50 ± 4 % compared to S1 (P < 0.01). After the conditioning step, the slowing of the 0–50 % rise time of ICa (S2) in the presence of PGE2 was significantly less than the slowing of the 0–50 % rise time of S1 (P < 0.01, Table 1). When Type 1 cells were subjected to the same conditioning step as Type 2 cells, the PGE2 (1 μm) inhibition of ICa elicited by the second test step (S2) was only reduced by 7 ± 3 % compared to S1, and there was no change in the rise time of S2 when compared with S1 (0–50 % rise time: S1, 1.04 ± 0.06 ms in control vs. 1.17 ± 0.07 ms in PGE2; S2, 1.05 ± 0.07 ms in control vs. 1.15 ± 0.07 ms in PGE2; n = 6). However, when the depolarizing conditioning step applied to Type 1 cells was increased to +130 mV and lengthened to 70 ms, PGE2 inhibition of ICa elicited by S2 was reduced by 39 ± 6 % compared to S1 (Table 1). The slowing of the 0–50 % rise time of ICa by PGE2, apparent in S1, was no longer apparent following the conditioning step (Table 1). Facilitation of S2 by the depolarizing conditioning step was also observed with baclofen, an agonist that activates Gi/o-coupled GABAB receptors (Table 1). Baclofen (30 μm) significantly decreased the ratio of S1:S2 (reduced inhibition in the second test pulse) and significantly slowed 0–50 % rise times of S1 in Type 1 cells (Table 1).
Table 1.
Effects of a depolarizing conditioning step on PGE2 modulation of HVA/Ca in mouse trigeminal neurons
| n | Control | 1 μm PGE2 | n | Control | 30 μm Baclofen | |
|---|---|---|---|---|---|---|
| Type 1 | ||||||
| Ratio S2:S1 peak amplitude | 7 | 1.01 ± 0.07 | 1.19 ±0.09* | 9 | 1.00 ± 0.05 | 1.18 ± 0.07*** |
| 0–50% rise time of S1 (ms) | 7 | 0.95 ± 0.07 | 1.11 ± 0.05* | 9 | 1.18 ±0.07 | 1.29 ± 0.10* |
| 0–50% rise time of S2 (ms) | 7 | 1.02 ± 0.08 | 1.05 ±0.08 | 9 | 1.11 ±0.05 | 1.14 ± 0.06 |
| Type 2 | ||||||
| Ratio S2:S1 peak amplitude | 8 | 1.07 ± 0.07 | 1.36 ± 0.09*** | |||
| 0–50% rise time of S1 (ms) | 8 | 1.69 ±0.08 | 2.35 ±0.17* | |||
| 0–50% rise time of S2 (ms) | 8 | 1.60 ±0.15 | 1.86 ± 0.18† | |||
Values are means and s.e.m. The S1:S2 ratio and 0–50% rise times were significantly greater than controls on indicated values (Student's paired, two tailed t test)
P < 0.05
P < 0.001.
Significantly different from S1 in PGE2, Student's unpaired t test,P <0.02.
Figure 5. PGE2 inhibition of HVA ICa in sensory neurons is relieved by a positive prepulse.
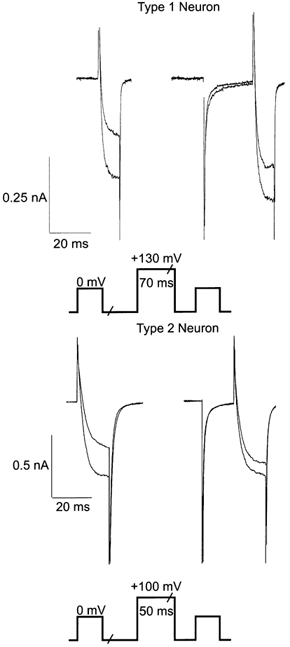
Different prepulse protocols were used for Type 1 and Type 2 neurons to achieve relief of ICa inhibition. Trigeminal neurons were voltage clamped at −80 mV and were stepped initially (S1) to a test potential of 0 mV. In Type 1 neurons, a 70 ms step to +130 mV was applied. Cells recovered for 20 ms before a second test pulse (S2) was applied. In Type 2 neurons, a 50 ms step to +100 ms was applied. Cells recovered for 15 ms before the second test pulse. A section of approximately 80 ms in the current trace has been omitted for clarity. Inhibition of HVA ICa by 1 μm PGE2 was relieved after a large positive prepulse in both Type 1 and Type 2 cells, but a greater depolarizing pulse was required to achieve relief in Type 1 neurons.
The effects of prostaglandin are mediated by EP3 receptors
PGE2 acts predominantly on prostanoid receptors of the EP type. The expression of the four cloned prostaglandin EP receptors in the mouse trigeminal ganglion was examined using RT-PCR. Messenger RNA was isolated from the trigeminal ganglion of eight male and five female animals, and PCR was performed using the primers described in Table 2. The primers selectively amplified mRNA fragments of the expected size (Fig. 6, Table 2), and the mRNA for all four prostaglandin EP receptors was found in all animals tested (Fig. 6).
Table 2.
Primer sequences used to amplify mRNA fragments from prostanoid EP receptors EP1–4 and the housekeeping enzyme hypoxanthine phosphoribosyl-transferase (HPRT)
| Receptor | Size (bp) | ||
|---|---|---|---|
| EP1 | F | GCTTAACCTGAGCCTAGCGGA | 294 |
| R | CGCAGTATACAGGCGAAGCAC | ||
| EP2 | F | CTCAACTACGGGGAGTACGTCC | 277 |
| R | AGGAGAATGAGGTGGTCCGTC | ||
| EP3 | F | CATGATGGTCACTGGCTTCGT | 238 |
| R | ACACTGTCATGGTTAGCCGCA | ||
| EP4 | F | CGTAGTATTGTGCAAGTCGCG | 215 |
| R | CAGATGATGCTGAGACCCGAC | ||
| HPRT | F | GCTACTGTAATGATCAGTCAACGGG | 394 |
| R | CAACATCAACAGGACTCCTCGTA |
F, the forward primer; R, the reverse; Size (bp), the length of the expected PCR product.
Figure 6. Trigeminal ganglion cells express 4 EP prostanoid receptor subtypes.
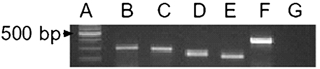
Total trigeminal ganglion mRNA was extracted and subjected to RT PCR utilizing the primers outlined in Table 1. The resulting PCR products were separated on a 2 % agarose gel stained with ethidium bromide, as illustrated. Lane A is a nucleotide size ladder in 100 bp increments, lanes B, C, D and E contain the amplified DNA fragments corresponding to EP1, EP2, EP3 and EP4 receptors respectively, lane F is the amplified DNA fragment corresponding to hypoxanthine phosphoribosyltransferase, a housekeeping enzyme, lane G contains all the PCR reaction components without added reverse transcription product. This is representative gel from the ganglia of 1 of 13 mice (8 male, 5 female).
To determine which EP receptor PGE2 was acting at to inhibit ICa, we examined the effects of selective EP receptor agonists on ICa in Type 1 and Type 2 cells. The effects of PGE2 were mimicked by the selective EP3 receptor agonist ONO-AE-248 (Fig. 7), but not by selective agonists for EP1 receptors (ONO-DI-004, 100 nm-3 μm), EP2 receptors (ONO-AE1–259, 300 nm-10 μm) or EP4 receptors (ONO-AE1–329, 100 nm-3 μm). At the highest concentrations tested the EP1, EP2 and EP4 receptor agonists did not affect ICa in any cell (n = 7–9 for each of Type 1 and Type 2 cells, data not shown).
Figure 7. The EP3 agonist ONO-AE-248 inhibits ICa in Type 1 and Type 2 trigeminal neurons.
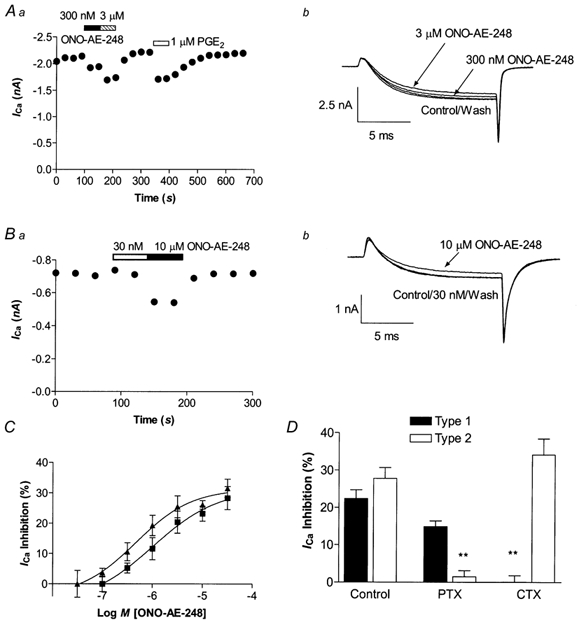
Representative time plots of ONO-AE-248 inhibition of peak ICa in a Type 1 (Aa) and Type 2 (Ba) neuron. ICa was elicited by stepping the membrane potential from −80 to 0 mV. Example traces from the Type 1 neuron (Ab) and Type 2 (Bb) neurons are shown in Aa and Ba. C, the inhibition of ICa by ONO-AE-248 in Type 1 (triangles) and Type 2 (squares) neurons was concentration dependent, with an EC50 of 580 nm and 1.4 μm, respectively. Each point represents the mean and s.e.m. of 6–10 cells. D, overnight treatment with PTX (100 ng ml−1) strongly reduced the ONO-AE-248 (10 μm) inhibition of ICa in Type 2 but not Type 1 cells, whereas treatment with CTX (250 ng ml−1) overnight blocked the ONO-AE-248 inhibition of ICa in Type 1 but not Type 2 cells (n = 6–15 for each).
In Type 1 neurons ONO-AE-248 inhibited ICa in 25 of 36 cells, with an EC50 of 1.4 μm (logEC50 −5.9 ± 0.1) to a maximum of 28 ± 2 % (Fig. 7C). In Type 2 cells ONO-AE-248 inhibited ICa in all 40 cells tested, with an EC50 of 580 nm (logEC50 −6.2 ± 0.1) to a maximum of 30 ± 2 % (Fig. 7C). The inhibition of ICa by ONO-AE-248 was also associated with significant slowing of the activation kinetics of ICa in both Type 1 and Type 2 cells. In Type 1 cells, 30 μm ONO-AE-248 resulted in a slowing of the 0–50 % rise time of ICa from 1.21 ± 0.15 ms to 1.38 ± 0.18 ms (n = 10, P < 0.02 paired t test). In Type 2 cells 30 μm ONO-248 slowed the 0–50 % rise time from 1.2 ± 0.1 ms to 1.55 ± 0.15 ms (n = 9, P < 0.02, paired t test). The inhibition of ICa by ONO-AE-248 was blocked by overnight CTX treatment in Type 1 but not Type 2 cells, whereas PTX pre-treatment blocked the effects of ONO-AE-248 in Type 2 cells but not in Type 1 cells (Fig. 7D). These data indicate that ONO-AE-248 inhibits ICa in a manner similar to that of PGE2.
PGE2 inhibits predominantly N- and P/Q-type ICa
Type 1 and Type 2 trigeminal sensory neurons both express predominantly N- and P/Q-type HVA ICa, with a lesser amount of R-type current, while Type 1 cells also have some L-type ICa (Borgland et al. 2001). We examined the inhibition of the different types of ICa in Type 1 and Type 2 sensory neurons by applying PGE2 in the presence of combinations of calcium channel antagonists. A predominantly N-type ICa was isolated by using a combination of the P/Q-type ICa antagonist ω-agatoxin IVA (500 nm) and the L-type ICa antagonist nimodipine (3 μm). When applied in the presence of these antagonists, PGE2 (1 μm) inhibited N-type ICa by 21 ± 3 % (n = 5) in Type 1 neurons and 30 ± 4 % (n = 9) in Type 2 neurons. A predominantly P/Q-type ICa was isolated using the N-type ICa antagonist ω-conotoxin GVIA (1 μm) and nimodipine (3 μm). In the presence of these antagonists PGE2 (1 μm), inhibited P/Q-type ICa by 19 ± 3 % (n = 6) in Type 1 cells and 15 ± 1 % (n = 6) in Type 2 neurons. The residual L-type and R-type ICa recorded in the presence of ω-agatoxin IVA (500 nm) and ω-conotoxin GVIA (1 μm) were not inhibited by PGE2 (1 μm) in either Type 1 or Type 2 neurons (n = 9).
DISCUSSION
In adult mouse trigeminal sensory neurons, PGE2 inhibited HVA ICa in almost all of the cells tested, but did so via two different G protein coupling mechanisms in electrophysiologically and pharmacologically distinct subpopulations of cells. Prostaglandins have been shown to inhibit ICa in rat sympathetic neurons (Ikeda, 1992; Ito et al. 2000), melanotrophs (Tanaka et al. 1998) and bovine adrenal chromaffin cells (Currie & Fox, 2000), but the only previous study to examine the actions of PGE2 on ICa in sensory neurons found that PGE2 increased the calcium current amplitude (Nicol et al. 1992). The reason(s) for the very different findings of this study and those of Nicol and colleagues (1992) are not known, but the earlier study was performed in very different cells — embryonic chicken sensory neurons.
PGE2 inhibited ICa via two different signal transduction pathways in different trigeminal neuron populations. In Type 1 trigeminal neurons PGE2-mediated inhibition of ICa was abolished by CTX pre-treatment and unaffected by PTX. In contrast, in Type 2 neurons inhibition of ICa was blocked by PTX treatment and unaffected by CTX treatment. These results imply that in Type 2 cells the PGE2 receptors are coupled predominantly through Gi/o proteins, whereas in Type 1 cells PGE2 receptors couple to calcium channels exclusively through Gs. Rapid, voltage-dependent inhibition of ICa via native PTX-insensitive G proteins has been observed in several cell types with agonists such as vasoactive intestinal polypeptide (VIP) (Zhu & Ikeda, 1994; Ehrlich & Elmslie, 1995), histamine and PGE2 (Currie & Fox, 2000). VIP inhibition of ICa in sympathetic neurons occurs via a CTX-sensitive, PKA-independent pathway with kinetic characteristics identical to PTX-sensitive pathways in the same cell (Zhu & Ikeda, 1994; Ehrlich & Elmslie, 1995). Similar results were obtained in this study, where the CTX-sensitive PGE2 inhibition of ICa appeared to be mediated via G protein βγ subunits, which also mediated the PTX-sensitive inhibition of ICa by baclofen (see below). In bovine adrenal chromaffin cells, both histamine and PGE2 inhibit ICa via a voltage-dependent, PTX-insensitive pathway, but the involvement of Gs, or other types of G protein, has not been addressed.
In both types of cells the inhibition of ICa appeared to be consistent with βγ-subunits of the G protein heterotrimers interacting with calcium channels. The rise time of ICa in the presence of PGE2 was slowed in both types of cell, and the magnitude of the PGE2-induced inhibition of ICa could be strongly reduced by a positive conditioning step; both these features are characteristic of the G protein βγ subunit-mediated pathway of calcium channel inhibition (Herlitze et al. 1996; Ikeda 1996; Zamponi & Snutch, 1998). Similarly, in sympathetic neurons and melanotrophs PGE2 couples to inhibition of ICa predominantly via a PTX-sensitive βγ subunit-mediated pathway (Ikeda 1992; Tanaka et al. 1998; Ito et al. 2000). The present results do not preclude the possibility that PGE2 modulates HVA ICa via other second messenger systems as well.
Different conditioning pulse protocols were required to relieve ICa inhibition by PGE2 in Type 1 versus Type 2 cells. In Type 1 cells a step to +130 mV was required to observe significant relief of PGE2-induced inhibition, while a step to +100 mV was sufficient in Type 2 cells. There are several possible explanations for this. Different G protein βγ subtypes that have a stronger affinity for calcium channels may be coupled to different G proteins in Type 1 versus Type 2 cells. Differential voltage-dependent effects on ICa of different G protein βγ subunits have been observed in rat superior cervical ganglion neurons, where G protein βγ subunits derived from Goa or Gi heterotrimers were not equally effective in promoting voltage-dependent inhibition of ICa (Delmas et al. 1999). Recombinant N-type and P/Q-type calcium channels are differentially modulated by different types of G protein β subtypes (Arnot et al. 2000), and so association of different βγ subunits with Gsα versus PTX-sensitive G proteins in sensory neurons may explain our observations. However, the identity of the calcium channels and their accessory subunits in Type 1 or Type 2 cells may also be responsible for the need for more robust depolarizations in Type 1 cells to obtain relief from PGE2 inhibition. Different splice variants of N-type or P/Q-type calcium channels have been identified and may be differentially affected by G protein βγ subunits (Lin et al. 1997; Bourinet et al. 1999; Delmas et al. 2000), leading to variable neurotransmitter modulation of otherwise similar channels in native neurons. An example of such variability which may be a result of alternative splicing of the α1B subunit is found in superior cervical ganglion neurons. In these cells, dendrites uniquely expressed an N-type calcium channel which had enhanced interactions with G protein βγ subunits (Delmas et al. 2000), which resulted in the dendrites of these neurons being ‘hypersensitive’ to neurotransmitter-mediated regulation of ICa when compared with the cell body. The dendritic N-type channel also showed reduced relief from G protein βγ inhibition for a given depolarizing step. Similar differences in G protein βγ subunit-mediated regulation of ICa can also occur when different calcium channel β-subunits associate with a given pore forming subunit (Canti et al. 2000; Feng et al. 2001). Although mouse trigeminal neurons appear to have similar amounts of N-type and P/Q-type calcium channels in Type 1 and Type 2 cells (Borgland et al. 2001), our studies cannot detect different splice variants of the α subunit of the channels, or the identity of the calcium channel β subunits they are associated with.
Although we found mRNA for all four types of EP prostanoid receptor in whole trigeminal ganglion, EP3 receptors appeared to mediate the effects of PGE2 in both types of trigeminal neuron. Of the selective EP receptor agonists tested, only the EP3 agonist ONO-AE-248 mimicked the PGE2 inhibition of ICa. Agonists selective for EP1 (ONO-DI-004), EP2 (ONO-AE1–259) or EP4 receptors (ONO-AE1–329) did not modulate ICa in any neurons examined. These agents have been reported to be potent and very selective agonists at native and recombinant mouse EP receptors (Zacherowski et al. 1999; Suzawa et al. 2000). The inhibition of ICa by ONO-AE-248 had similar operational characteristics to that caused by PGE2 in that it was associated with a significant slowing of the activation of ICa and was largely abolished by CTX treatment in Type 1 cells and PTX treatment in Type 2 cells. ONO-AE-248 was considerably less potent than PGE2 in inhibiting ICa. This is consistent with previous findings demonstrating that ONO-AE-248 inhibited forskolin-stimulated adenylyl cyclase activity in cells expressing recombinant murine EP3 receptors at least 10-fold less potently than PGE2 (Zacharowski et al. 1999). Mouse EP3 receptor mRNA can undergo multiple alternative splicing events to produce proteins which can couple to either only Gi/Go (EP3α and EP3β, Sugimoto et al. 1993) or to both Gi/Go and Gs (EP3γ, Irie et al. 1993). The relative expression of these isoforms in mouse sensory neurons has not been established, but the results of the present study are consistent with the expression of at least the EP3γ isoform in trigeminal neurons.
The presence of multiple EP receptors in mouse trigeminal ganglion is generally consistent with other reports (Oida et al. 1995; Narumiya et al. 1999), although the presence of EP2 receptor mRNA has not to our knowledge been reported in mouse. All four EP receptor types are found in rat dorsal root ganglion neurons although one study found only one of the rat EP3 splice variants (Southall & Vasko, 2001), while another found all three (Donaldson et al. 2001). In cultured rat dorsal root ganglion neurons, the Gs-coupled EP3c receptor subtype (together with EP4 receptors), was found to be necessary for full expression of some of the biochemical correlates of PGE2-mediated sensitization, namely elevation of cAMP and enhanced release of substance P and calcitonin gene-related peptide (Southall & Vasko, 2001).
The present study identified inhibition of HVA ICa as a novel mechanism by which PGE2 can modify the activity of virtually all sensory neurons. In many sensory neurons PGE2 directly enhances excitability (Ingram & Williams, 1996; Gold et al. 1996; Evans et al. 1999) and sensitizes the cells to a variety of stimuli (Pitchford & Levine, 1991; Cui & Nicol, 1995; Lopshire & Nicol, 1998). These effects occur predominantly via processes that involve activation of adenylyl cyclase via Gs (Ingram & Williams, 1996; England et al. 1996; Evans et al. 1999). The adenylyl cyclase-dependent sensitization by PGE2 is likely to occur in the Type 1 trigeminal sensory neurons, which are opioid and capsaicin sensitive, and in which PGE2 inhibits ICa via Gs. Although often considered simply an inhibitory effect, the reduction of calcium entry via HVA ICa may also enhance sensory neuronal excitability by indirectly inhibiting calcium-dependent after-hyperpolarizations, which can allow neurons to attain higher frequencies of action potentials.
We have also identified a significant population of trigeminal sensory neurons in which PGE2 is unlikely to act as a sensitizing agent, at least when acting through EP3 receptors. In these Type 2 neurons PGE2 appears to couple predominantly through inhibitory G proteins. Although we have not established whether other EP receptor subtypes act on other channels to modulate the activity of Type 2 neurons, these neurons also lack one of the major substrates for PGE2-mediated sensitization, VR1 receptors (Lopshire & Nicol, 1998; Borgland et al. 2001). We speculate that the rapid elevations of PGE2 in the spinal cord that can follow a peripheral noxious stimulus (Yang et al. 1996) may act to dampen sensory input from this population of neurons by inhibiting ICa in the spinal terminals of these cells. In a preliminary study in the substantia gelatinosa of the spinal trigeminal nucleus of the rat, we have reported that PGE2 inhibited spontaneous miniature glutamatergic synaptic currents (Jennings et al. 2000). In contrast, and consistent with the excitatory effects of PGE2 on sensory neurons, another study found a PGE2-induced enhancement of glutamatergic synaptic currents in mouse lumbar dorsal horn neurons (Minami et al. 1999). These two studies, together with the data reported here, indicate that PGE2 can act in ways other than simply as a sensitizing agent for sensory neurons, and appears to differentially modulate the sensory information coming from distinct populations of sensory neurons.
Acknowledgments
We thank ONO Pharmaceuticals for their kind donation of the EP receptor agonists used in this study, and Dr Elena Bagley for her comments on the manuscript.
REFERENCES
- Abdulla FA, Smith PA. Ectopic alpha2-adrenoceptors couple to N-type Ca2+ channels in axotomized rat sensory neurons. Journal of Neuroscience. 1997;17:1633–41. doi: 10.1523/JNEUROSCI.17-05-01633.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnot MI, Stotz SC, Jarvis SE, Zamponi GW. Differential modulation of N-type α1B and P/Q-type α1A calcium channels by different G protein β subunit isoforms. Journal of Physiology. 2000;527:203–212. doi: 10.1111/j.1469-7793.2000.00203.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baccaglini PI, Hogan PG. Some rat sensory neurons in culture express characteristics of differentiated pain sensory cells. Proceedings of the National Academy of Sciences of the USA. 1983;80:594–598. doi: 10.1073/pnas.80.2.594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borgland SL, Connor M, Christie MJ. Nociceptin inhibits calcium channel currents in a subpopulation of small nociceptive trigeminal ganglion neurons in mouse. Journal of Physiology. 2001;536:35–47. doi: 10.1111/j.1469-7793.2001.t01-1-00035.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourinet E, Soong TW, Sutton K, Slaymaker S, Mathews E, Monteil A, Zamponi GW, Nargeot J, Snutch TP. Splicing of a1A subunit gene generates phenotypic variants of P- and Q-type calcium channels. Nature Neuroscience. 1999;2:407–421. doi: 10.1038/8070. [DOI] [PubMed] [Google Scholar]
- Canti C, Bogdanov Y, Dolphin AC. Interaction between G proteins and accessory β subunits in the regulation of α1B calcium channels in Xenopus oocytes. Journal of Physiology. 2000;527:419–432. doi: 10.1111/j.1469-7793.2000.t01-1-00419.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chahl LA, Iggo A. The effects of bradykinin and prostaglandin E1 on rat cutaneous nerve activity. British Journal of Pharmacology. 1977;59:343–347. doi: 10.1111/j.1476-5381.1977.tb07498.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui M, Nicol GD. Cyclic AMP mediates the prostaglandin E2-induced potentiation of bradykinin excitation in rat sensory neurons. Neuroscience. 1995;66:459–466. doi: 10.1016/0306-4522(94)00567-o. [DOI] [PubMed] [Google Scholar]
- Currie KPM, Fox AP. Voltage-dependent, pertussis toxin insensitive inhibition of calcium currents by histamine in bovine adrenal chromaffin cells. Journal of Neurophysiology. 2000;83:1435–1442. doi: 10.1152/jn.2000.83.3.1435. [DOI] [PubMed] [Google Scholar]
- Delmas P, Abogadie FC, Buckley NJ, Brown DA. Calcium channel gating and modulation by transmitters depend on cellular compartmentalization. Nature Neuroscience. 2000;3:670–678. doi: 10.1038/76621. [DOI] [PubMed] [Google Scholar]
- Delmas P, Abogadie FC, Milligan G, Buckley NJ, Brown DA. βγ dimers derived from Go and Gi proteins contribute different components of adrenergic inhibition of Ca2+ channels in rat sympathetic neurons. Journal of Physiology. 1999;518:23–36. doi: 10.1111/j.1469-7793.1999.0023r.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donaldson LF, Humphrey PS, Oldfield S, Giblett S, Grubb BD. Expression and regulation of prostaglandin E receptor subtype mRNAs in rat sensory ganglia and spinal cord in response to peripheral inflammation. Prostaglandins and Other Lipid Mediators. 2001;63:109–122. doi: 10.1016/s0090-6980(00)00101-5. [DOI] [PubMed] [Google Scholar]
- Eckert SP, Taddese A, McClesky EW. Isolation and culture of rat sensory neurons having distinct sensory modalities. Journal of Neuroscience Methods. 1997;77:183–190. doi: 10.1016/s0165-0270(97)00125-8. [DOI] [PubMed] [Google Scholar]
- Ehrlich I, Elmslie KS. Neurotransmitters acting via different G proteins inhibit N-type calcium current by an identical mechanism in rat sympathetic neurons. Journal of Neurophysiology. 1995;74:2251–2257. doi: 10.1152/jn.1995.74.6.2251. [DOI] [PubMed] [Google Scholar]
- England S, Bevan S, Docherty RJ. PGE2 modulates the tetrodotoxin-resistant sodium current in neonatal rat dorsal root ganglion neurones via the cyclic AMP-protein kinase A cascade. Journal of Physiology. 1996;495:429–440. doi: 10.1113/jphysiol.1996.sp021604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans AR, Vasko MR, Nicol GD. The cAMP transduction cascade mediates the PGE2-induced inhibition of potassium currents in rat sensory neurones. Journal of Physiology. 1999;516:163–178. doi: 10.1111/j.1469-7793.1999.163aa.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Z-P, Arnot MI, Doering CJ, Zamponi GW. Calcium channel β subunits differentially regulate the inhibition of N-type channels by individual G β isoforms. Journal of Biological Chemistry. 2001;276:45051–45058. doi: 10.1074/jbc.M107784200. [DOI] [PubMed] [Google Scholar]
- Ferreira SH. Prostaglandins, aspirin-like drugs and analgesia. Nature New Biology. 1972;240:200–203. doi: 10.1038/newbio240200a0. [DOI] [PubMed] [Google Scholar]
- Ferreira SH, Nakamura M. Prostaglandin hyperalgesia. Prostaglandins. 1979;18:179–209. doi: 10.1016/0090-6980(79)90103-5. [DOI] [PubMed] [Google Scholar]
- Fowler JC, Wonderlin WF, Weinreich D. Prostaglandins block a Ca2+-dependent slow spike after hyperpolarization independent of effects on Ca2+ influx in visceral afferent neurons. Brain Research. 1985;345:345–349. doi: 10.1016/0006-8993(85)91014-5. [DOI] [PubMed] [Google Scholar]
- Greaves MW, Sondergaard J, McDonald-Gibson W. Recovery of prostaglandins in human cutaneous inflammation. British Medical Journal. 1971;2:258–260. doi: 10.1136/bmj.2.5756.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold MS, Levine JD, Correa AM. Modulation of TTX-R INa by PKC and PKA and their role in PGE2-induced sensitization of rat sensory neurons in vitro. Journal of Neuroscience. 1998;18:10345–10355. doi: 10.1523/JNEUROSCI.18-24-10345.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold MS, Reichling DB, Shuster MJ, Levine JD. Hyperalgesic agents increase a tetrodotoxin-resistant Na+ current in nociceptors. Proceedings of the National Academy of Sciences of the USA. 1996;93:1108–1112. doi: 10.1073/pnas.93.3.1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Archiv. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Handwerker HO. Influences of algogenic substances and prostaglandins on the discharges of unmyelinated cutaneous nerve fibers identified as nociceptors. In: Bonica JJ, Albe-Fessard D, editors. Advances in Pain Research and Therapy. Vol. 1. New York: Raven Press; 1976. pp. 41–45. [Google Scholar]
- Herlitze S, Garcia DE, Mackie K, Hille B, Scheuer T, Catterall WA. Modulation of Ca2+ channels by G protein βγ subunits. Nature. 1996;380:258–262. doi: 10.1038/380258a0. [DOI] [PubMed] [Google Scholar]
- Hingtgen CM, Waite KJ, Vasko MR. Prostaglandins facilitate peptide release from rat sensory neurons by activating the adenosine 3′5′-cyclic monophosphate transduction cascade. Journal of Neuroscience. 1995;15:5411–5419. doi: 10.1523/JNEUROSCI.15-07-05411.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hori Y, Endo K, Takahashi T. Presynaptic inhibitory action of enkephalin on excitatory transmission in superficial dorsal horn of rat spinal cord. Journal of Physiology. 1992;450:673–685. doi: 10.1113/jphysiol.1992.sp019149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda SR. Prostaglandin modulation of Ca2+ channels in rat sympathetic neurones is mediated by guanine nucleotide binding proteins. Journal of Physiology. 1992;458:339–359. doi: 10.1113/jphysiol.1992.sp019421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda SR. Voltage-dependent modulation of N-type calcium channels by G protein βγ subunits. Nature. 1996;380:255–258. doi: 10.1038/380255a0. [DOI] [PubMed] [Google Scholar]
- Ingram SL, Williams JT. Opioid inhibition of Ih via adenylyl cyclase. Neuron. 1994;13:179–186. doi: 10.1016/0896-6273(94)90468-5. [DOI] [PubMed] [Google Scholar]
- Ingram SL, Williams JT. Modulation of the hyperpolarization-activated current (Ih) by cyclic nucleotides in guinea-pig primary afferent neurons. Journal of Physiology. 1996;492:97–106. doi: 10.1113/jphysiol.1996.sp021292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irie A, Sugimoto Y, Namba T, Harazono A, Honda A, Watabe A, Negishi M, Narumiya S, Ichikawa A. Third isoform of the prostaglandin-E-receptor EP3 subtype with different C-terminal tail coupling to both stimulation and inhibition of adenylate cyclase. European Journal of Biochemistry. 1993;217:313–316. doi: 10.1111/j.1432-1033.1993.tb18248.x. [DOI] [PubMed] [Google Scholar]
- Ito Y, Murai Y, Ishibashi H, Onoue H, Akaike N. The prostaglandin E series modulates high-voltage-activated calcium channels probably through the EP3 receptor in rat paratracheal ganglia. Neuropharmacology. 2000;39:181–190. doi: 10.1016/s0028-3908(99)00142-2. [DOI] [PubMed] [Google Scholar]
- Jennings EA, Vaughan CW, Christie MJ. Prostaglandin E2 inhibits synaptic transmission in rat substantia gelatinosa. Proceedings of Australian Neuroscience Society. 2000;11:170. [Google Scholar]
- Lin ZX, Haus S, Edgerton J, Lipscombe D. Identification of functionally distinct isoforms of the N-Type Ca2+ channel in rat sympathetic ganglia and brain. Neuron. 1997;18:153–166. doi: 10.1016/s0896-6273(01)80054-4. [DOI] [PubMed] [Google Scholar]
- Lopshire JC, Nicol GD. The cAMP transduction cascade mediates the prostaglandin E2 enhancement of the capsaicin-elicited current in rat sensory neurons: whole-cell and single-channel studies. Journal of Neuroscience. 1998;18:6081–6092. doi: 10.1523/JNEUROSCI.18-16-06081.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mense S. Sensitization of group IV muscle receptors to bradykinin by 5-hydroxytryptamine and prostaglandin E2. Brain Research. 1981;225:95–105. doi: 10.1016/0006-8993(81)90320-6. [DOI] [PubMed] [Google Scholar]
- Minami T, Okuda-Ashitaka E, Hori Y, Sakuma S, Sugimoto T, Sakimura K, Mishina M, Ito S. Involvement of primary afferent C-fibers in touch evoked pain (allodynia) induced by prostaglandin E2. European Journal of Neuroscience. 1999;11:1849–1856. doi: 10.1046/j.1460-9568.1999.00602.x. [DOI] [PubMed] [Google Scholar]
- Narumiya S, Sugimoto Y, Ushikubi F. Prostanoid receptors: structures, properties, and functions. Physiological Reviews. 1999;79:1193–1226. doi: 10.1152/physrev.1999.79.4.1193. [DOI] [PubMed] [Google Scholar]
- Nicol GD, Klingberg DK, Vasko MR. Prostaglandin E2 increases calcium conductance and stimulates release of substance P in avian sensory neurons. Journal of Neuroscience. 1992;12:1917–1927. doi: 10.1523/JNEUROSCI.12-05-01917.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicol GD, Vasko MR, Evans AR. Prostaglandins suppress an outward potassium current in embryonic rat sensory neurons. Journal of Neurophysiology. 1997;77:167–176. doi: 10.1152/jn.1997.77.1.167. [DOI] [PubMed] [Google Scholar]
- Oida H, Namba T, Sugimoto Y, Ushikubi F, Ohishi HH, Icikawa A, Narumiya S. In situ hybridization studies of prostacyclin receptor mRNA expression in various mouse organs. British Journal of Pharmacology. 1995;116:2828–2837. doi: 10.1111/j.1476-5381.1995.tb15933.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitchford S, Levine JD. Prostaglandins sensitize nociceptors in cell culture. Neuroscience Letters. 1991;131:105–108. doi: 10.1016/0304-3940(91)90444-x. [DOI] [PubMed] [Google Scholar]
- Southall MD, Vasko MR. Prostaglandin receptor subtypes, EP3C and EP4, mediate the prostaglandin E2-induced cAMP production and sensitization of sensory neurons. Journal of Biological Chemistry. 2001;276:16083–16091. doi: 10.1074/jbc.M011408200. [DOI] [PubMed] [Google Scholar]
- Sugimoto Y, Negishi M, Hayashi Y, Namba T, Honda A, Watabe A, Hirata M, Narumiya S, Ichikawa A. Two isoforms of the EP3 receptor with different carboxy-terminal domains: identical ligand binding properties and different coupling properties with Gi proteins. Journal of Biological Chemistry. 1993;268:2712–2718. [PubMed] [Google Scholar]
- Suzawa T, Miyaura C, Inada M, Maruyama T, Sugimoto Y, Ushikubi F, Ichikawa A, Narumiya S, Suda T. The role of prostaglandin E receptor subtypes (EP1, EP2, EP3, and EP4) in bone resorption: an analysis using specific agonists for the respective EPs. Endocrinology. 2000;141:1554–1559. doi: 10.1210/endo.141.4.7405. [DOI] [PubMed] [Google Scholar]
- Tanaka K, Shibuya I, Kabashima N, Ueta Y, Yamashita K. Inhibition of voltage-dependent calcium channels by prostaglandin E2 in rat melanotrophs. Endocrinology. 1998;139:4801–4810. doi: 10.1210/endo.139.12.6383. [DOI] [PubMed] [Google Scholar]
- Vasko MR, Campbell WB, Waite KJ. Prostaglandin E2 enhances bradykinin-stimulated release of neuropeptides from rat sensory neurons in culture. Journal of Neuroscience. 1994;14:4987–4997. doi: 10.1523/JNEUROSCI.14-08-04987.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White G, Lovinger DM, Weight FF. Transient low-threshold Ca2+ triggers burst firing afterdepolarizing potential in adult mammalian neuron. Proceedings of the National Academy of Sciences of the USA. 1989;86:6802–6806. doi: 10.1073/pnas.86.17.6802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis AL. Parallel assay of prostaglandin-like activity in rat inflammatory exudate by means of cascade superfusion. Journal of Pharmacy and Pharmacology. 1969;21:126–128. doi: 10.1111/j.2042-7158.1969.tb08213.x. [DOI] [PubMed] [Google Scholar]
- Xiao WH, Bennett GJ. Synthetic ω-conopeptides applied to the site of nerve injury suppress neuropathic pain in rats. Journal of Pharmacology and Experimental Therapeutics. 1995;274:666–672. [PubMed] [Google Scholar]
- Yang L-C, Marsala M, Yaksh TL. Characterization of time course of spinal amino acids, citrulline and PGE2 release after carrageenan/kaolin-induced knee joint inflammation: a chronic microdialysis study. Pain. 1996;67:345–354. doi: 10.1016/0304-3959(96)03106-5. [DOI] [PubMed] [Google Scholar]
- Yoshida S, Matsuda Y, Samejima A. Tetrodotoxin-resistant sodium and calcium components of action potentials in dorsal root ganglion cells of the adult mouse. Journal of Neurophysiology. 1978;41:1096–1106. doi: 10.1152/jn.1978.41.5.1096. [DOI] [PubMed] [Google Scholar]
- Zacharowski K, Olbrich A, Piper J, Hafner G, Kondo K, Thiemermann C. Selective activation of the prostanoid EP3 receptor reduces myocardial infarct size in rodents. Arteriosclerosis. Thrombosis and Vascular Biology. 1999;19:2141–2147. doi: 10.1161/01.atv.19.9.2141. [DOI] [PubMed] [Google Scholar]
- Zamponi GW, Snutch TP. Decay of prepulse facilitation of N type calcium channels during G protein inhibition is consistent with binding of a single G βγ subunit. Proceedings of the National Academy of Sciences of the USA. 1998;95:4035–4039. doi: 10.1073/pnas.95.7.4035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Ikeda SR. VIP inhibits N-type Ca2+ channels of sympathetic neurons via a pertussis toxin-insensitive but cholera toxin sensitive pathway. Neuron. 1994;13:657–699. doi: 10.1016/0896-6273(94)90033-7. [DOI] [PubMed] [Google Scholar]