

Abstract
Modulation of the steady-state inactivation and current amplitude by the γ1 subunit of the murine skeletal muscle L-type Ca2+ channel were investigated using the whole-cell patch-clamp technique. Transient expression of the γ1 subunit, but not of the γ2 (stargazin) protein, in primary cultured myotubes from γ1-deficient mice shifted the steady-state inactivation approximately −15 mV, thereby restoring wildtype (WT) steady-state inactivation and current amplitude. The increased Ca2+ current amplitude in γ1-deficient cells was abolished in myotubes from animals of 4 weeks and older whereas the positive shift in steady-state inactivation was independent of mouse age. Raising intracellular cAMP levels using the membrane-permeant analogue 8-Br-cAMP led to an increase in Ca2+ current amplitude in WT cells to the level in γ1-deficient myotubes. There was no effect on the current amplitude in γ1-deficient cells or on the steady-state inactivation in either genotype. Rp-cAMPS, a competitive inhibitor of cAMP-dependent protein kinase, had no effect on the WT Ca2+ current amplitude and steady-state inactivation, but diminished the current amplitude in γ1-deficient myotubes without affecting the steady-state inactivation in these cells. These data show that the increased Ca2+ influx in myotubes lacking the γ1 subunit, due to right-shifted steady-state inactivation and increased L-type Ca2+ current amplitude, is determined by the γ1 subunit. The effect on current amplitude depends on the age of the mice and its cAMP-dependent modulation appears to be controlled by the γ1 subunit.
The skeletal muscle L-type Ca2+ channel consists of the α1S subunit together with the auxiliary β1, α2δ and the γ1 subunits. The γ1 subunit is unique to skeletal muscle and has been shown to be important for some of the biophysical properties of the channel (Freise et al. 2000) but its role in excitation-contraction coupling is less clear (Ursu et al. 2001). We have recently shown that the presence of the γ1 subunit reduces Ca2+ influx in primary cultured skeletal muscle myotubes (Freise et al. 2000). In the absence of the γ1 subunit, the L-type Ca2+ current is increased, presumably due to a higher open probability, and the steady-state inactivation is shifted to more depolarised potentials thereby allowing an increased Ca2+ influx (Freise et al. 2000) in agreement with results obtained with an independently produced γ1-deficient mouse (Ahern et al. 2001).
Since most of the skeletal muscle L-type Ca2+ channels primarily act as voltage sensors (Schwartz et al. 1985), it is conceivable that the γ1 subunit is implicated in gating currents. Charge movement measurements were however unaltered in primary cultured myotubes from fetal wildtype (WT) and γ1-deficient mice (Ahern et al. 2001). T-type Ca2+ channels which are also present in skeletal muscle cells and which activate at test potentials around −40 mV, were observed in some but not all myotubes, regardless of the genotype. These channels were not altered by the γ1-deletion (Freise et al. 2000; Berthier et al. 2001), it is therefore unlikely that the γ1 subunit is part of the T-type Ca2+ channel in skeletal muscle cells.
Expression of Ca2+ channel subunits is not only tissue specific but also developmentally regulated. In rabbit skeletal muscle, α1S, β, α2δ and γ1 transcripts increase 4 weeks after birth (Brillantes et al. 1994) which corresponds to the increase in L-type Ca2+ current density and charge movement seen in the first 4 weeks (Beam & Knudson, 1988b). In contrast, the α1S protein decreases in aged rats and mice (Renganathan et al. 1997; Wang et al. 2000). Similar alterations in Ca2+ channel current during development and ageing have been observed in cardiac myocytes (Brillantes et al. 1994; Liu et al. 2000).
In this study, we investigated whether the changes in the L-type Ca2+ current steady-state inactivation and amplitude in γ1-deficient myotubes are both dependent on the γ1 subunit. We show that transient expression of the skeletal γ1 subunit but not of the γ-like protein stargazin (also called γ2, Letts et al. 1998) in neonatal γ1-deficient myotubes restored WT L-type Ca2+ current steady-state inactivation. In myotubes cultured from mice which were 4 weeks old or older, the current amplitude between the two genotypes was equivalent, however, the steady-state inactivation remained shifted. We also tested whether in γ1-deficient myotubes L-type currents can be further modulated by cAMP. Part of this study has been published in abstract form (Held et al. 2001).
METHODS
Cell culture
All procedures were carried out according to the guidelines of the Animal Welfare Committee of the Universität des Saarlandes. Primary cultured skeletal muscle myotubes from newborn mice were prepared with minor modifications as described previously (Beam & Knudson, 1988a; Freise et al. 2000). Pairs of litter-matched inbred mice were mated to give rise to either WT (+/+) or homozygous γ1-deficient (−/−) offspring. One- to three-day-old mice (unless stated otherwise) were killed by decapitation or by cervical dislocation, limb muscles were minced and incubated with 2 mg ml−1 collagenase I (Sigma, Germany). The cell suspension was centrifuged at 200 g for 9–10 min, the pellet was resuspended in culture medium supplemented with 10 % fetal calf serum, 10 % horse serum, 100 IU ml−1 penicillin and 100 μg ml−1 streptomycin. After 2 days, the medium was replaced by medium with 10 % horse serum, 100 IU ml−1 penicillin and 100 μg ml−1 streptomycin. Cells were used between 5–10 days in vitro.
Electrophysiology
Ca2+ channel currents in skeletal muscle myotubes were recorded as described before (Freise et al. 2000) using the whole-cell patch-clamp configuration of the patch-clamp technique (Hamill et al. 1981). The bath solution contained (mm): TEA-Cl 146, CaCl2 10, MgCl2 1, glucose 10, Hepes 10, pH 7.4 (CsOH). Pipettes were made from borosilicate glass and had resistances between 1.5 and 3.5 MΩ. The pipette solution consisted of (mm): caesium aspartate 145, MgCl2 5, EGTA 20, Mg-ATP 5, Hepes 10, pH 7.2 (CsOH). Ca2+ currents were activated every 5 s by step depolarisation from a holding potential of −90 mV to test potentials from −70 mV to +70 mV in 10 mV increments. L-type Ca2+ channel current was measured at the end of the 400 ms depolarisation. To measure steady-state inactivation, cells were depolarised for 5 s to various prepulse potentials from −100 mV to +20 mV in 20 mV increments. Subsequently, a depolarisation to +20 mV for 400 ms was applied at the end of which the L-type Ca2+ channel current amplitude was measured. Steady-state inactivation curves, normalised to the current density after a prepulse to −100 mV, were averaged and fitted with a Boltzman equation:
with V1/2 being the voltage of half-inactivation, k the slope factor and A the initial current ratio. A P/4 protocol was used in all measurements for linear leak and capacitance subtraction.
Transfection
To obtain the recombinant dicistronic expression plasmids pdi-γ1 and pdi-γ2 carrying the entire protein-coding regions of the murine γ1 (Freise et al. 2000) and γ2 (stargazin, Letts et al. 1998), respectively, and that of GFP (Prasher et al. 1992), the consensus sequence for initiation of translation in vertebrates (Kozak, 1987) was introduced immediately 5′ of the respective translation initiation codon. The resulting cDNA was subcloned in the pCAGGS vector, downstream of the chicken actin promoter (Niwa et al. 1991). The internal ribosomal entry site derived from the encephalomyocarditis virus (Kim et al. 1992) followed by the GFP cDNA containing a ser-65-thr mutation, was then cloned 3′ to the γ1 and γ2 cDNA, respectively. As controls, the same vector was used but without the γ sequences. Transfections were carried out using SuperFect (Qiagen, Germany). Cells were incubated for 2 h at 37 °C with 2 μl ml−1 SuperFect and 2 μl ml−1 vector DNA per 35 mm Petri dish. Transfection efficacy as determined by the number of green fluorescent myotubes 3–5 days after transfection, was approximately 5–10 %.
Materials
8-Br-cAMP was obtained from Sigma, Germany; Rp- and Sp-cyclic 3′,5′- hydrogen phosphorothioate adenosine triethylammonium salt (Rp-cAMPS and Sp-cAMPS, respectively), and cBIMPS (5,6-dichloro-1-β-d-ribofuranosylbenzimidazole-3′, 5′-cyclic monophosphorothioate, Sp-isomer) were from BioLog, Bremen, Germany. The protein kinase inhibitor peptide (PKI)6–22 amide was purchased from Calbiochem. The mPKI 6–24 containing a d-arginine at position 18 and a cyclohexylmethylester group blocking the lateral chain of the aspartic acid at position 24 (Fernandez et al. 1991) was synthesised by Dr W. Nastainczyk (Institut für Medizinische Biochemie and Molekularbiologie, Universität des Saarlandes, Homburg, Germany), the peptide sequence was confirmed by sequence analysis and mass spectroscopy. All other reagents were obtained from Sigma.
RESULTS
Expression of the γ1 subunit in WT and γ1-deficient myotubes
Previously, we have shown that the lack of the skeletal muscle γ subunit (γ1) in cultured myotubes increases the L-type Ca2+ channel current and shifts the steady-state inactivation to more hyperpolarised potentials (Freise et al. 2000). Therefore, more Ca2+ ions will enter the cell upon depolarisation. To confirm that these two effects are due to the lack of the γ1 subunit, the γ1 subunit together with the green fluorescent protein (GFP) was coexpressed in the γ1-deficient myotubes using the dicistronic expression vector pdi-γ1. The internal ribosomal entry site sequence allows simultaneous translation of the γ1 and GFP from one transcript. Thus, transfected cells can be detected unequivocally by the development of green fluorescence. Three to five days after transfection, L-type Ca2+ channel currents at the end of the 400 ms depolarisation and steady-state inactivation were measured and compared with the respective currents in non-transfected cells. The steady-state inactivation was shifted to hyperpolarised potentials when the γ1 subunit was expressed in γ1-deficient myotubes thus resembling the inactivation pattern observed in WT cells (Fig. 1B). The voltage of half-inactivation (V1/2) of the averaged curve was −14.8 mV in WT cells (n = 27), 0.4 mV in γ1-deficient cells (n = 36), the slope factor k was 13.2 mV in WT and 8.0 mV in γ1-deficient myotubes. After transfection of the γ1 subunit, V1/2 was −20.9 mV with k being 15.9 mV (n = 8). Expression of the γ1 subunit in γ1-deficient cells reduced the current amplitude to levels observed in WT cells, without shifting the voltage dependence of activation. The peak current densities (at +20 mV) in γ1-deficient myotubes were −26.5 ± 1.5 pA pF−1 (n = 29) and −17.8 ± 2.9 pA pF−1 (n = 9) in those transiently expressing the γ1 subunit which is not significantly different from the current density in WT cells at the same potential (-20.0 ± 1.4 pA pF−1, n = 30). This is illustrated by the representative current traces at −40 mV and +20 mV in Fig. 1A and by the corresponding current-voltage (I-V) relationship in Fig. 1C. In summary, these results indicate that both effects on skeletal muscle L-type Ca2+ currents observed in γ1-deficient myotubes are directly due to the lack of the γ1 protein and, accordingly can be restored by expression of γ1 in these cells.
Figure 1. Expression of the γ1 subunit in γ1-deficient myotubes restores WT L-type Ca2+ current amplitude and steady-state inactivation.
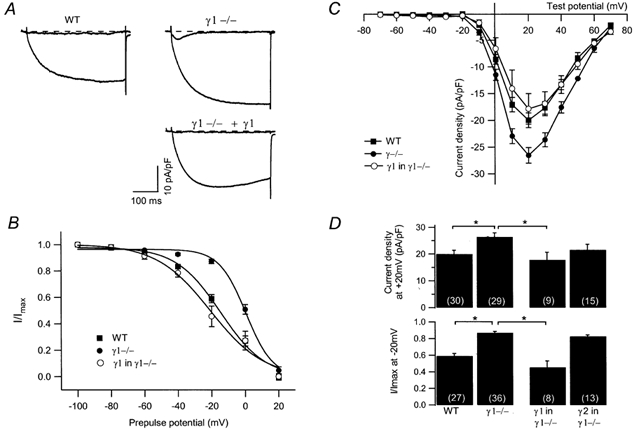
A, representative current traces during a 400 ms depolarisation at −40 mV and at +20 mV from a WT and a γ1-deficient myotube, and from a γ1-deficient cell transfected with the γ1 subunit. The dashed line indicates zero. At −40 mV, a T-type Ca2+ channel is activated in the γ1-deficient cell. B, average steady-state inactivation (normalised to −100 mV prepulse potential) from WT (filled squares, n = 27), γ1-deficient cells (filled circles, n = 36) and from γ1-transfected γ1-deficient cells (open circles, n = 8). Data were fitted with a Boltzman equation. C, average I-V relationships of WT (filled squares, n = 30), γ1−/− (filled circles, n = 29) and γ1-transfected γ1-deficient (open circles, n = 9) myotubes. D, bar graphs showing the current density at +20 mV (top) and the steady-state inactivation after a prepulse of −20 mV (bottom) for WT and γ1-deficient myotubes, γ1-deficient myotubes expressing the γ1or the γ2 protein. Asterisks indicate statistical significance (P < 0.05), values in parentheses indicate number of cells.
To further substantiate this conclusion, we performed two types of control experiments. First, γ1-deficient cells were transfected with the expression vector lacking the DNA and second, the neuronal γ-like protein stargazin which has recently been implicated to represent the γ2 subunit, was expressed in γ1-deficient cells. In control transfections with the vector lacking the γ1 sequence, no effect of GFP expression alone was observed on the steady-state inactivation curve in cells lacking the γ1 subunit (V1/2, 7.2 mV; k, 7.0 mV; n = 10; data not shown), indicating that restoration of the steady-state inactivation was due to the expression of the γ1 subunit and not of GFP. Expression of the structurally related γ2 in γ1-deficient myotubes using the same dicistronic vector described above, had also no effect on the steady-state inactivation curve (Fig. 1D). V1/2 determined from averaged curves was 0.4 (n = 36) and 1.3 mV (n = 13) in the absence and presence of the γ2 subunit, the slope factor k was 8.0 (n = 36) and 6.8 mV (n = 13), respectively. In summary, these results confirm that restoration of wildtype steady-state inactivation properties of ICa is due to the specific action of the γ1 subunit.
GFP expression alone induced a decrease in current density at +20 mV from −26.9 ± 2.3 pA pF−1 (n = 15) in non-transfected cells to −22.6 ± 2.1 pA pF−1 (n = 12; data not shown) which, over the voltage range −10 to +10 mV, was statistically significant (P < 0.05). Likewise, but not significantly, expression of the γ2 protein did decrease the peak Ca2+ current at +20 mV (γ1−/−, −26.5 ± 1.5 pA pF−1, n = 29; γ2 in γ1−/−, −21.6 ± 2.1 pA pF−1, n = 15), whereas at positive potentials from +50 to +70 mV, expression of γ2 in γ1-deficient myotubes resulted in a reduction of L-type Ca2+ current amplitude (P < 0.05). This reduction in current may be due to the GFP expressed as the current amplitude in the presence of γ2 is almost identical to GFP expression alone. Probably, the actions on steady-state inactivation and on current amplitude are independent properties of the γ1 subunit. Expression of the γ1 subunit in γ1-deficient myotubes re-establishes WT L-type Ca2+ current steady-state inactivation, whereas L-type current amplitude might not be influenced.
Age-dependent alterations in L-type Ca2+ channel current in WT and γ1-deficient myotubes
During ageing of mice, a decline in the number of skeletal muscle L-type Ca2+ channels has been described (Renganathan et al. 1998; Wang et al. 2000). In addition, the Ca2+ current amplitude was not found to be enhanced in γ1-deficient myotubes cultured from adult mice (Ursu et al. 2001). This prompted us to investigate whether L-type Ca2+ channel current magnitude and steady-state inactivation are altered during postnatal development. Accordingly, myotubes were cultured from mice 2 weeks, 4 weeks or 4 months of age. These cells grew slower and were therefore kept in high serum medium for 4 days instead of 2 days to allow more time for cell proliferation. Overall there were fewer cells, although most were well differentiated, some forming long strands of fused muscle cells. The data are summarised as a bar graph in Fig. 2A showing the current density at +20 mV for WT and γ1-deficient cells. Myotubes from 2-week-old WT mice had an L-type Ca2+ channel amplitude at +20 mV of −18.7 ± 1.7 pA pF−1 (n = 19), those from γ1-deficient cells of −28.4 ± 1.5 pA pF−1 (n = 22). The current amplitudes of cells from 2-week-old WT and γ1-deficient animals are significantly different between −10 mV and +60 mV. At the age of 4 weeks, however, the differences in L-type Ca2+ current amplitude were no longer present, the amplitude in γ1-deficient cells had reached approximately neonatal WT levels (at +20 mV: WT, −19.9 ± 1.9 pA pF−1, n = 19; γ1-deficient, −21.8 ± 1.5 pA pF−1; n = 21). To ensure that the decrease of L-type amplitude in γ1-deficient myotubes is constant, myotubes from 4-month-old mice were also tested. In WT cells, the current amplitude at +20 mV was slightly but significantly smaller than in neonatal cells (−15.4 ± 1.6 pA pF−1, n = 17, P < 0.05). In γ1-deficient myotubes, the current amplitude (-17.6 ± 1.9, n = 20) was not significantly different from WT cells at the same age or from neonatal WT mice. Thus, the L-type Ca2+ current amplitude in WT myotubes appears to remain constant from birth to adulthood whereas the current in γ1-deficient cells declines after the first 2 weeks of life to WT levels. This is in agreement with results by Ursu et al. (2001) measuring Ca2+ current in myotubes cultured from 2- to 6-month-old mice.
Figure 2. Age dependence of L-type Ca2+ current amplitude and steady-state inactivation.
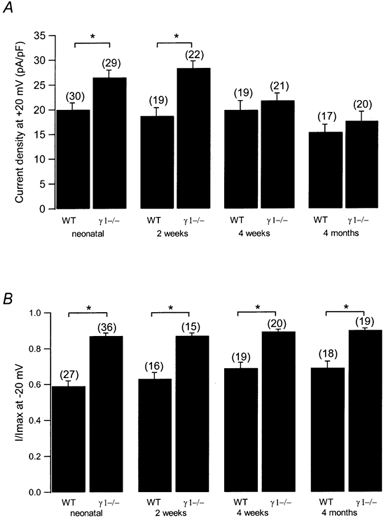
A, current densities at +20 mV are plotted for WT and γ1-deficient myotubes from neonatal, 2-week-old, 4-week-old and 4-month-old mice. B, normalised steady-state inactivation after a 5 s prepulse to −20 mV for WT and γ1-deficient myotubes from neonatal, 2-week-old, 4-week-old and 4-month-old mice. Asterisks indicate statistical significance (P < 0.05), values in parentheses indicate number of cells.
Similarly, the steady-state inactivation was measured as before in neonatal myotubes. In contrast to the changing L-type Ca2+ current amplitude, the steady-state inactivation remained shifted to more depolarised potentials in γ1-deficient cells at all ages examined. In WT myotubes, the V1/2 of the average steady-state inactivation was −14.8, −9.8, −6.7 and −5.4 mV for cells from neonatal, 2-week-old, 4-week-old and 4-month-old mice, respectively, and 0.4, −2.6, 1.8 and 4.4 mV from γ1-deficient mice at the same ages. Also, the slope factor k remained in the same range for WT and γ1-deficient myotubes, respectively (for WT cells: neonatal, 13.2 mV (n = 27); 2 week old, 14.5 mV (n = 16); 4 week old, 13.9 mV (n = 19); 4 month old, 15.3 mV (n = 18); for γ1-deficient cells: neonatal, 8.0 mV (n = 36); 2 week old, 6.8 mV (n = 15); 4 week old, 8.6 mV (n = 20); 4 month old, 9.7 mV (n = 19)). Figure 2B illustrates the average normalised current after a prepulse to −20 mV for 5 s. These results are also in agreement with Ursu et al. (2001) and indicate that L-type Ca2+ current amplitude and steady-state inactivation are regulated by γ1 independently from each other.
Effects of increased cAMP levels using 8-Br-cAMP
Isoprenaline and cAMP enhance 45Ca2+ influx in chick skeletal myotubes (Schmid et al. 1985) and stimulate the L-type Ca2+ channel current in frog muscle fibres (Arreola et al. 1987), as well as in the immortalised mouse skeletal muscle myoblast line 129CBl3 (Johnson et al. 1997). Therefore, we examined, whether the increase in L-type Ca2+ current amplitude in primary myotubes lacking the γ1 subunit from neonatal mice may be related to cAMP-dependent processes. To increase the cAMP levels, 100 μm of the membrane-permeant derivative 8-Br-cAMP was included in the bath solution. After a few minutes, L-type Ca2+ channel currents were measured. As seen in the I-V relationship depicted in Fig. 3A, in WT myotubes, the peak L-type Ca2+ channel amplitude at +20 mV increased by approximately 40 % from −20.0 ± 1.4 pA pF−1 (n = 30) in the absence to −25.0 ± 1.6 pA pF−1 (n = 16) in the presence of 8-Br-cAMP. In γ1-deficient cells, the current amplitude was not changed in the presence of 8-Br-cAMP (in the absence of 8-Br-cAMP, −26.5 ± 1.5 pA pF−1, n = 29; in the presence of 8-Br-cAMP, −27.6 ± 2.8 pA pF−1, n = 18; Fig. 3A). The current densities at +20 mV are summarised in Fig. 3D (top panel). Representative current traces in Fig. 3B illustrate these findings. Thus, only in WT but not in γ1-deficient myotubes, can the L-type Ca2+ channel current amplitude be increased significantly in the presence of 8-Br-cAMP. Cyclic BIMPS and Sp-cAMPS, membrane-permeant cAMP derivatives known to activate preferentially the type II cAMP-dependent protein kinase (cBIMPS) or to activate the cAMP- and in parallel to inhibit cGMP-dependent protein kinases (Sp-cAMPS) (Hofmann et al. 1985), had no effect on peak ICa at +20 mV when added to the bath at concentrations of 100 μm each. These findings may indicate that cAMP-dependent current modulation occurs via the type I cAMP-dependent protein kinase which is the predominant isoform in skeletal muscle (Hofmann et al. 1975). Accordingly, cAMP-dependent Ca2+ current potentiation has been shown not to be altered in mice which are deficient in the type II regulatory subunit of the PKA (Burton et al. 1997).
Figure 3. Effects of 8-Br-cAMP on L-type Ca2+ current amplitude and steady-state inactivation.
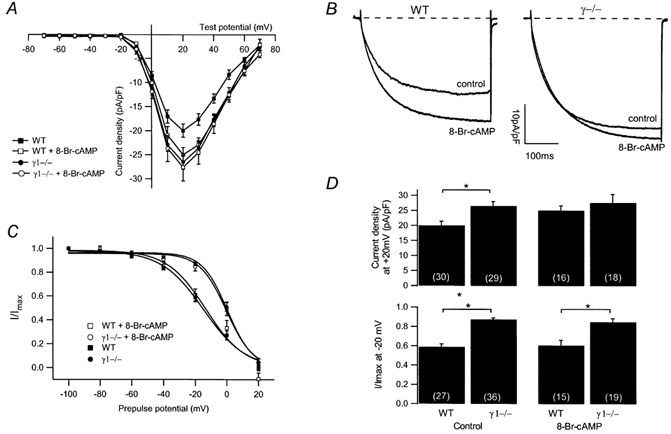
A, averaged I-V relationships in the presence and absence of 100 μm 8-Br-cAMP from WT (filled squares, without 8-Br-cAMP, n = 30; open squares, with 8-Br-cAMP, n = 16) and γ1-deficient (filled circles, without 8-Br-cAMP, n = 29; open circles, with 8-Br-cAMP, n = 18) myotubes. B, representative current traces at +20 mV in the absence (control) or presence of 100 μm 8-Br-cAMP from a WT and a γ1-deficient myotube. Traces of WT and γ1-deficient cells in the absence of 8-Br-cAMP same as in Fig. 1A. C, normalised steady-state inactivation for WT (filled squares, n = 27) and γ1-deficient cells (filled circles, n = 36) under control conditions and in the presence of 8-Br-cAMP (100 μm) (WT, open squares, n = 16; γ1−/−, open circles, n = 18). D, bar graph summarising the effect of 100 μm 8-Br-cAMP on the current density at +20 mV (top) and on the steady-state inactivation after a prepulse of −20 mV (bottom) for WT and γ1-deficient myotubes. Asterisks indicate statistical significance (P < 0.05), values in parentheses indicate number of cells.
In cardiac cells, the steady-state inactivation is shifted to more hyperpolarised potentials by cAMP-dependent processes, presumably via phosphorylation (Tiaho et al. 1991; Petit-Jacques & Hartzell, 1996). To investigate whether cAMP might influence steady-state inactivation in γ1-deficient myotubes, 100 μm 8-Br-cAMP was added to the bath. Averaged normalised steady-state inactivation curves in Fig. 3C compare WT and γ1-deficient myotubes in the absence and presence of 8-Br-cAMP, respectively. V1/2 obtained from the averaged steady-state inactivation curves in WT cells was −14.8 mV with k being 13.2 mV (n = 27) and −13.3 mV with k being 12.3 mV (n = 15) in the absence and presence of 8-Br-cAMP, respectively. Similarly, in γ1-deficient myotubes V1/2 and k were unaltered (in the absence of 8-Br-cAMP: V1/2, 0.4 mV; k, 8.0 mV, n = 36; in the presence of 8-Br-cAMP: V1/2, −0.3 mV; k, 8.4 mV, n = 19). Figure 3D (bottom panel) further illustrates that cAMP has no effect on the steady-state inactivation after a prepulse to −20 mV. Although there is a modulatory effect in WT cells on the L-type Ca2+ channel amplitude, there is no effect on the steady-state inactivation in any cell group. These findings again support the conclusion that in skeletal muscle the Ca2+ channel amplitude and the steady-state inactivation are regulated independently from each other.
Effects of Rp-cAMPS, an cAMP antagonist
The L-type Ca2+ channel current in γ1-deficient myotubes was not modulated by increased cAMP levels, suggesting that in these cells cAMP might already have modulated the current to a maximal extent and could therefore not further increase the Ca2+ influx. With this assumption, the membrane-permeant inhibitor of cAMP-dependent protein kinase Rp-cAMPS should lead to a reduction of the current amplitude in γ1-deficient cells. Rp-cAMPS prevents activation of the cAMP-dependent protein kinase by cAMP. Accordingly, under these conditions, basal phosphorylation should be decreased due to endogenous phosphatase activity and thereby dephosphorylation facilitated. Rp-cAMPS (100 μm) was included in the bath solution and after a preincubation of cells for at least 15 min, L-type Ca2+ channel currents and steady-state inactivation were measured. For this set of experiments, cells from the same preparations were also used without Rp-cAMPS as controls. In the absence of the cAMP antagonist, the peak current density of WT cells at +20 mV was −20.0 ± 1.4 pA pF−1 (n = 30). In the presence of Rp-cAMPS, this was slightly but not significantly decreased (-18.9 ± 1.2 pA pF−1, n = 16) as seen in the I-V relationship in Fig. 4A and the representative current traces in Fig. 4B. In γ1-deficient myotubes however, the current densities were decreased to a level similar to those observed in WT cells (γ1−/−: in the absence of Rp-cAMPS, −26.5 ± 1.5 pA pF−1, n = 29; in the presence of Rp-cAMPS, −21.6 ± 1.7 pA pF−1, n = 20) (Fig. 4A and B). To inhibit cAMP-dependent protein kinase we also perfused cells with two variants of the specific peptide inhibitor of the kinase (Cheng et al. 1985), PKI 6–22 amide and mPKI (Fernandez et al. 1991). These peptides have been chemically modified by an amide group (PKI 6–22 amide) or by incorporation of a d-amino acid and a cyclohexylmethylester group in the aspartic acid at the C-terminus to increase resistance upon proteolytic enzymes. In the presence of either peptide at 100 μm within the pipette solution, there was no significant change of Ca2+ current amplitude in WT or γ1 deficient myotubes when measured at +20 mV 60 s and 3 min after starting the perfusion. The lack of effect could well be explained by diffusion problems of large peptides into the myoblasts from the patch pipette. It has been shown that equilibration of large molecules from the patch pipette into chromaffin cells can take up to 10 min (Pusch & Neher, 1988). In the elongated skeletal muscle myotubes, full equilibration might even take considerably longer, thus explaining the lack of peptide effect
Figure 4. Effects of Rp-cAMPS on L-type Ca2+ current amplitude and steady-state inactivation.
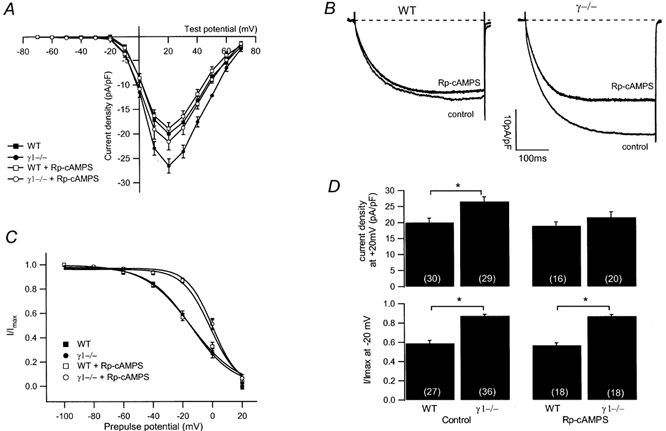
A, averaged I-V relationships in the presence and absence of 100 μm Rp-cAMPS from WT (filled squares, without Rp-cAMPS, n = 30; open squares, with Rp-cAMPS, n = 17) and γ1-deficient (filled circles, without Rp-cAMPS, n = 29; open circles, with Rp-cAMPS, n = 20) myotubes. B, representative current traces at +20 mV in the absence (control) or presence of 100 μm Rp-cAMPS from a WT and a γ1-deficient myotube. Traces of WT and γ1-deficient cells in the absence of Rp-cAMPS same as in Fig. 1A,C, normalised steady-state inactivation for WT (filled squares, n = 27) and γ1-deficient cells (filled circles, n = 36) under control conditions and in the presence of Rp-cAMPS (100 μm) (WT, open squares, n = 18; γ1−/−, open circle, n = 18). D, bar graph summarising the effect of 100 μm Rp-cAMPS on the current density at +20 mV (top) and on the steady-state inactivation after a prepulse of −20 mV (bottom) for WT and γ1-deficient myotubes. Asterisks indicate statistical significance (P < 0.05), values in parentheses indicate number of cells.
cAMP had no effect on the steady-state inactivation of the L-type Ca2+ channel and accordingly both parameters were not changed by Rp-cAMPS in WT or in γ1-deficient myotubes (Fig. 4C). In WT myotubes, in the absence of Rp-cAMPS, V1/2 was −14.8 mV with the slope factor k being 13.2 mV (n = 27), in the presence of the PKA inhibitor, V1/2 was −14.6 mV and k was 15.7 mV (n = 18). In γ1-deficient cells, V1/2 was 0.4 (n = 36) and 0.3 mV (n = 18), with the slope factor k being 8.0 (n = 36) and 8.5 mV (n = 18) in the absence and presence of Rp-cAMPS, respectively. The effect of Rp-cAMPS on current densities at +20 mV and the steady-state inactivation after a 5 s prepulse to −20 mV is summarised in Fig. 4D. Obviously, the current density is decreased in γ1-deficient myotubes but not in WT cells, and in terms of current density a WT current phenotype is restored in the presence of Rp-cAMPS. The steady-state inactivation, however, is not altered by Rp-cAMPS and appears independent from the current amplitude as observed above.
DISCUSSION
Expression of the γ1 subunit in γ1-deficient and WT cells
The role of the skeletal muscle L-type Ca2+ channel γ subunit is, in contrast to the pore-forming α1S and the β subunits, not well defined. The generation of mice lacking this subunit should facilitate these investigations as, in contrast to heterologous expression studies using non-muscle cells, all other muscle cell components are present. Also, the difficulty to express the α1S subunit in heterologous expression studies, where the cardiac α1C subunit has often been used instead (Wei et al. 1991), is avoided. In the absence of the γ1 subunit, the L-type Ca2+ channel current is enhanced and the steady-state inactivation is shifted to more depolarised potentials, resulting in an increased Ca2+ influx (Freise et al. 2000). The enhanced Ca2+ influx is coupled to an increase of the Ca2+ released from the sarcoplasmic reticulum, however, EC coupling in skeletal muscle fibres is not affected (Ursu et al. 2001). To obtain further insight into the mechanisms by which the γ1 subunit alters current density and steady-state inactivation properties, we transiently expressed the γ1 subunit in primary cultured γ1-deficient myotubes. The skeletal muscle γ1 subunit clearly restored WT steady-state inactivation whereas its effect on current density might be influenced by the coexpressed GFP. These results indicate that at least the effect on the steady-state inactivation directly depends on the γ1 subunit; whereas the increase of current density might indirectly be coupled to the lack of γ1 expression (see below).
Altered L-type channel properties in older mice
γ1-Deficient mice show no obvious defects in movement or muscle contraction when compared with WT animals (Freise et al. 2000; Ursu et al. 2001). In addition, Ursu et al. (2001) using myotubes cultured from 2- to 6-month-old mice, found no difference in L-type Ca2+ current amplitude between WT and γ1-deficient cells. Therefore we asked whether the larger Ca2+ influx might change during development and whether current amplitude and steady-state inactivation are independent from one another. Indeed, the L-type Ca2+ current amplitude in γ1-deficient cells declined to WT levels within the first 4 weeks of life whereas the WT L-type Ca2+ current amplitude only slightly declined after 4 months of age. However, the steady-state inactivation was not altered in either WT or γ1-deficient cells during development. These findings parallel results obtained for the steady-state inactivation of the cardiac L-type Ca2+ current which is not changed during development and ageing (Katsube et al. 1996; Liu et al. 2000) as in skeletal myotubes here. Similarly, the maximal current amplitude has been shown to decrease in aged cardiac myocytes (Katsube et al. 1996) as observed here in WT skeletal muscle cells. Thus, contributions of the γ1 subunit to the Ca2+ current amplitude and to the steady-state inactivation appear to be independent mechanisms.
Stargazin cannot substitute γ1
Recently, several γ1-like gene products, γ2 to γ5, have been identified and it has been shown that they may slightly affect time-dependent and steady-state inactivation when coexpressed with the neuronal P/Q-type α1A subunit (Letts et al. 1998; Klugbauer et al. 2000; Rousset et al. 2001). We show here that γ2 (also called stargazin), in contrast to γ1, is not capable of restoring the L-type Ca2+ current or the steady-state inactivation in WT myotubes. Stargazin has also been shown to mediate the delivery of AMPA-receptors to the plasma membrane and further into the synapse (Chen et al. 2000) and it has been questioned whether it indeed constitutes a subunit of voltage-dependent Ca2+ channels. Assuming that stargazin is correctly targeted to the plasma membrane as anticipated from previous studies (Klugbauer et al. 2000; Rousset et al. 2001), it might not colocalise with the skeletal muscle L-type Ca2+ channel complex or if it does, it cannot substitute for the γ1 subunit.
In mice lacking the γ1 subunit, the L-type Ca2+ current amplitude might decrease with age possibly by regulatory mechanisms in order to moderate the overall Ca2+ influx. Alternatively, Ca2+-buffering proteins might be up-regulated or preferentially located near the mouth of the channel in the γ1-deficient myotubes. In γ1-deficient cells, the current magnitude decreases between 2 and 4 weeks after birth whereas in WT muscle cells, the L-type Ca2+ current declines only when the animal becomes much older. Compensatory mechanisms such as up- or down-regulation of a regulatory protein or changes in gene expression of α1S, β, α2δ and γ1 mRNA during development may occur (Brillantes et al. 1994). Accordingly, decreased density of L-type Ca2+ channels in ageing rat muscle fibres has been observed using the dihydropyridine isradipine to determine the density of the DHP receptor (Renganathan et al. 1997) and by measuring charge movement in mice (Wang et al. 2000).
cAMP modulation of the L-type Ca2+ channel
Cyclic AMP-dependent modulation of L-type Ca2+ channels following β-adrenergic stimulation is well studied in cardiac myocytes (Brum et al. 1983; Tsien et al. 1986; McDonald et al. 1994). cAMP enhance 45Ca2+ influx in chick skeletal myotubes (Schmid et al. 1985) and stimulate the L-type Ca2+ channel current in frog skeletal muscle fibres (Arreola et al. 1987), in the immortalised mouse skeletal muscle myoblast line 129CB3 (Johnson et al. 1997) and in colchicin-treated rat myoballs (Sculptoreanu et al. 1993). In this study we show that the L-type Ca2+ current in primary skeletal muscle myotubes from neonatal mice is enhanced in the presence of 8-Br-cAMP added to the bath. In myotubes lacking the γ1 subunit, 8-Br-cAMP has no effect on the current whereas a reduction of the current in these cells is observed in the presence of Rp-cAMPS. cAMP acts on cAMP-dependent protein kinase to modulate Ca2+ channel activity. Correspondingly, kinase activation by cAMP can be blocked in the presence of Rp-cAMPS. The apparent restoration of the WT current phenotype in γ1-deficient myotubes in terms of current density by Rp-cAMPS and the lack of effect in the presence of cAMP may implicate that in the absence of the γ1 subunit, the L-type Ca2+ channel is already fully phosphorylated by cAMP-dependent protein kinase under non-stimulated conditions. The α1S and the β1 subunit of the skeletal muscle Ca2+ channel have been shown to be readily phosphorylated by cAMP-dependent protein kinase in vitro (Curtis & Catterall, 1985; Flockerzi et al. 1986; O'Callahan & Hosey, 1988; Röhrkasten et al. 1988; Chang et al. 1991; Zhao et al. 1994; Rotman et al. 1995). To ascertain whether cAMP-dependent phosphorylation underlies the current increase in response to 8-Br-cAMP in myotubes from neonatal mice, it has to be studied whether the phosphorylation state of these subunits is altered in skeletal muscle in vivo from γ1-deficient mice compared with those from WT animals.
In summary, the level of γ1 subunit regulates the amount of Ca2+ influx by shifting the steady-state inactivation curve to the left and in newborn mice, by also increasing L-type Ca2+ current amplitude. Both these effects of the γ1 subunit are independent of each other as shown by the differential age-dependent changes and the differential modulation by cAMP.
Acknowledgments
We wish to thank Dr S. Philipp for providing the pdi-vector, Drs A. Cavalié and M. Bödding for helpful discussion and K. Fischer, I. Schramm, A. Brendemühl, U. Soltek and M. Simon-Thomas for expert technical assistance. This work was supported, in part, by the Deutsche Forschungsgemeinschaft and Fonds der Chemischen Industrie.
REFERENCES
- Ahern CA, Powers PA, Biddlecome GH, Roethe L, Vallejo P, Mortenson L, Strube C, Campbell KP, Coronado R, Gregg RG. Modulation of L-type Ca2+ current but not activation of Ca2+ release by the gamma1 subunit of the dihydropyridine receptor of skeletal muscle. BioMedCentral Physiology. 2001;1:8. doi: 10.1186/1472-6793-1-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arreola J, Calco J, Garcia MC, Sanchez JA. Modulation of calcium channels of twitch skeletal muscle fibres of the frog by adrenaline and cyclic adenosine monophosphate. Journal of Physiology. 1987;393:307–330. doi: 10.1113/jphysiol.1987.sp016825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beam KG, Knudson CM. Calcium currents in embryonic and neonatal mammalian skeletal muscle. Journal of General Physiology. 1988a;91:781–798. doi: 10.1085/jgp.91.6.781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beam KG, Knudson CM. Effect of postnatal development on calcium currents and slow charge movement in mammalian muscle. Journal of General Physiology. 1988b;91:799–815. doi: 10.1085/jgp.91.6.799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berthier C, Powers PA, Gregg RG, Coronado R, Strube C. Absence of regulation of skeletal muscle T-type calcium channel by dihydropyridine receptor subunits in vivo. Biophysical Journal. 2001;80:546. [Google Scholar]
- Brillantes A-MB, Bezprovannaya S, Marks A. Developmental and tissue-specific regulation of rabbit skeletal and cardiac muscle calcium channels involved in excitation-contraction coupling. Circulation Research. 1994;75:503–510. doi: 10.1161/01.res.75.3.503. [DOI] [PubMed] [Google Scholar]
- Brum G, Flockerzi V, Hofmann F, Osterrieder W, Trautwein W. Injection of catalytic subunit of cAMP-dependent kinase into isolated cardiac myocytes. Pflügers Archiv. 1983;398:147–154. doi: 10.1007/BF00581064. [DOI] [PubMed] [Google Scholar]
- Burton KA, Johnson BD, Hausken ZE, Westenbroek RE, Idzerda RL, Scheuier T, Scott JD, Catterall WA, McKnight GS. Type II regulatory subunits are not required for the anchoring-dependent modulation of Ca2+ channel activity by cAMP-dependent protein kinase. Proceedings of the National Academy of Sciences of theUSA. 1997;94:11067–11072. doi: 10.1073/pnas.94.20.11067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CF, Gutierrez LM, Mundina-Weilenmann C, Hosey MM. Dihydropyridine-sensitive calcium channel from skeletal muscle. II. Functional effects of differential phosphorylation of channel subunits. Journal of Biological Chemistry. 1991;266:16395–16400. [PubMed] [Google Scholar]
- Chen L, Chetkovitch DM, Petralia RS, Sweeney NT, Kawasaki Y, Wenthold RJ, Bred DS, Nicoll RA. Stargazin regulates synaptic targeting of AMPA receptors by two distinct mechanisms. Nature. 2000;408:936–943. doi: 10.1038/35050030. [DOI] [PubMed] [Google Scholar]
- Cheng HC, Kemp BE, Pearson RB, Smith AJ, Misconi L, Van Patten SM, Walsh DA. A potent synthetic peptide inhibitor of the cAMP-dependent protein kinase. Journal of Biological Chemistry. 1986;261:989–992. [PubMed] [Google Scholar]
- Curtis BM, Catterall WA. Phosphorylation of the calcium antagonist receptor of the voltage-sensitive calcium channel by cAMP-dependent protein kinase. Proceedings of the National Academy of Sciences of the USA. 1985;82:2528–2532. doi: 10.1073/pnas.82.8.2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez A, Mery J, Vandromme M, Basset M, Cavadore J-C, Lamb NJC. Effective intracellular inhibition of the cAMP-dependent protein kinase by microinjection of a modified form of the specific inhibitor peptide PKI in living fibroblasts. Experimental Cell Research. 1991;195:468–477. doi: 10.1016/0014-4827(91)90398-e. [DOI] [PubMed] [Google Scholar]
- Flockerzi V, Oeken H-J, Hofmann F, Pelzer D, Cavalié A, Trautwein W. Purified dihydropyridine-binding site from skeletal muscle t-tubules is a functional calcium channel. Nature. 1986;323:66–68. doi: 10.1038/323066a0. [DOI] [PubMed] [Google Scholar]
- Freise D, Held B, Wissenbach U, Pfeifer A, Trost C, Himmerkus N, Schweig U, Freichel M, Biel M, Hofmann F, Hoth M, Flockerzi V. Absence of the γ subunit of the skeletal muscle dihydropyridine receptor increases L-type Ca2+ currents and alters channel inactivation properties. Journal of Biological Chemistry. 2000;275:14476–14481. doi: 10.1074/jbc.275.19.14476. [DOI] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Archiv. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Held B, Freise D, Hoth M, Flockerzi V. Modulation of the skeletal muscle L-type Ca2+ channel current by cAMP and the channel's γ1 subunit. Pflügers Archiv. 2001;441:P6–4. [Google Scholar]
- Hofmann F, Beavo JA, Bechtel PJ, Krebs EG. Comparison of adenosine 3′:5′-monophosphate-dependent protein kinases from rabbit skeletal and bovine heart muscle. Journal of Biological Chemistry. 1975;250:7795–7801. [PubMed] [Google Scholar]
- Hofmann F, Gensheimer HP, Landgraf W, Hullin R, Jastorff B. Diastereomers of adenosine 3′,5′-monothionophosphate (cAMP[S]) antagonize the activation of cGMP-dependent protein kinase. European Journal of Biochemistry. 1985;150:85–88. doi: 10.1111/j.1432-1033.1985.tb08991.x. [DOI] [PubMed] [Google Scholar]
- Johnson BD, Brousal JP, Peterson BZ, Gallombardo PA, Hockermann GH, Lai Y, Scheuer T, Catterall WA. Modulation of the cloned skeletal muscle L-type Ca2+ channel by anchored cAMP-dependent protein kinase. Journal of Neuroscience. 1997;17:1243–1255. doi: 10.1523/JNEUROSCI.17-04-01243.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsube Y, Yokoshiki H, Nguyen L, Sperelakis N. Differences in isoproterenol stimulation of Ca2+ current of rat ventricular myocytes in neonatal compared to adult. European Journal of Pharmacology. 1996;317:391–400. doi: 10.1016/s0014-2999(96)00745-5. [DOI] [PubMed] [Google Scholar]
- Kim DG, Kang HM, Jang SK, Shin HS. Construction of a bifunctional mRNA in the mouse by using the internal ribosomal entry site of the encephalomyocarditis virus. Molecular and Cellular Biolgy. 1992;12:3636–3643. doi: 10.1128/mcb.12.8.3636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klugbauer N, Dai S, Specht V, Lacinova L, Marais E, Bohn G, Hofmann F. A family of γ-like calcium channel subunits. FEBS Letters. 2000;470:189–197. doi: 10.1016/s0014-5793(00)01306-5. [DOI] [PubMed] [Google Scholar]
- Kozak M. At least six nuscleotides preceding the AUG initiator codon enhance translation in mammalian cells. Journal of Molecular Biology. 1987;196:947–950. doi: 10.1016/0022-2836(87)90418-9. [DOI] [PubMed] [Google Scholar]
- Letts VA, Felix R, Biddlecome GH, Arikkath J, Mahaffey CL, Valenzuela A, Bartlett FS, Mori Y, Campbell KP, Frankel WN. The mouse stargazer gene encodes a neuronal Ca2+-channel γ subunit. Nature Genetics. 1998;19:340–347. doi: 10.1038/1228. [DOI] [PubMed] [Google Scholar]
- Liu SJ, Wyeth RP, Melchert RB, Kennedy RH. Ageing-associated changes in whole cell K+ and L-type Ca2+ currents in rat ventricular myocytes. American Journal of Physiology. 2000;279:H889–900. doi: 10.1152/ajpheart.2000.279.3.H889. [DOI] [PubMed] [Google Scholar]
- McDonald TF, Pelzer S, Trautwein W, Pelzer D. Regulation and modulation of calcium channels in cardiac, skeletal, and smooth muscle cells. Physiological Reviews. 1994;74:365–507. doi: 10.1152/physrev.1994.74.2.365. [DOI] [PubMed] [Google Scholar]
- Niwa H, Yamamura K, Miyazaki J. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene. 1991;108:193–199. doi: 10.1016/0378-1119(91)90434-d. [DOI] [PubMed] [Google Scholar]
- O'Callahan CM, Hosey MM. Multiple phosphorylation sites in the 165-kilodalton peptide associated with dihydropyridine-sensitive calcium channels. Biochemistry. 1988;27:6071–6077. doi: 10.1021/bi00416a036. [DOI] [PubMed] [Google Scholar]
- Petit-Jacques J, Hartzell HC. Effect of arachidonic acid on the L-type calcium current in frog cardiac myocytes. Journal of Physiology. 1996;493:67–81. doi: 10.1113/jphysiol.1996.sp021365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasher DC, Eckenrode VK, Ward WW, Prendergast FG, Cormier MJ. Primary structure of the Aequorea victoria green-flourescent protein. Gene. 1992;111:229–233. doi: 10.1016/0378-1119(92)90691-h. [DOI] [PubMed] [Google Scholar]
- Pusch M, Neher E. Rates of diffusional exchange between small cells and a measuring patch pipette. Pflügers Archiv. 1988;411:204–211. doi: 10.1007/BF00582316. [DOI] [PubMed] [Google Scholar]
- Renganathan M, Messi ML, Delbono O. Dihydropyridine receptor-ryanodine receptor uncoupling in aged skeletal muscle. Journal of Membrane Biology. 1997;157:247–253. doi: 10.1007/s002329900233. [DOI] [PubMed] [Google Scholar]
- Röhrkasten A, Meyer HE, Nastainczyk W, Sieber M, Hofmann F. cAMP-dependent protein kinase rapidly phosphorylates serine-687 of the skeletal muscle receptor for calcium channel blockers. Journal of Biological Chemistry. 1988;263:15325–15329. [PubMed] [Google Scholar]
- Rotman EI, Murphy BJ, Catterall WA. Sites of selective cAMP-dependent phosphorylation of the L-type calcium channel α1 subunit from intact rabbit skeletal muscle myotubes. Journal of Biological Chemistry. 1995;270:16371–16377. doi: 10.1074/jbc.270.27.16371. [DOI] [PubMed] [Google Scholar]
- Rousset M, Cens T, Restituito S, Barrere C, Black J L III, McEnery MW, Charnet P. Functional roles of γ2, γ3 and γ4, three new Ca2+ channel subunits, in P/Q-type Ca2+ channel expressed in Xenopus oocytes. Journal of Physiology. 2001;523:583–593. doi: 10.1111/j.1469-7793.2001.0583e.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid A, Renaud J-F, Lasdunski Short term and long term effects of β-adrenergic effectors and cyclic AMP on nitrendipine-sensitive voltage-dependent Ca2+ channels of skeletal muscle. Journal of Biological Chemistry. 1985;260:13041–13046. [PubMed] [Google Scholar]
- Schwartz LM, McCleskey EW, Almers W. Dihydropyridine receptors in muscle are voltage-dependent but most are not functional calcium channels. Nature. 1985;314:747–751. doi: 10.1038/314747a0. [DOI] [PubMed] [Google Scholar]
- Sculptoreanu A, Scheuer T, Catterall WA. Voltage-dependent potentiation of L-type Ca2+ channel due to phosphorylation by cAMP-dependent protein kinase. Nature. 1993;364:240–243. doi: 10.1038/364240a0. [DOI] [PubMed] [Google Scholar]
- Tiaho F, Nargeot J, Richard S. Voltage-dependent regulation of L-type cardiac Ca channels by isoproterenol. Pflügers Archiv. 1991;419:596–602. doi: 10.1007/BF00370301. [DOI] [PubMed] [Google Scholar]
- Tsien RW, Bean BP, Hess P, Lansman JB, Nilius B, Nowycky MC. Mechanisms of calcium channel modulation by β-adrenergic agents and dihydropyridine calcium agonists. Journal of Molecular and Cellular Cardiology. 1986;18:691–710. doi: 10.1016/s0022-2828(86)80941-5. [DOI] [PubMed] [Google Scholar]
- Ursu D, Sebille S, Dietze B, Freise D, Flockerzi V, Melzer W. Excitation-contraction coupling in skeletal muscle of a mouse lacking the DHP receptor subunit γ1. Journal of Physiology. 2001;533:367–377. doi: 10.1111/j.1469-7793.2001.0367a.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z-M, Messi ML, Delbono O. L-type Ca2+ channel movement and intracellular Ca2+ in skeletal muscle fibres from ageing mice. Biophysical Journal. 2000;78:1947–1954. doi: 10.1016/S0006-3495(00)76742-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wie X, Perez-Reyes E, Lacerda AE, Schuster G, Brown AM, Birnbaumer L. Heterologous regulation of the cardiac Ca2+ channel α1 subunit by skeletal muscle β and γ subunits. Journal of Biological Chemistry. 1991;266:21943–21947. [PubMed] [Google Scholar]
- Zhao X, Gutierrez LM, Chang CF, Hosey MM. The α1-subunit of skeletal muscle L-type Ca channels is the key target for regulation by A-kinase and protein phosphatase-1c. Biochemical and Biophysical Research Communications. 1994;198:166–173. doi: 10.1006/bbrc.1994.1024. [DOI] [PubMed] [Google Scholar]