

Abstract
Butyrate is the principal source of energy for colonic epithelial cells, and has profound effects on their proliferation, differentiation and apoptosis. Transport of butyrate across the colonocyte luminal membrane is mediated by the monocarboxylate transporter 1 (MCT1). We have examined the regulation of expression of human colonic MCT1 by butyrate, in cultured colonic epithelial cells (AA/C1). Treatment with sodium butyrate (NaBut) resulted in a concentration- and time-dependent upregulation of both MCT1 mRNA and protein. At 2 mm butyrate, the magnitude of induction of mRNA (5.7-fold) entirely accounted for the 5.2-fold increase in protein abundance, and was mediated by both activation of transcription and enhanced mRNA stability. The other monocarboxylates found naturally in the colon, acetate and propionate, had no effect. The properties of butyrate uptake by AA/C1 cells were characteristic of MCT1. Induction of the MCT1 protein resulted in a corresponding increase in the maximal rate of butyrate transport. The Vmax for uptake of [U-14C]butyrate was increased 5-fold following pre-incubation with 2 mm NaBut, with no significant change in the apparent Km. In conclusion, this study is the first to show substrate-induced regulation of human colonic MCT1. The basis of this regulation is a butyrate-induced increase in MCT1 mRNA abundance, resulting from the dual control of MCT1 gene transcription and stability of the MCT1 transcript. We suggest that butyrate-induced increases in the expression and resulting activity of MCT1 serve as a mechanism to maximise intracellular availability of butyrate, to act both as a source of energy and to influence processes maintaining cellular homeostasis in the colonic epithelium.
Butyrate, a monocarboxylate commonly referred to as a short-chain fatty acid, is a naturally occurring product of colonic microbial fermentation of dietary carbohydrates that are not hydrolysed in the small intestine (Macfarlane & Cummings, 1991). It is present in the colon in millimolar concentrations and is the principal energy source for colonic epithelial cells (Cummings, 1984). In addition, butyrate is important for maintaining homeostasis of the normal colonic mucosa (Bugaut & Bentejac, 1993; Treem et al. 1994) and exerts a number of biological effects on cultured mammalian cells (Kruh, 1982). These effects include the inhibition of proliferation, and the induction of both differentiation and apoptosis (Hague et al. 1995; Hague & Paraskeva, 1995). In many cases, these effects are correlated with changes in expression of various genes associated with these processes. In colonic cells, these genes include p21Waf/Cip1 (Archer et al. 1998; Bai & Merchant, 2000), cyclin D3 (Siavoshian et al. 2000), bcl2 and bak (Hague et al. 1997).
At the colonic luminal pH (pH 7), butyrate (pKa, 4.7) dissociates almost entirely to the butyrate anion (Phillips & Devroede, 1979). This charged species cannot cross the luminal membrane by free diffusion, and therefore requires a specific transport system. Accordingly, it has been shown that butyrate is transported across the colonic luminal membrane by a carrier-mediated mechanism (Mascolo et al. 1991; Ritzhaupt et al. 1998a). More recently, we have demonstrated that the protein that mediates this transport is the monocarboxylate transporter, MCT1. Indeed, we have shown that: (i) MCT1 is expressed in human colon; (ii) the functional protein is located on the luminal membrane of the colonic absorptive cells, and (iii) it transports butyrate (Ritzhaupt et al. 1998b).
MCT1 is a member of the family of monocarboxylate transporters (MCTs), which mediate the transport of monocarboxylates across the plasma membrane of a variety of cell types (Garcia et al. 1994b; Halestrap & Price, 1999). Although in the lumen of the colon the major monocarboxylates are normally acetate, propionate and butyrate (Cummings, 1984), in other tissues a range of metabolically important monocarboxylates including lactate, pyruvate, and the ketone bodies acetoacetate and β-hydroxybutyrate are transported by MCTs (Garcia et al. 1994a; Jackson & Halestrap, 1996; Tamai et al. 1999; Halestrap & Price, 1999). There are also diversities in the mechanisms of transport of monocarboxylates by MCTs expressed in different tissues. In hepatocytes (Jackson & Halestrap, 1996), erythrocytes (Poole & Halestrap, 1991) and cardiac myocytes (Wang et al. 1996), the mechanism of transport is via proton-monocarboxylate symport; in kidney it is sodium coupled (Poole & Halestrap, 1993); and in the colon it is by an anion-exchange mechanism (Ritzhaupt et al. 1998a). Amongst the MCT isoforms, MCT1 is expressed most widely, and has broad substrate specificity (Halestrap & Price, 1999). As such, in addition to its role in butyrate transport in the colon, MCT1 is likely to play a central role in metabolism and metabolic communication in many tissues (Halestrap & Price, 1999).
Intestinal mucosal cells are exposed to a luminal composition that varies significantly with diet (Karasov & Diamond, 1987). Under such conditions, it is not surprising that the expression and activity of many nutrient transport proteins are adaptive to changes in substrate levels (Ferraris & Diamond, 1989; Lescale-Maty et al. 1993; Shirazi-Beechey, 1995). The adaptation may be either positively (Dyer et al. 1997; Ferraris & Diamond, 1997; Walker et al. 1998) or negatively (Diamond & Karasov, 1987; Hattenhauer et al. 1999) correlated to levels of their specific dietary substrates. The former pattern of regulation serves to match absorption of metabolisable nutrients to altered dietary composition in a manner that ensures economy of biosynthetic costs, whilst at the same time maximising dietary energy (or other) gain (Diamond & Karasov, 1987). The latter pattern of regulation is thought to ensure adequate absorption of essential nutrients, and to minimise effects of nutrients with potential toxicity when in excess (Diamond & Karasov, 1987; Wessling-Resnick, 2000).
Although it is becoming increasingly apparent that the colon is an important site for the salvage of nutrients that escape digestion in the small intestine (Macfarlane & Cummings, 1991; Danielsen & Jackson, 1992; Ugawa et al. 2001), relatively little is known of the mechanisms by which dietary components modulate the expression of colonic nutrient transporters. Moreover, despite the vital roles played by butyrate in the colon, very little is known of the factors regulating its transport into colonic epithelial cells. Indeed, it is only recently that a specific transport protein for butyrate has been identified in the colon (Ritzhaupt et al. 1998a, b). In the present study, we have examined the regulation of the expression of human colonic MCT1 in response to monocarboxylates found naturally in the colon. We show that in the colonic epithelial cell line, AA/C1, expression of the butyrate transporter, MCT1, is subject to upregulation by butyrate. This upregulation involves both transcriptional and post-transcriptional mechanisms, and is reflected functionally as an increase in butyrate uptake. These results provide a critical step towards a detailed analysis of the regulatory mechanisms controlling the expression of MCT1, and hence butyrate transport, in the human colon.
Part of this work was presented as an ‘invited lecture’ at the Experimental Biology Meeting, Orlando, FL, USA, April 2001, in the session entitled ‘Adaptive Response of Epithelial Solute Transporters’ and the American Chemical Society, Agricultural and Food Division, Chicago, IL, USA, August 26–30 2001, in the session entitled ‘Nutrigenomics’.
METHODS
Cells and cell culture
The colonic epithelial cell line, AA/C1, was kindly provided by Professor C. Paraskeva, University of Bristol, UK. The cells are derived from a non-tumourigenic human colonic adenoma (Williams et al. 1990). Routinely, AA/C1 cells were maintained at 37 °C in Dulbecco's modified Eagle's medium (Sigma, UK), supplemented with 20 % (v/v) fetal bovine serum, 100 μg ml−1 streptomycin, 100 units ml−1 penicillin, 1 μg ml−1 hydrocortisone sodium succinate, 2 mm glutamine and 0.2 units ml−1 insulin. To verify that they express MCT1, we employed reverse transcription PCR (RT-PCR) to amplify a 545 bp fragment of the MCT1 coding region. The product was cloned, sequenced, and found to be identical to that of MCT1 previously isolated from human colon (Ritzhaupt et al. 1998b). For treatment with monocarboxylates, sodium acetate (NaAc), sodium propionate (NaProp) or sodium butyrate (NaBut) was added to the culture medium to a final concentration of 0–5 mm, and re-supplemented daily. Unless otherwise stated, for harvesting, cells were washed twice in phosphate-buffered saline (PBS) before being scraped from the culture flasks, collected by centrifugation and frozen at −80 °C until use.
Northern analysis
Total RNA was isolated from frozen cells using the RNeasy Kit (Qiagen, UK), according to the manufacturer's recommendations. RNA samples (10 μg per lane) were fractionated through 1 % (w/v) denaturing agarose gels and transferred to uncharged nylon membrane (Duralon, Stratagene, UK) in the presence of 3 m sodium chloride and 0.3 m sodium citrate (20 × SSC). The RNA was fixed to the membranes by UV light irradiation. RNA integrity and equality of loading/transfer was assessed by methylene blue staining of the membranes. The membranes were pre-hybridised for 3 h at 42 °C with ULTRAhyb hybridisation solution (Ambion, USA) in a rotating hybridisation incubator. The membranes were then hybridised for 16 h in the same buffer containing 1 × 106 c.p.m. ml−1 of the appropriate cDNA probe labelled with [α-32P]dCTP using an Oligolabelling kit (Amersham Pharmacia Biotech). Details of MCT1 and β-actin probes are described below (see ‘Nuclear run-on assays’). Subsequently, the membranes were washed three times at 55 °C in a solution consisting of 0.1 × SSC and 0.1 % (w/v) SDS. The washed membranes were exposed to Biomax-MS film (Kodak) for 24 h at −80 °C and subjected to autoradiography. The relative amounts of mRNA were estimated by scanning densitometry of the autoradiograms (Phoretix ID quantifier, Non-linear Dynamics).
Electron microscopic examination of the polarity of AA/C1 cells
Cells were grown to confluence in 35 mm plates and fixed overnight in 2 ml 0.1 m phosphate buffer (pH 7.2) containing 2.5 % (w/v) glutaraldehyde and 4 % (w/v) paraformaldehyde, before incubation in 1 % osmium tetroxide for 5 min. The cells were dehydrated in ethanol and embedded in Taab epoxy resin prior to sectioning (60–90 nm). Sections were counterstained with Reynolds lead citrate solution and uranyl acetate for 5 min and examined by transmission microscopy (Hitachi H600).
Isolation and characterisation of luminal membranes
Luminal membranes were prepared from frozen AA/C1 cells using a combination of cation precipitation and differential centrifugation techniques, as described previously (Shirazi-Beechey et al. 1990; Ritzhaupt et al. 1998a). The final membrane fraction was resuspended in a small volume (20–50 μl) of a buffer consisting of 300 mm mannitol, 20 mm Hepes-Tris pH 7.5 and 0.1 mm magnesium sulphate. Protein concentration of the membranes was determined according to the Biorad technique as described previously (Tarpey et al. 1995). To solubilise the luminal membrane proteins for subsequent Western analysis, the purified luminal membranes were resuspended in sample buffer (62.5 mm Tris-HCl pH 6.8, 0.1 % (w/v) glycerol, 2 % (w/v) SDS, 0.05 % (w/v) β-mercaptoethanol, 0.00125 % (w/v) bromophenol blue) and heated at 65 °C for 3 min. The luminal membrane origin and the purity of the membranes were assessed by the determination of the levels of marker proteins characteristic of the luminal and basolateral membranes, as described previously (Ritzhaupt et al. 1998a).
Antibodies
The antibody to MCT1 was raised in rabbits against the synthetic peptide CQKDTEGGPKEEESPV (corresponding to the C-terminus region of human MCT1), according to the procedure described by Lachman et al. (1986). Animals were humanely killed at the end of the experiment in accordance with UK legislation. The monoclonal antibodies to villin and the Na+−K+−2Cl− cotransporter were purchased from Binding Site (UK) and Developmental Studies Hybridoma Bank (Iowa, USA), respectively.
Western analysis
The abundance of MCT1, villin and the Na+−K+−2Cl− cotransporter in both cellular homogenates and luminal membrane samples was determined by Western blotting. The protein components of AA/C1 cellular homogenates and luminal membranes (5 μg per lane) were separated on 8 % (w/v) polyacrylamide gels containing 0.1 % (w/v) SDS before being electrotransferred to nitrocellulose membrane (Trans-blot, Bio-Rad, UK). Non-specific protein binding sites were blocked by immersion of the nitrocellulose in PBS-TM (PBS, 0.05 % (v/v) Tween 20, 2 % (w/v) non-fat dry milk) for 1 h at room temperature. The nitrocellulose was then incubated at room temperature for 1 h with the appropriate primary antibodies (1 : 5000 in PBS-TM for MCT1, 1 : 1000 for villin and 1 : 10 000 for Na+−K+−2Cl− cotransporter). Horseradish peroxidase-conjugated secondary antibodies (Dako AC, Denmark) were used at a dilution of 1 : 2000. The 40 kDa MCT1 immunoreactive band was specifically blocked when the primary antibody was pre-incubated with the immunising peptide. Immunoblots were developed using the enhanced chemiluminescence (ECL) system (Amersham International, UK) and exposed to Biomax-MR film (Kodak). Band intensities were quantified by scanning densitometry.
Characterisation of butyrate uptake
AA/C1 cells were grown to confluence in 24-well plates, washed twice in PBS and incubated in equilibration buffer (280 mm mannitol, 10 mm Hepes/Tris pH 7.0) at 37 °C for 30 min. For transport studies, 500 μl of uptake buffer consisting of 278 mm mannitol, 10 mm Mes/Tris pH 6.0 and 1 mm [U-14C]butyrate (specific radioactivity 50 nCi per reaction) was added to each well. Following incubation at 25 °C for appropriate lengths of time (15 s to 30 min), the uptake buffer was aspirated and the cells washed twice with 1 ml of ice-cold stop buffer (10 mm Mes/Tris pH 6.0 and 280 mm mannitol). The cells were incubated with 100 % ethanol for 30 min and the lysate collected for scintillation counting. One hundred microlitres of 0.1 m sodium hydroxide was added to each well, and the solubilised protein quantified by the Biorad technique as described previously (Tarpey et al. 1995). Replacing 278 mm mannitol in the incubation medium with 139 mm of either NaCl or KCl resulted in similar rates and patterns of uptake. The assay was carried out at pH 6 as this is the pH at which optimum activity was observed.
For competition studies, the monocarboxylates NaAc, NaProp and sodium lactate (NaLac) were used at a final concentration of 10 mm. For studies employing MCT1 inhibitors, α-cyano 4-hydroxycinnamate (4-CHC) and p-chloromercuribenzoate (pCMB) were added to a final concentration of 5 and 1 mm, respectively. For studies examining the concentration dependence of butyrate uptake, unlabelled NaBut (0.5–20 mm) was added to the standard uptake buffer containing constant tracer amounts of [U-14C]butyrate. Osmolarity of the uptake buffers was adjusted with varying concentrations of mannitol. Initial rates of uptake were measured over a period of 15 s, and the Vmax and Km values calculated by linear regression analysis from Eadie-Hofstee plots.
Nuclear run-on assays
All steps in the isolation of nuclei were performed at 4 °C. Confluent AA/C1 cells were washed three times in ice-cold PBS, scraped from the culture flasks and collected by centrifugation. The pelleted cells were resuspended in 4 ml lysis solution (10 mm Tris-HCl (pH 7.4), 10 mmNaCl, 3 mm MgCl2 and 0.25 % Nonidet P-40), and incubated on ice for 5 min. Following cell lysis, crude nuclei were collected by centrifugation at 500 g for 5 min (Sorvall, HS-3000 rotor), resuspended in 1 ml of lysis solution, and pelleted by a 5 min centrifugation at 1500 r.p.m. in a bench-top microfuge (Eppendorf, model 5415C). The nuclei were then resuspended in 1 ml of lysis solution, visualised by microscopy and counted. The nuclei were once again collected by centrifugation and resuspended at a concentration of 5 × 107 nuclei in 300 μl of transcription solution (5 mm Tris-HCl pH 8.0, 2.5 mm MgCl2, 150 mm KCl, plus 0.25 mm each of ATP, GTP and CTP, and 250 μCi [α-32P]UTP (3000 Ci mmol−1, 10 μCi μl−1)).
In vitro elongation of nascent RNA was performed by incubation at 30 °C for 20 min. The reactions were stopped by addition of 30 μl of 10 mm CaCl2, 20 μl of DNase I (8.5 mg ml−1) and incubation at 37 °C for 15 min. The mixtures were deproteinised by treatment with 20 μg ml−1 proteinase K for 45 min at 37 °C, followed by two phenol-chloroform extractions. The labelled RNA was precipitated by addition of one volume of 5 m ammonium acetate and two volumes of isopropanol. The precipitated material was collected by centrifugation, resuspended in 100 μl of TE buffer (10 mm Tris-HCl, 1 mm EDTA), and hybridised to slot-blotted cDNA probes as described below. The cDNA probes were: (i) a 545 bp fragment of the MCT1 coding region, prepared by RT-PCR as described previously (Ritzhaupt et al. 1998b), (ii) an 884 bp human β-actin cDNA probe, prepared by PCR using primers BA1 (5′-ATCCTGACCCTGAAGTACC-3′) and BA2 (5′-GATCCACATCTGCTGGAAGG-3′), and (iii) a 1 kb Eco RI/ Xho I restriction fragment of the cyclin-dependent kinase inhibitor (p21) cDNA (American Tissue Culture Collection, USA). Each cDNA probe (7.5 μg) was denatured by boiling for 2 min before immobilisation on a Hybond+ nylon membrane using a slot-blot apparatus (Scotlab, UK). Prior to hybridisation with radiolabelled transcripts, the membranes were incubated for 4 h at 65 °C in 5 ml of ULTRAhyb hybridisation solution. Hybridisation was performed for 36 h at 65 °C in 3 ml of fresh ULTRAhyb solution containing 1 × 107 c.p.m. of the labelled transcripts. Membranes were washed twice for 30 min at 65 °C with 2 × SSC buffer. To remove non-hybridised RNA, the membranes were washed at 37 °C for 15 min in the same solution containing 0.4 μg ml−1 RNase A. Finally, the membranes were washed twice in 2 × SSC at room temperature for 15 min, and exposed to Biomax-MS film at −80 °C for 5 days. The relative amounts of 32P-labelled RNA bound to the cDNA probes were estimated densitometrically and the values were normalised to the β-actin signals.
Statistical analysis
Data are presented as means ± s.e.m of n experiments. Vmax and Km values were obtained by linear regression analysis of Eadie-Hofstee plots.
RESULTS
Butyrate induction of MCT1
We have previously shown that MCT1 is expressed in human colon, that the MCT1 protein is located on the colonocyte luminal membrane, and that it transports butyrate (Ritzhaupt et al. 1998a, b). Given that many nutrient transporters expressed in the intestinal epithelium are subject to nutrient-induced regulation (Shirazi-Beechey, 1996; Ferraris & Diamond, 1997), we sought to investigate whether the expression of MCT1 in the colon is regulated by the monocarboxylates acetate, propionate and butyrate. For these studies, we employed the human colonic epithelial cell line, AA/C1. These adherent cells are derived from non-tumourigenic adenoma, grow as a monolayer, possess many properties of normal human colonic absorptive epithelial cells (Williams et al. 1990; Williams et al. 1993; also see our data below) and express MCT1 (see Methods). Prior to their use in the study of MCT1 expression, the state of polarity of AA/C1 cells was examined by both electron microscopy and by Western analysis of the levels of marker proteins in the isolated luminal membranes (Fig. 1A and B, respectively). Figure 1A shows a typical electronmicrograph of AA/C1 cells, demonstrating that they possess well-defined brush border membranes displaying numerous microvilli. Immunoblotting of isolated AA/C1 luminal membranes (Fig. 1B) demonstrated (i) negligible levels of the Na+−K+−2Cl− cotransporter protein (a recognised marker of the basolateral membrane; Greger, 2000), (ii) 20-fold enrichment in the abundance of villin (a classical colonic luminal membrane marker; Robine et al. 1985), compared with the original cellular homogenates, and (iii) co-localisation of villin and MCT1 proteins. These data indicate that the isolated membranes originated from the luminal membrane of AA/C1 cells and that MCT1 is predominantly located within this domain. Furthermore, the location of MCT1 on the luminal membrane of AA/C1 cells correlates with its expression in human colonic tissue, as assessed by Western blotting (Ritzhaupt et al. 1998b) and immunocytochemistry (D. W. Lambert, I. S. Wood & S. P. Shirazi-Beechey, manuscript submitted).
Figure 1. Examination of membrane polarity in AA/C1 cells.
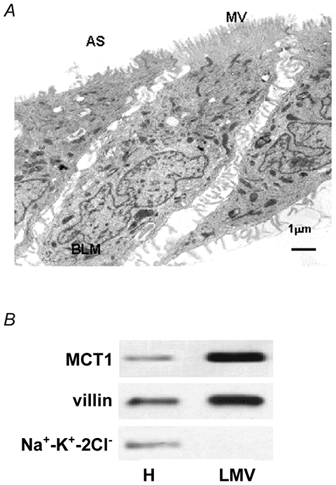
A, a representative electron micrograph of a confluent monolayer of AA/C1 cells. AS, apical surface; BLM, basolateral membrane; MV, microvilli. B, a representative immunoblot for the villin, Na+−K+−2Cl− and MCT1 proteins in AA/C1 luminal membranes (LMV) and cellular homogenates (H). Electron microscopy and Western analysis were performed as described in Methods.
Having demonstrated the AA/C1 cell line to be a suitable in vitro model with which to study the regulation of MCT1 expression, we examined the potential effect of the three monocarboxylates, acetate, propionate and butyrate, on the levels of MCT1 mRNA and protein. The effect of 5 mm NaAc, NaProp and NaBut on the expression of MCT1 mRNA and protein is presented in Fig. 2A. Neither NaAc nor NaProp had any significant effect on MCT1 expression compared with untreated control cells (Fig. 2A, lanes b and c). However, treatment with NaBut resulted in upregulation of both MCT1 mRNA and protein (Fig. 2A, lane d). Treatment with increasing concentrations of NaBut (0–5 mm) revealed the effects to be dose dependent, with the increases maximal at 5 mm NaBut (Fig. 2B). The similarity in the magnitude of the increases in MCT1 mRNA (6.7 ± 0.4-fold) and protein (6.2 ± 0.5-fold) suggests that treatment with butyrate leads to an increase in abundance of the MCT1 transcript, and that this in turn gives rise to a corresponding increase in MCT1 protein.
Figure 2. Effect of monocarboxylates on MCT1 expression.
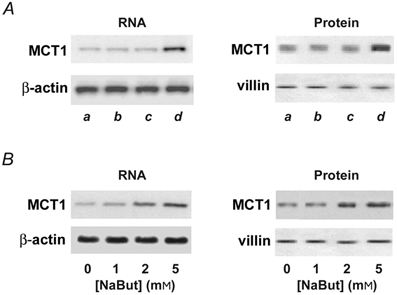
A, effect of NaAc, NaProp and NaBut on MCT1 expression. Confluent AA/C1 cells were either left untreated (a), or treated with 5 mm NaAc (b), 5 mm NaProp (c) or 5 mm NaBut (d) for 48 h. Total RNA and luminal membranes were isolated and subjected to Northern blotting (left panel) or Western blotting (right panel) for MCT1. B, dose-response for induction of MCT1 by NaBut. Confluent AA/C1 cells were either grown for 48 h in the presence of 1–5 mm NaBut or left untreated as controls. Blots shown are representative of 3 experiments. β-Actin and villin served as internal loading controls.
We next examined the kinetics of the induction by NaBut. For these studies, we employed NaBut at the minimum concentration (2 mm) required to give a significant upregulation. MCT1 mRNA induction began at 12 h and continued to increase throughout the period of study (Fig. 3A). Induction of MCT1 protein followed a similar but delayed time course; the initial increase was observed at 24 h (Fig. 3B). The abundance of the β-actin and villin loading controls remained essentially unchanged.
Figure 3. Kinetics of induction of MCT1 expression by NaBut.
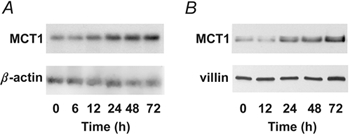
A, time course of induction of the MCT1 transcript. B, time course of induction of the MCT1 protein. Confluent AA/C1 cells were treated with 2 mm NaBut for increasing amounts of time (0–72 h), and MCT1 mRNA and protein abundance was examined by Northern and Western analysis, respectively. β-Actin and villin served as internal loading controls.
Effect of MCT1 induction on butyrate transport
To examine whether the butyrate induction of MCT1 abundance was reflected functionally as an increase in butyrate transport, uptake studies were performed using radiolabelled butyrate. As a requisite to these studies, we first examined the basic characteristics of the transport of butyrate into AA/C1 cells. [U-14C]Butyrate uptake was linear up to 2 min (Fig. 4A, inset) and pH sensitive, with optimal uptake at pH 6 (data not shown). In addition, the uptake was inhibited by a range of monocarboxylates, and the MCT1 inhibitors, 4-CHC and pCMB (Fig. 4B). These characteristics are consistent with those previously reported for MCT1 in human colonic tissue (Ritzhaupt et al. 1998a). To examine the effect of induction of the MCT1 protein on subsequent [U-14C]butyrate uptake, we determined the rate of uptake in AA/C1 cells that had either been pre-treated with unlabelled 2 mm NaBut for 30 h, or maintained under standard conditions for the same period. As shown in Fig. 5, prior exposure of AA/C1 cells to NaBut resulted in a 5-fold increase in the rate of butyrate uptake (Vmax = 310 ± 41 nmol min−1 mg−1) relative to controls (Vmax = 65 ± 8.5 nmol min−1 mg−1). The Km remained essentially unchanged at 9.1 ± 0.8 mm for untreated control cells and 9.8 ± 1.2 mm for cells pre-treated with NaBut.
Figure 4. Characterisation of butyrate transport into AA/C1 cells.
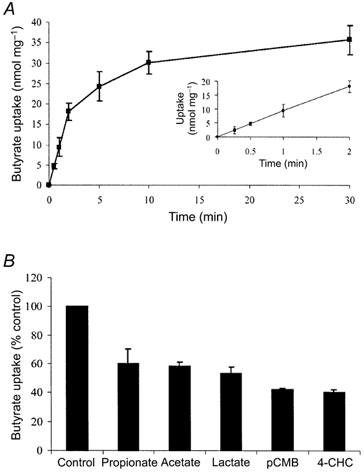
Transport of butyrate into confluent AA/C1 cells was assessed as described in Methods. A, time course of uptake of [U-14C]butyrate. B, effect of monocarboxylates and MCT1 inhibitors on butyrate uptake. Data are presented as the mean ± s.e.m of 6 experiments.
Figure 5. Concentration dependence of butyrate uptake in control and butyrate pre-treated cells.
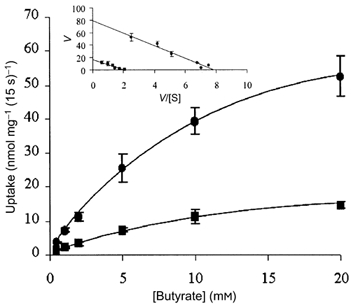
AA/C1 cells grown to confluence in 24-well plates were either pre-treated with 2 mm butyrate (•) for 30 h or left untreated as controls (▪). Concentration-dependent [U-14C]butyrate uptake was determined for 15 s at pH 6. Inset, Eadie-Hoftsee plot used to determine Km and Vmax values. V, velocity; [S], concentration of the substrate. Values are means ± s.e.m. of 6 experiments.
Transcriptional induction of MCT1 expression by butyrate
The steady-state abundance of a specific mRNA species is a direct reflection of the relative rates of transcription and transcript decay (Day & Tuite, 1998). Accordingly, regulation of mRNA abundance can result from a change in either (or both) of these processes. Therefore we sought to investigate the specific molecular basis of the butyrate-induced increase in MCT1 mRNA. We first analysed the effect of NaBut specifically on MCT1 gene transcription. Nuclear run-on reactions were performed using nuclei isolated from AA/C1 cells that had either been treated with 2 mm NaBut for 30 h or maintained in standard culture conditions for the same period. The radiolabelled nascent RNA transcripts were isolated and hybridised to immobilised MCT1 and β-actin cDNA probes. As a positive control for transcriptional induction by NaBut, a cDNA fragment of p21Waf1/Cip1 (p21) was also included on the nylon membrane. p21 is a cyclin-dependent kinase inhibitor that has been shown to be regulated at the level of transcription by NaBut (Nakano et al. 1997; Hassig et al. 1997). Analysis of the resulting hybridisation signals revealed that NaBut increased transcription of MCT1 and p21 approximately 3-fold and 2-fold, respectively (Fig. 6). These results indicate that induction of MCT1 mRNA by NaBut is, at least in part, due to an increase in transcription. However, given that the magnitude of the observed transcriptional induction accounts for only about half of the overall increase in MCT1 mRNA levels previously detected by Northern analysis, these data also indicate that additional mechanisms may contribute to the induction.
Figure 6. Nuclear run-on assay demonstrates butyrate-induced transcription of MCT1.
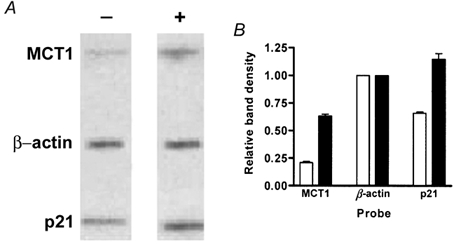
AA/C1 colonic epithelial cells were either incubated for 30 h in the presence of 2 mm NaBut or left untreated as controls. Nuclei were isolated and nuclear run-on reactions were performed as described in Methods. The reaction products were purified and hybridised to immobilised cDNA probes. A, a representative autoradiogram of the hybridising material. -, control; +, NaBut treated. MCT1, human MCT1 cDNA; p21, human p21 cDNA; β-actin, human β-actin cDNA. B, combined densitometric analyses of autoradiograms obtained from hybridisation with labelled RNA from butyrate-treated (▪) or control cells (□) in 3 independent experiments. Error bars represent s.e.m. The results were obtained by adding equal amounts of incorporated radioactivity to each blot, and normalisation to the β-actin signal, which was assigned a value of 1.
To investigate this further, we examined the effect of the inhibition of transcription on the butyrate-induced increase in MCT1 protein as measured by Western blotting. AA/C1 cells were treated with 2 mm NaBut in the presence of the transcriptional inhibitor actinomycin D. Parallel control experiments were performed using untreated cells, and cells treated with either NaBut or inhibitor alone. Although MCT1 protein abundance was increased by treatment with NaBut (Fig. 7, lane 2), this increase was significantly inhibited (60 ± 4.2 %) by the simultaneous presence of actinomycin D (Fig. 7, lane 3). Whilst these data further support a role for transcription, the failure of the inhibition of transcription to completely abolish the induction also indicates the involvement of other post-transcriptional control mechanisms.
Figure 7. Inhibition of butyrate induction of MCT1 by actinomycin D.
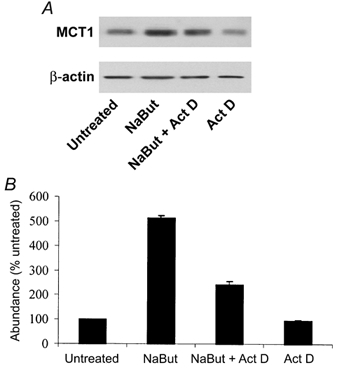
Effect of the inhibition of transcription on butyrate induction of MCT1 protein. Confluent AA/C1 cells were maintained in standard culture conditions (lane 1) or treated for 30 h with 2 mm NaBut either alone (lane 2) or in the presence of 5 μg ml−1 of the transcriptional inhibitor actinomycin D (Act D, lane 3). A parallel control experiment was performed using cells treated with inhibitor alone (lane 4). A, a representative immunoblot. B, combined densitometic analyses from 3 independent experiments. Error bars represent s.e.m.
Effect of butyrate on MCT1 mRNA stability
To examine whether the increase in MCT1 mRNA induced by butyrate is confined to the activation of transcription or also involves an increase in mRNA stability, we investigated the effect of butyrate on the half-life of the MCT1 transcript. To this end, AA/C1 cells were treated with 2 mm butyrate for 30 h followed by addition of the transcriptional inhibitor actinomycin D. The decay in MCT1 mRNA after inhibition of RNA synthesis was measured by Northern analysis as a function of time (Fig. 8A). In parallel, control experiments were performed with cells maintained in the absence of butyrate. In Fig. 8B, the levels of MCT1 mRNA are plotted as a function of time after actinomycin D addition, allowing the estimation of the relative half-life of the MCT1 transcript. The mean values obtained were 4.8 h for MCT1 from the control cells and 9.7 h from the cells treated with butyrate. The data indicate that, in addition to increasing transcription, butyrate also stabilises MCT1 mRNA by a factor of about 2. Taken together, this 2-fold increase in transcript stability and the 3-fold increase in transcription correlate well with the overall increase (5.7-fold at 2 mm NaBut) in MCT1 mRNA levels detected by Northern analysis.
Figure 8. Analysis of the effect of butyrate on the half-life of the MCT1 transcript.
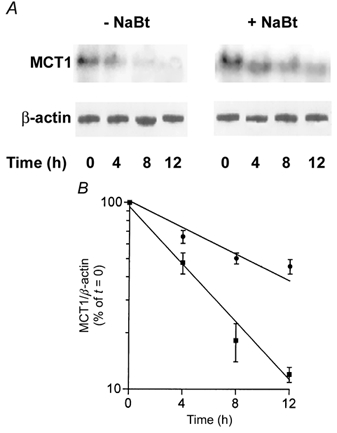
Decay in MCT1 transcript abundance following the addition of the transcription inhibitor, actinomycin D. AA/C1 cells were incubated in the presence or absence of 2 mm NaBut for 30 h prior to the addition of actinomycin D (5 μg ml−1). RNA was isolated at the time of actinomycin D addition (t = 0) or at 4, 8 and 12 h thereafter, and analysed by Northern analysis for MCT1 and β-actin. A, a representative autoradiogram. B, combined densitometric scan data from 3 independent experiments expressed graphically as a percentage of MCT1/β-actin at t = 0, in the form of means ± s.e.m. ▪, untreated control cells; •, butyrate-treated cells.
DISCUSSION
We have previously provided evidence that transport of butyrate into the absorptive cells lining the lumen of the colon is mediated by the monocarboxylate transporter, MCT1 (Ritzhaupt et al. 1998b). In this study, we have investigated the regulation of MCT1 expression by the three principal monocarboxylates found in the lumen of the colon. Whilst acetate and propionate had no effect, butyrate significantly and similarly enhanced the levels of both MCT1 mRNA and protein, and this was reflected functionally as an increase in butyrate transport. The similarity in the magnitude of the increases in MCT1 mRNA and protein suggests that the primary mechanism of regulation is the control of mRNA abundance, and by extension, that translational and post-translational regulation of MCT1 plays little or no part in the induction. Similarly, the close agreement in the enhanced levels of butyrate uptake and abundance of the MCT1 protein suggests that the primary route for butyrate-regulated uptake into AA/C1 cells is via MCT1. Furthermore, the demonstration that the kinetic basis for the enhanced butyrate uptake is an increase in Vmax (with no significant change in the apparent Km) indicates that the response results from an increase in the number of MCT1 molecules, rather than in affinity for butyrate.
Given that many of the cellular effects of butyrate are concentration dependent (Hague et al. 1993; Archer et al. 1998), the ability of butyrate to exert these effects may be dependent upon its intracellular concentration. By extension, factors affecting the intracellular accumulation of butyrate have the potential to influence its availability to modulate processes such as proliferation and apoptosis. Accordingly, it has been proposed that one such factor is the rate of removal of butyrate from the cytoplasm via β-oxidation (Jass, 1985), and this hypothesis has received direct experimental support (Singh et al. 1997). We suggest that a more significant consideration is the rate of butyrate uptake, and that regulation of MCT1 may therefore be an important determinant of the specific response to butyrate. Thus, in addition to economically maximising the calorific potential of luminal butyrate, an upregulation of MCT1 may play an important role in maintaining homeostasis in the colonic epithelium.
Concerning the mode of regulation of MCT1, the nuclear run-on data show that butyrate significantly increases MCT1 gene transcription. However, the discrepancy between the increase in the rate of transcription and the overall increase in mRNA levels indicates that other post-transcriptional mechanisms also contribute to the induction. This is supported by the inability of the inhibition of transcription to completely prevent the induction, and confirmed by the demonstration that butyrate also increases the half-life of the MCT1 transcript. Similar effects of butyrate, namely modulation of both transcription and mRNA stability, have been reported for the expression of tissue-type plasminogen activator mRNA in human endothelial cells (Arts et al. 1995), and carcinoembryonic-antigen in colorectal carcinoma cells (Saini et al. 1990). The specific significance of the dual control of MCT1 mRNA levels is unknown; however, it may provide a more efficient regulation than might otherwise be possible using either mechanism alone.
Modulation of gene transcription by butyrate is generally attributed to its ability to induce hyperacetylation of histones by the inhibition of histone deacetylase (Sealy & Chalkley, 1978; Candido et al. 1978; Kruh, 1982). Hyperacetylation is thought to result in a more ‘relaxed’ chromatin structure that facilitates transcription factor access to their DNA binding motifs, leading to transcriptional activation (Grunstein, 1997). Besides having a direct effect via histone acetylation, butyrate may also affect transcription by the induction of transcription factors, which in turn activate expression of other genes (Nishina et al. 1993). In either case, the identification of the mechanism by which butyrate enhances MCT1 transcription will require the isolation and characterisation of the MCT1 gene promoter. Similarly, further study of the mechanisms by which butyrate regulates stability requires the identification of proteins that interact with the MCT1 transcript. Interestingly, we have recently isolated the 3′-UTR of the MCT1 transcript and have identified potential regulatory elements (authors' unpublished data). Work is in progress to characterise these elements and identify regulatory protein factors.
It has been reported that MCT1 has the ability to transport a number of monocarboxylates in addition to butyrate; these include acetate and propionate (Tamai et al. 1999; Juel & Halestrap, 1999). In addition, there are some reports that both acetate and propionate can have similar, but less potent, effects to butyrate on colonic epithelial cells (Hague et al. 1995; Tran et al. 1998). The finding that neither acetate nor propionate affected MCT1 expression may therefore be a consequence of their lesser potency compared with butyrate. However, preliminary studies in our laboratory, using higher concentrations of acetate and propionate, indicate this not to be the case (authors' unpublished data). Nevertheless, the demonstration that butyrate uptake into AA/C1 cells was inhibited by both acetate and propionate raises the possibility that these could act as indirect regulators by influencing subsequent butyrate-induced upregulation of MCT1 expression. The potential significance of this is that the relative amounts of acetate, propionate and butyrate differ according to the specific source of fermentable substrate in the colon (Macfarlane & Cummings, 1991).
The central position of butyrate in colonic metabolism and the maintenance of tissue homeostasis makes an understanding of the regulation of its transport across the colonic luminal membrane of particular importance. Indeed, it has been reported that impairment of butyrate transport in the colon may play a role in the development of disease states such as ulcerative colitis and Crohn's disease (Treem et al. 1994), and that expression of MCT1 declines in human colonic tissue during the transition from normality to malignancy (Ritzhaupt et al. 1998b). A better understanding of the regulation of MCT1 expression is therefore likely to have important clinical implications. The work presented here is the first evidence that the expression and activity of colonic MCT1 is regulated by its substrate, butyrate. These findings will provide an important foundation for further studies on the regulation of MCT1 expression in the colon and its subsequent effect on the butyrate-induced expression of genes maintaining homeostasis.
Acknowledgments
We are grateful to Professor C. Paraskeva (University of Bristol) and to Dr K. M. Rocha (University of Sao Paulo) for the provision of AA/C1 cells and the p21 cDNA, respectively. The financial support of the Wellcome Trust, Biotechnology and Biological Sciences Research Council, the University of Liverpool and Tenovus Cancer Fund is gratefully acknowledged.
REFERENCES
- Archer SY, Meng S, Shei A, Hodin RA. p21(WAF1) is required for butyrate-mediated growth inhibition of human colon cancer cells. Proceedings of the National Academy of Sciences of the USA. 1998;95:6791–6796. doi: 10.1073/pnas.95.12.6791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arts J, Lansink M, Grimbergen J, Toet KH, Kooistra T. Stimulation of tissue-type plasminogen activator gene expression by sodium butyrate and trichostatin A in human endothelial cells involves histone acetylation. Biochemical Journal. 1995;310:171–176. doi: 10.1042/bj3100171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai L, Merchant JL. Transcription factor ZBP-89 cooperates with histone acetyltransferase p300 during butyrate activation of p21/waf1 transcription in human cells. Journal of Biological Chemistry. 2000;275:30725–30733. doi: 10.1074/jbc.M004249200. [DOI] [PubMed] [Google Scholar]
- Bugaut M, Bentejac M. Biological effects of short-chain fatty acids in nonruminant mammals. Annual Review of Nutrition. 1993;13:217–241. doi: 10.1146/annurev.nu.13.070193.001245. [DOI] [PubMed] [Google Scholar]
- Candido EP, Reeves R, Davie JR. Sodium butyrate inhibits histone deacetylation in cultured cells. Cell. 1978;14:105–113. doi: 10.1016/0092-8674(78)90305-7. [DOI] [PubMed] [Google Scholar]
- Cummings JH. The importance of SCFA in man. Scandanavian Journal of Gastroenterology. 1984;19:89–99. [PubMed] [Google Scholar]
- Danielsen M, Jackson AA. Limits of adaptation to a diet low in protein in normal man: urea kinetics. Clinical Science. 1992;83:103–108. doi: 10.1042/cs0830103. [DOI] [PubMed] [Google Scholar]
- Day DA, Tuite MF. Post-transcriptional gene regulatory mechanisms in eukaryotes: an overview. Journal of Endocrinology. 1998;157:361–371. doi: 10.1677/joe.0.1570361. [DOI] [PubMed] [Google Scholar]
- Diamond JM, Karasov WH. Adaptive regulation of intestinal nutrient transporters. Proceedings of the National Academy of Sciences of the USA. 1987;84:2242–2245. doi: 10.1073/pnas.84.8.2242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyer J, Hosie KB, Shirazi-Beechey SP. Nutrient regulation of human intestinal sugar transporter (SGLT1) expression. Gut. 1997;41:56–59. doi: 10.1136/gut.41.1.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferraris RP, Diamond JM. Specific regulation of intestinal nutrient transporters by their dietary substrates. Annual Review of Physiology. 1989;51:125–141. doi: 10.1146/annurev.ph.51.030189.001013. [DOI] [PubMed] [Google Scholar]
- Ferraris RP, Diamond JM. Regulation of intestinal sugar transport. Physiological Reviews. 1997;77:257–301. doi: 10.1152/physrev.1997.77.1.257. [DOI] [PubMed] [Google Scholar]
- Garcia CK, Goldstein JL, Pathak RK, Anderson RG, Brown MS. Molecular characterization of a membrane transporter for lactate, pyruvate, and other monocarboxylates: implications for the Cori cycle. Cell. 1994a;76:865–873. doi: 10.1016/0092-8674(94)90361-1. [DOI] [PubMed] [Google Scholar]
- Garcia CK, Li X, Luna J, Francke U. cDNA cloning of the human monocarboxylate transporter 1 and chromosomal localization of the SLC16A1 locus to 1p13. 2-p12. Genomics. 1994b;23:500–503. doi: 10.1006/geno.1994.1532. [DOI] [PubMed] [Google Scholar]
- Greger R. Role of CFTR in the colon. Annual Review of Physiology. 2000;62:467–491. doi: 10.1146/annurev.physiol.62.1.467. [DOI] [PubMed] [Google Scholar]
- Grunstein M. Histone acetylation in chromatin structure and transcription. Nature. 1997;389:349–352. doi: 10.1038/38664. [DOI] [PubMed] [Google Scholar]
- Hague A, Diaz GD, Hicks DJ, Krajewski S, Reed JC, Paraskeva C. bcl-2 and bak may play a pivotal role in sodium butyrate-induced apoptosis in colonic epithelial cells; however overexpression of bcl-2 does not protect against bak-mediated apoptosis. International Journal of Cancer. 1997;72:898–905. doi: 10.1002/(sici)1097-0215(19970904)72:5<898::aid-ijc30>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- Hague A, Elder DJ, Hicks DJ, Paraskeva C. Apoptosis in colorectal tumour cells: induction by the short chain fatty acids butyrate, propionate and acetate and by the bile salt deoxycholate. International Journal of Cancer. 1995;60:400–406. doi: 10.1002/ijc.2910600322. [DOI] [PubMed] [Google Scholar]
- Hague A, Manning AM, Hanlon KA, Huschtscha LI, Hart D, Paraskeva C. Sodium butyrate induces apoptosis in human colonic tumour cell lines in a p53-independent pathway: implications for the possible role of dietary fibre in the prevention of large-bowel cancer. International Journal of Cancer. 1993;55:498–505. doi: 10.1002/ijc.2910550329. [DOI] [PubMed] [Google Scholar]
- Hague A, Paraskeva C. The short-chain fatty acid butyrate induces apoptosis in colorectal tumour cell lines. European Journal of Cancer Prevention. 1995;4:359–364. doi: 10.1097/00008469-199510000-00005. [DOI] [PubMed] [Google Scholar]
- Halestrap A, Price NT. The proton-linked monocarboxylate transporter (MCT) family: structure, function and regulation. Biochemical Journal. 1999;343:281–299. [PMC free article] [PubMed] [Google Scholar]
- Hassig CA, Tong JK, Schreiber SL. Fiber-derived butyrate and the prevention of colon cancer. Chemistry and Biology. 1997;4:783–789. doi: 10.1016/s1074-5521(97)90111-3. [DOI] [PubMed] [Google Scholar]
- Hattenhauer O, Traebert M, Murer H, Biber J. Regulation of small intestinal Na-P(i) type IIb cotransporter by dietary phosphate intake. American Journal of Physiology. 1999;277:G756–762. doi: 10.1152/ajpgi.1999.277.4.G756. [DOI] [PubMed] [Google Scholar]
- Jackson VN, Halestrap AP. The kinetics, substrate, and inhibitor specificity of the monocarboxylate (lactate) transporter of rat liver cells determined using the fluorescent intracellular pH indicator, 2′,7′-bis(carboxyethyl)-5(6)-carboxyfluorescein. Journal of Biological Chemistry. 1996;271:861–868. doi: 10.1074/jbc.271.2.861. [DOI] [PubMed] [Google Scholar]
- Jass JR. Diet, butyric acid and differentiation of gastrointestinal tract tumours. Medical Hypotheses. 1985;18:113–118. doi: 10.1016/0306-9877(85)90043-x. [DOI] [PubMed] [Google Scholar]
- Juel C, Halestrap AP. Lactate transport in skeletal muscle - role and regulation of the monocarboxylate transporter. Journal of Physiology. 1999;517:633–642. doi: 10.1111/j.1469-7793.1999.0633s.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karasov WH, Diamond JM. Adaptation of intestinal nutrient transport. In: Johnson LR, editor. Physiology of the Gastrointestinal Tract. New York: Raven Press; 1987. pp. 1489–1497. [Google Scholar]
- Kruh J. Effects of sodium butyrate, a new pharmacological agent, on cells in culture. Molecular and Cellular Biochemistry. 1982;42:65–82. doi: 10.1007/BF00222695. [DOI] [PubMed] [Google Scholar]
- Lachman PJ, Strangeways L, Vyakarnam A, Evan GI. Raising antibodies by coupling peptides to PPD and immunizing BCG-sensitised animals. Ciba Foundation Symposia. 1986;119:25–57. doi: 10.1002/9780470513286.ch3. [DOI] [PubMed] [Google Scholar]
- Lescale-Maty L, Dyer J, Scott D, Wright EM, Shirazi-Beechey SP. Regulation of ovine intestinal Na+/glucose cotransporter (SGLT1) is dissociated from mRNA abundance. Biochemical Journal. 1993;291:435–440. doi: 10.1042/bj2910435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macfarlane GT, Cummings JH. The colonic flora, fermentation, and large bowel digestive function. In: Phillips SF, Pemberton JH, Shorter RG, editors. The Large Intestine; Physiology, Pathophysiology and Disease. New York: Raven Press Ltd; 1991. pp. 51–92. [Google Scholar]
- Mascolo N, Rajendran VM, Binder HJ. Mechanism of short-chain fatty acid uptake by apical membrane vesicles of rat distal colon. Gastroenterology. 1991;101:331–338. doi: 10.1016/0016-5085(91)90008-9. [DOI] [PubMed] [Google Scholar]
- Nakano K, Mizuno T, Sowa Y, Orita T, Yoshino T, Okuyama Y, Fujita T, Ohtani-Fujita N, Matsukawa Y, Tokino T, Yamagishi H, Oka T, Nomura H, Sakai T. Butyrate activates the WAF1/Cip1 gene promoter through Sp1 sites in a p53-negative human colon cancer cell line. Journal of Biological Chemistry. 1997;272:22199–22206. doi: 10.1074/jbc.272.35.22199. [DOI] [PubMed] [Google Scholar]
- Nishina Y, Suni T, Iwai SA, Kosaka M, Nishimune Y. Induction of jun genes during the reversible changes induced with sodium butyrate on the differentiation of F9 cells. Experimental Cell Research. 1993;208:492–497. doi: 10.1006/excr.1993.1271. [DOI] [PubMed] [Google Scholar]
- Phillips SF, Devroede GJ. International Review of Physiology. Baltimore: University Park Press; 1979. pp. 263–290. [PubMed] [Google Scholar]
- Poole RC, Halestrap A. Reversible and irreversible inhibition, by stilbenedisulphonates, of lactate transport into rat erythrocytes - identification of some new high-affinity inhibitors. Biochemical Journal. 1991;275:307–312. doi: 10.1042/bj2750307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poole RC, Halestrap A. Transport of lactate and other monocarboxylates across the mammalian plasma membranes. American Journal of Physiology. 1993;264:C761–782. doi: 10.1152/ajpcell.1993.264.4.C761. [DOI] [PubMed] [Google Scholar]
- Ritzhaupt A, Ellis A, Hosie KB, Shirazi-Beechey SP. The characterization of butyrate transport across pig and human colonic luminal membrane. Journal of Physiology. 1998a;507:819–830. doi: 10.1111/j.1469-7793.1998.819bs.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritzhaupt A, Wood IS, Ellis A, Hosie KB, Shirazi-Beechey SP. Identification and characterization of a monocarboxylate transporter (MCT1) in pig and human colon: its potential to transport l-lactate as well as butyrate. Journal of Physiology. 1998b;513:719–732. doi: 10.1111/j.1469-7793.1998.719ba.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robine S, Huet C, Moll R, Sahuguillo-Merino C, Coudrier E, Zweibaum A, Louvard D. Can villin be used to identify malignant and undifferentiated normal digestive epithelial cells. Proceedings of the National Academy of Sciences of the USA. 1985;82:8488–8492. doi: 10.1073/pnas.82.24.8488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saini K, Steele G, Thomas P. Induction of carcinoembryonic-antigen-gene expression in human colorectal carcinoma by sodium butyrate. Biochemical Journal. 1990;272:541–544. doi: 10.1042/bj2720541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sealy L, Chalkley R. The effect of sodium butyrate on histone modification. Cell. 1978;14:115–121. doi: 10.1016/0092-8674(78)90306-9. [DOI] [PubMed] [Google Scholar]
- Shirazi-Beechey SP. Molecular biology of intestinal glucose transport. Nutrition Research Reviews. 1995;8:27–41. doi: 10.1079/NRR19950005. [DOI] [PubMed] [Google Scholar]
- Shirazi-Beechey SP. Intestinal sodium-dependent D-glucose co-transporter: dietary regulation. Proceedings of the Nutrition Society. 1996;55:167–178. doi: 10.1079/pns19960018. [DOI] [PubMed] [Google Scholar]
- Shirazi-Beechey SP, Davies AG, Tebbutt K, Dyer J, Ellis A, Taylor CJ, Fairclough P, Beechey RB. Preparation and properties of brush-border membrane vesicles from human small intestine. Gastroenterology. 1990;98:1–12. doi: 10.1016/0016-5085(90)90288-c. [DOI] [PubMed] [Google Scholar]
- Siavoshian S, Segain JP, Kornprobst M, Bonnet C, Cherbut C, Galmiche JP, Blottiere HM. Butyrate and trichostatin A effects on the proliferation/differentiation of human intestinal epithelial cells: induction of cyclin D3 and p21 expression. Gut. 2000;46:507–514. doi: 10.1136/gut.46.4.507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh B, Halestrap AP, Paraskeva C. Butyrate can act as a stimulator of growth or inducer of apoptosis in human colonic epithelial cell lines depending on the presence of alternative energy sources. Carcinogenesis. 1997;18:1265–1270. doi: 10.1093/carcin/18.6.1265. [DOI] [PubMed] [Google Scholar]
- Tamai I, Sai Y, Ono A, Kido Y, Yabuchi H, Takanaga H, Satoh E, Ogihara T, Amano O, Izeki S, Tsuji A. Immunohistochemical and functional characterization of pH-dependent intestinal absorption of weak organic acids by the monocarboxylic acid transporter MCT1. Journal of Pharmacy and Pharmacology. 1999;51:1113–1121. doi: 10.1211/0022357991776804. [DOI] [PubMed] [Google Scholar]
- Tarpey PS, Wood IS, Shirazi-Beechey SP, Beechey RB. Amino acid sequence and the cellular location of the Na +-dependent D-glucose symporters (SGLT1) in the ovine intestinal enterocyte and the parotid acinar cell. Biochemical Journal. 1995;312:293–300. doi: 10.1042/bj3120293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran CP, Familari M, Parker LM, Whitehead RH, Giraud AS. Short-chain fatty acids inhibit intestinal trefoil factor gene expression in colon cancer cells. American Journal of Physiology. 1998;275:G85–94. doi: 10.1152/ajpgi.1998.275.1.G85. [DOI] [PubMed] [Google Scholar]
- Treem WR, Ahsan N, Shoup M, Hyams JS. Fecal short-chain fatty acids in children with inflammatory bowel disease. Journal of Pediatric Gastroenterology and Nutrition. 1994;18:159–164. doi: 10.1097/00005176-199402000-00007. [DOI] [PubMed] [Google Scholar]
- Ugawa S, Sunouchi Y, Ueda T, Takahashi E, Saishin Y, Shimada S. Characterisation of a mouse colonic system Bo+ amino acid transporter related to amino acid absorption in colon. American Journal of Physiology - Gastrointestinal and Liver Physiology. 2001;281:G365–370. doi: 10.1152/ajpgi.2001.281.2.G365. [DOI] [PubMed] [Google Scholar]
- Walker D, Thwaites DT, Simmons NL, Gilbert HJ, Hirst BH. Substrate upregulation of the human small intestinal peptide transporter, hPepT1. Journal of Physiology. 1998;507:697–706. doi: 10.1111/j.1469-7793.1998.697bs.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Levi AJ, Halestrap AP. Substrate and inhibitor specificities of the monocarboxylate transporters of single rat heart cells. American Journal of Physiology. 1996;270:H476–484. doi: 10.1152/ajpheart.1996.270.2.H476. [DOI] [PubMed] [Google Scholar]
- Wessling-Resnick M. Iron transport. Annual Review of Nutrition. 2000;20:129–151. doi: 10.1146/annurev.nutr.20.1.129. [DOI] [PubMed] [Google Scholar]
- Williams AC, Hague A, Manning AM, van der Stappen JWJ, Paraskeva C. In vitro models of human colorectal cancer. Cancer Surveys. 1993;16:15–28. [PubMed] [Google Scholar]
- Williams AC, Harper SJ, Paraskeva C. Neoplastic transformation of a human colonic epithelial cell line: in vitro evidence for the adenoma to carcinoma sequence. Cancer Research. 1990;50:4724–4730. [PubMed] [Google Scholar]