

Abstract
Glycine receptors (GlyRs) are transmitter-gated channels that mediate fast inhibitory neurotransmission in the spinal cord and brain. The GlyR β subunit contains a putative tyrosine phosphorylation site whose functional role has not been determined. To examine if protein tyrosine kinases (PTKs) regulate the function of GlyRs, we analysed whole-cell currents activated by applications of glycine to CA1 hippocampal neurons and spinal neurons. The role of a putative site for tyrosine phosphorylation at position 413 of the β subunit was examined using site-directed mutagenesis and expression of recombinant (α1βY413F) receptors in human embryonic kidney (HEK 293) cells. Lavendustin A, an inhibitor of PTKs, depressed glycine-evoked currents (IGly) in CA1 neurons and spinal neurons by 31 % and 40 %, respectively. In contrast, the intracellular application of the exogenous tyrosine kinase, cSrc, enhanced IGly in CA1 neurons by 56 %. cSrc also accelerated GlyR desensitization and increased the potency of glycine 2-fold (control EC50 = 143 μm; cSrc EC50 = 74 μm). Exogenous cSrc, applied intracellularly, upregulated heteromeric α1β receptors but not homomeric α1 receptors. Substitution mutation of the tyrosine to phenylalanine at position β-413 prevented this enhancement. Furthermore, a selective inhibitor of the Src family kinases, PP2, down-regulated wild-type α1β but not α1βY413F receptors. Together, these findings indicate that GlyR function is upregulated by PTKs and this modulation is dependent on the tyrosine-413 residue of the β subunit.
Reversible phosphorylation of protein targets by receptor and non-receptor PTKs was initially shown to influence signalling pathways involved in cell maturation and ontogeny. Recently, tyrosine phosphorylation was shown to rapidly regulate the function of ligand-gated channels involved in fast synaptic transmission. In particular, the Src family of endogenous PTKs upregulate inhibitory GABAA receptors (Moss et al. 1995; Wan et al. 1997) and excitatory NMDA receptor function (Ali & Salter, 2001). GlyRs are transmitter-gated anion channels with similar structural and biophysical properties as GABAA receptors; however, the functional consequence of GlyR modulation by tyrosine phosphorylation has not been previously examined.
Glycine is a major inhibitory transmitter in the spinal cord, brainstem and other parts of the brain. Activation of ionotropic GlyRs increases chloride conductance, hyperpolarizes the membrane and reduces neuronal excitability (for review see Legendre, 2001). Glycinergic inhibition regulates motor and sensory pathways involved in physiological processes such as locomotion, the coordination of spinal reflexes and nociception. Adult GlyRs are composed of α1–4 (48–50 kDa) and β (58 kDa) subunits that form hetero-oligomeric complexes (3α:2β). Only the α subunits form functional homomeric channels that contain binding sites for agonists and competitive antagonists (Grenningloh et al. 1990; Pribilla et al. 1992; Handford et al. 1996). The β subunit appears to serve a regulatory role by influencing receptor anchoring (Meyer et al. 1995), the allosteric modulation by pharmacological agents (Pribilla et al. 1992; Handford et al. 1996) and post-translational modifications including receptor phosphorylation (Grenningloh et al. 1990).
Cyclic AMP-dependent protein kinase (PKA), protein kinase C (PKC) and calcium-dependent calmodulin kinase II (CaMKII) have been shown to change the amplitude of IGly. Furthermore, PKA and PKC phosphorylate the α subunit (for review see Legendre, 2001). Although a putative consensus site for tyrosine phosphorylation was identified on the large intracellular loop of the GlyR β subunit at position Y-413 (Grenningloh et al. 1990), the effects of PTKs on the function of native and recombinant receptors have not been reported. The objective of this study was to determine if inhibitors of PTKs or the exogenous cellular Src gene product, cSrc, influence IGly in hippocampal neurons and recombinant human GlyRs. Since GlyRs mediate the majority of fast inhibitory synaptic transmission in the spinal cord, GlyRs in spinal neurons were also examined. Here we show that PTKs upregulate GlyR function through a process involving β tyrosine-413.
METHODS
Various GlyR α1–4 subunit isoforms are expressed in distinct regional patterns in the CNS and are developmentally regulated. Since the isoform of the α subunit and spliced variants influence regulation by second messenger systems, two different experimental preparations were used to examine the effects of PTKs on GlyRs; these include CA1 neurons acutely isolated from postnatal rat hippocampus and cultured spinal neurons from fetal mice. Experiments were performed with the approval of the Animal Care Committee of the University of Toronto.
CA1 neurons were acutely isolated from hippocampal slices as previously described (Xiong et al. 1999). Briefly, Wistar rats (∼14 days of age) were anaesthetized with halothane then killed by decapitation. Hippocampi were microdissected and cut into 400–500 μm slices then subjected to papain digestion (6.5 units ml−1, Sigma, St Louis, MO, USA). Slices were rinsed in enzyme-free extracellular solution and electrophysiological recordings were undertaken in neurons ∼15 min after isolation by mechanical trituration. Cultures of spinal neurons were prepared from Swiss white mice, using procedures previously described for hippocampal cultures (MacDonald et al. 1989) with additional 20 min incubation with trypsin-EDTA. Briefly, fetal pups (∼14 days in utero) were removed from mice that were killed by cervical dislocation. Neurons were cultured at 35.6 °C in a 5 % CO2 −95 % air environment and recordings were made 15–21 days after plating, corresponding to postnatal days ∼P9-P15. HEK 293 cells were transiently transfected with human GlyR α1, α1β (1:1) or α1βY413F (1:1) cDNAs (Valenzuela et al. 1998) using the lipid method (Invitrogen, Carlsbad, CA, USA). The conservative single substitution of β tyrosine-413 to phenylalanine-413 (Y413F) was undertaken to examine the influence of this putative tyrosine phosphorylation site on GlyR function. According to Grenningloh et al. (1990), the predicted site for tyrosine phosphorylation was ELSNYDCYG. The Y413F point mutation was made using the protocol supplied in the Stratagene QuikChange Site-Directed Mutagenesis kit (La Jolla, CA, USA) and verified by double-stranded DNA sequencing. Recordings were undertaken 24–48 h after transfection of the cDNAs.
Whole-cell IGly were recorded at a holding potential of −60 mV at room temperature. Pipettes were prepared using a two-step puller (Narishige Scientific Instrument Lab, Tokyo, Japan, PP-83). A multi-barrelled perfusion system (solution exchange time of ∼4 ms) was used to rapidly exchange the extracellular solutions. Glycine was applied at 2 min intervals to ensure sufficient time for GlyRs to recover from desensitization. Currents were filtered at 2 kHz, sampled at 500 μs per point, and analysed using pCLAMP6 software (Axon Instruments Inc., Foster City, CA, USA). Series resistance was monitored and cells with greater than a 20 % change in resistance were rejected.
The external solution contained (mm): 140 NaCl, 1.3 CaCl2, 5.4 KCl, 2 MgCl2, 25 Hepes, TTX (0.3 μm) and 33 glucose, at a pH of 7.4. Patch pipettes were filled with (mm): 63 CsCl, 70 CsF, 10 Hepes, 11 EGTA, 10 TEA-Cl, 2 MgCl2, 1 CaCl2 and 3.4 KATP, at a pH of 7.3. Identical results were obtained when CsCl replaced CsF for some experiments. Picrotoxin (50 mm) and strychnine (10 mm) were prepared in ethanol and distilled water, respectively, whereas the PTKs inhibitor, 5-amino-[(N-2,5-dihydroxybenzyl)-N′-2-hydroxybenzyl]salicylic acid (lavendustin A, 10 mm, Calbiochem, La Jolla, CA, USA) and its inactive analogue, 5-amino-(N,N′-bis-2-hydroxybenzyl)salicylic acid (lavendustin B) were dissolved in DMSO. In other experiments, active cSrc (30 U ml−1, Upstate Biotechnology, Lake Placid, NY, USA) or cSrc that was inactivated by boiling at 100 °C for 1 h (inactive cSrc) were included in the pipette solution, while the selective inhibitor of the Src family of PTKs, 4-amino-5-(4-chlorophenyl)-7-(t-butyl)pyrazolo[3,4-d]pyrimidine (PP2, 0.5 mm, Calbiochem) or its inactive analogue, 4-amino-7-phynelpyrazol[3,4-d]pyrimidine (PP3) were dissolved in DMSO.
Whole-cell currents evoked by glycine gradually declined or ‘ran down’ to a stable amplitude over 8–10 min. In CA1 neurons, this decline in amplitude was similar at 10 min whether currents were activated by saturating glycine (1 mm: 70 ± 7 % of the maximum current amplitude (Imax), n = 6) or sub-saturating concentrations of glycine (30 μm: 75 ± 5 % of Imax, n = 4). Similarly, in spinal neurons, glycine (45 μm)-evoked currents declined to 71 ± 7 % of Imax (n = 4) within 10 min. The amplitude of IGly was stable prior to the application of drugs that were intended to influence IGly. When inhibitors of PTKs or exogenous Src were applied intracellularly, currents were compared to controls at the same point in time. Concentration-response plots for glycine-activated currents were fitted to the equation: Imax/I = 1+([Gly]/EC50)n where Imax is maximal current, EC50 is the glycine concentration [Gly] that activates 50 % of the maximal response and n is the Hill coefficient. Results are expressed as means ± s.e.m. Statistical analysis was performed using Student's t test, Mann-Whitney test and two-way or one-way analysis of variance (ANOVA) with Dunnett's multiple comparison post hoc test, as appropriate (GraphPad Prism 3.02, GraphPad Software Inc.). P values < 0.05 were considered significant.
RESULTS
GlyRs are downregulated by inhibitors of PTKs and enhanced by exogenous cSrc
Initially, concentration-response plots for IGly in CA1 and spinal neurons were compared as the potency of glycine is influenced by GlyR subunit composition. The EC50 values for peak and steady-state currents in CA1 neurons were 134 ± 18 μm (n = 6) and 43 ± 8 μm (n = 4), respectively (Fig. 1A and B). In spinal neurons, the EC50 values were significantly less for peak (42 ± 6 μm, n = 8) and steady-state (18 ± 4 μm, n = 6) currents (P < 0.05, Fig. 1C and D). In subsequent experiments, concentrations of glycine that approximated the EC50 for peak current were used to activate IGly in CA1 (140 μm) and spinal (45 μm) neurons, unless indicated otherwise.
Figure 1. Concentration dependence of IGly in CA1 and spinal neurons. IGly is reduced by lavendustin A.
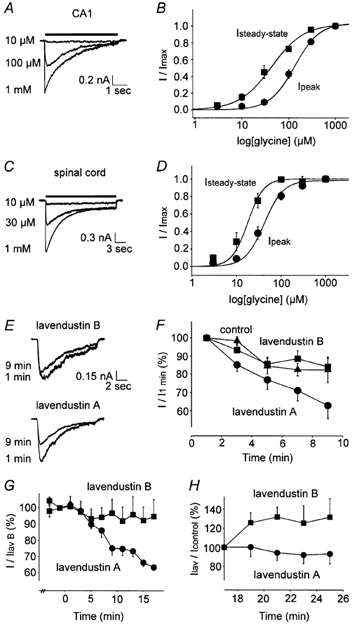
A, superimposed traces of IGly recorded in CA1 neurons. B, dose-response relationships indicate EC50 values for peak (•) and steady-state currents (▪) of 134 ± 18 μm (n = 6) and 43 ± 8 μm (n = 4) and Hill coefficients of 1.6 ± 0.1 and 1.3 ± 0.1, respectively. C and D, in spinal neurons, the EC50 and Hill coefficients for peak and steady-state currents were 42 ± 6 μm, 2.0 ± 0.4 (n = 8) and 18 ± 4 μm, 2.5 ± 0.5 (n = 6), respectively. E and F, peak IGly (140 μm) in CA1 neurons with control (▴, n = 4), lavendustin B (▪, n = 7) or lavendustin A (•, P < 0.01, n = 7) in the pipette solution. G and H, peak IGly (140 μm) in CA1 neurons with lavendustin A applied extracellularly (P < 0.01, n = 4) following pre-treatment with lavendustin B, as compared to neurons continuously exposed to lavendustin B (n = 5). Recovery of IGly following washout of lavendustin A (n = 3) and lavendustin B (P < 0.01, n = 3) is shown. Currents were normalized to the peak amplitude measured immediately prior to drug washout.
Next, the effects of lavendustin A, a membrane-permeable inhibitor of PTKs, and its inactive analogue, lavendustin B, on IGly in CA1 neurons were examined. Since genistein, another inhibitor of PTKs, directly blocked GABAA receptors when applied extracellularly (Dunne et al. 1998; Huang et al. 1999) but not intracellularly (Moss et al. 1995), lavendustin A (1 μm) or lavendustin B (1 μm) were added to the pipette solution. Lavendustin A caused a 21 ± 9 % (P < 0.01, n = 7, at 9 min) greater reduction in peak amplitude compared to lavendustin B, whereas lavendustin B had no effect compared to control (lavendustin B 84 ± 5 % of Imax, n = 7; control 82 ± 7 % of Imax, n = 4; Fig. 1E and F). Next, lavendustin A (10 μm) and lavendustin B (10 μm) were applied extracellularly. These compounds were shown not to directly block GlyRs in hypothalamic neurons (Huang & Dillon, 2000) or recombinant GABAA receptors (Huang et al. 1999). However, lavendustin B caused a rapid, reversible depression of IGly by 39 ± 3 % (n = 24) and this reduction was similar whether lavendustin B was pre-applied for 60 s or co-applied with glycine. Lavendustin A also caused a rapid inhibition of IGly by 33 ± 2 % (n = 12); however, this was followed by a gradual irreversible decline in current amplitude. We attributed the rapid, reversible inhibition by lavendustin A and lavendustin B to direct channel blockade whereas the slower irreversible reduction (observed only with lavendustin A) was due to inhibition of PTKs. We next examined whether lavendustin A reduced IGly in neurons pre-treated with lavendustin B and observed a 31 ± 11 % (n = 4) reduction in current amplitude (P < 0.01, Fig. 1G). In spinal neurons, lavendustin A reduced IGly by 40 ± 8 % (P < 0.05, n = 4, at 15 min) in cells pre-treated with lavendustin B. Moreover, IGly increased by 32 ± 11 % (P < 0.01, n = 3, over 8 min) following the washout of lavendustin B whereas IGly decreased by 8 ± 9 % (n = 3) following the washout of lavendustin A (Fig. 1H), suggesting that the effects of lavendustin A were, in part, tyrosine kinase specific.
To determine if members of the Src family of PTKs upregulate GlyR function, cSrc or inactive cSrc were applied intracellularly. Currents were first evoked by 1 mm glycine to mimic saturating concentrations at the synapse (Legendre, 1998). The maximum peak currents with cSrc or inactive cSrc in the pipette solution were 1.12 ± 0.48 nA (n = 6) and 1.37 ± 0.36 nA (n = 6), respectively. cSrc (30 U ml−1) enhanced IGly by 25 ± 14 % at 13 min (P < 0.05, n = 6) while inactive cSrc had no effect (n = 6); IGly declined to 64 ± 7 % (n = 6) of the initial response in the presence of inactive cSrc, similar to control (62 ± 6 %, n = 4, Fig. 2A–C).
Figure 2. cSrc enhances IGly in CA1 neurons.
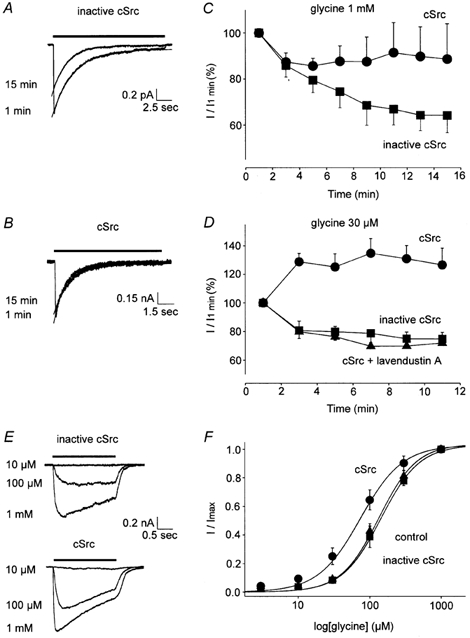
A and B, superimposed traces of IGly activated by a saturating [Gly] at 1 and 15 min. C, IGly evoked by 1 mm glycine with cSrc (•) or inactive cSrc (▪) applied intracellularly are shown (P < 0.05, each group n = 6). D, IGly activated by sub-saturating [Gly]= 30 μm with cSrc (•, P < 0.01), inactive cSrc (▪) or cSrc plus lavendustin A (▴) in the pipette solution (each group n = 4). E, traces of IGly with inactive or active cSrc in the pipette solution. F, the [glycine]-response plots with cSrc (•, EC50 = 74 ± 14 μm, P < 0.05, n = 5), inactive cSrc (▪) (EC50 = 143 ± 23 μm, n = 3) and control intracellular solution (▴, EC50 = 134 ± 18 μm, n = 6) are shown.
The effect of cSrc on IGly evoked by sub-saturating concentrations of glycine (30 μm) was examined in CA1 neurons. IGly increased by 56 ± 11 % with cSrc in the pipette solution (P < 0.01, n = 4, Fig. 2D, at 9 min) and the addition of lavendustin A (10 μm) to the pipette solution blocked the increase by cSrc (n = 4, Fig. 2D). The 2-fold greater enhancement (56 % versus 25 %, P < 0.01) of IGly evoked by a sub-saturating compared to saturating [Gly] suggests that cSrc increases the potency of glycine. Consequently, [Gly]-response plots were constructed with active or inactive cSrc in the pipette solution. While inactive cSrc failed to influence the [Gly]-response plot, active cSrc shifted the curve to the left, consistent with an increase in glycine potency (P < 0.05, Fig. 2E and F). Since receptor phosphorylation may also influence desensitization, we examined the effect of cSrc on the time course of GlyR desensitization in CA1 neurons. IGly rapidly reached a peak then slowly decayed close to the baseline level during a 5 s application of 1 mm glycine (Fig. 1A). This decay was well described by a single exponential function (Fig. 2A and B). cSrc reduced the time constant of desensitization, τ, by 30 % (cSrc τ = 2.01 ± 0.29 s, inactive cSrc τ = 2.85 ± 0.35 s, P < 0.05, n = 6, at 15 min), suggesting that tyrosine phosphorylation enhances GlyR desensitization.
Tyrosine phosphorylation of recombinant GlyRs expressed in HEK 293 cells
To examine the specific role of α and β subunits, recombinant GlyR subunits were expressed in HEK 293 cells. First, [picrotoxin]-inhibition plots were examined to ensure the functional expression of heteromeric α1β GlyRs as the β subunit reduced the sensitivity of heteromeric receptors to picrotoxin (Pribilla et al. 1992; Handford et al. 1996) and increased the amplitude of IGly (Bormann et al. 1993). The IC50 for picrotoxin inhibition of current activated by 100 μm glycine was lower in cells transfected with α1 subunits compared to α1β subunits (98 ± 41μm, n = 7 versus 1060 ± 313 μm, n = 6, P < 0.05). Also, Imax was greater for α1β heteromeric receptors (2.8 ± 0.6 nA, n = 12) compared to α1 receptors (1.6 ± 0.7 nA, n = 11, P < 0.05), consistent with the functional expression of the β subunit.
Since a putative tyrosine phosphorylation site resides on the β subunit, we compared the sensitivity of homomeric α1 and heteromeric α1β receptors to exogenous cSrc. IGly (1 mm) in recombinant GlyRs gradually ran down over 8–10 min. cSrc (30 U ml−1) reduced the run-down of IGly recorded from α1β receptors by 15 ± 6 % (P < 0.01, n = 12, Fig. 3A and B) but not α1 receptors (n = 12, Fig. 3C and D, at 13 min). The effects of cSrc on desensitization were examined by measuring the steady-state to peak current ratio (Iss/Ip) and τdesensitization. For α1β GlyRs, the Iss/Ip ratio was reduced from 0.46 ± 0.09 to 0.32 ± 0.10 in the presence of active compared to inactive cSrc (P < 0.01, n = 9, at 1 min). In contrast, the Iss/Ip ratio was similar with active and inactive cSrc for α1 GlyRs (0.47 ± 0.08, n = 10 versus 0.53 ± 0.09, n = 7 at 1 min, respectively). These values did not change over time. Moreover, cSrc accelerated τdesensitization of α1β receptors (cSrc τ = 2.34 ± 0.49 s, n = 10; inactive cSrc τ = 6.38 ± 1.33 s, n = 11; P < 0.05, at 13 min) but not α1 receptors (cSrc τ = 4.42 ± 0.98 s, n = 12; inactive cSrc τ = 4.61 ± 1.20 s, n = 11; at 13 min), indicating that tyrosine kinases enhance desensitization of α1β but not α1 GlyRs.
Figure 3. cSrc reduces the rundown of α1β GlyR-mediated currents.
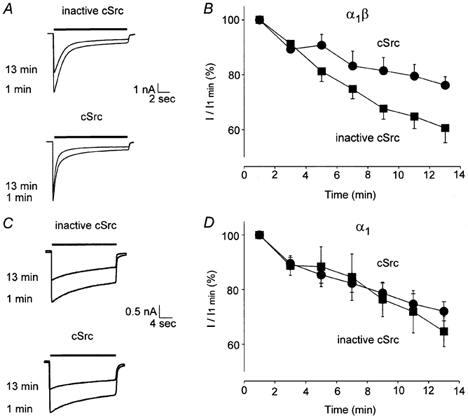
A and B, peak IGly (1 mm) from α1β GlyRs with cSrc (•) and inactive cSrc (▪) applied intracellularly (P < 0.01, each group n = 12). C and D, IGly (1 mm) for α1 GlyRs with cSrc (n = 12) or inactive cSrc (n = 11) applied intracellularly.
The specific role of β-Y413 in mediating the sensitivity to PTKs was next examined (Fig. 4A). The maximal amplitude of current recorded from α1βY413F receptors (3.08 ± 0.67 nA, n = 9) was not different from wild-type α1β receptors (3.02 ± 0.74 nA, n = 6). Also, consistent with the functional expression of the mutant βY413F subunit, IGly from α1βY413F receptors was less sensitive to inhibition by picrotoxin (1 mm) compared to homomeric α1 receptors as measured by percentage inhibition of control (α1β 46 ± 9 %, n = 5; α1βY413F 47 ± 10 %, n = 8 versus α1 79 ± 9 %, n = 5, P < 0.05).
Figure 4. Point mutation of βY413 abolishes GlyR sensitivity to cSrc.
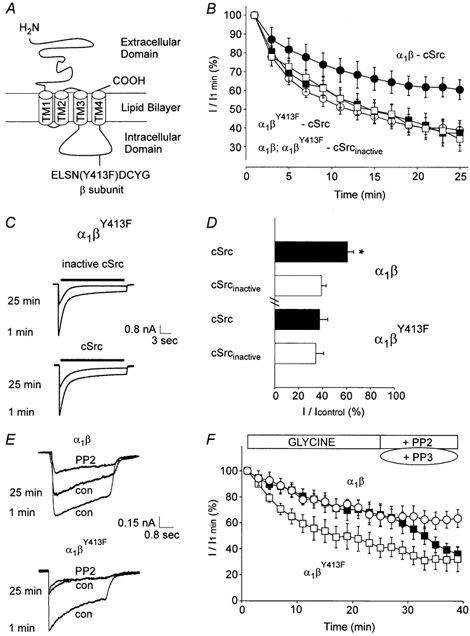
A, the location of tyrosine-413 of the GlyR β subunit. B, the time course of IGly from α1β receptors with active (•, n = 6) or inactive cSrc (○, n = 6) applied intracellularly. Also, IGly from α1βY413F receptors with active (▪, n = 10) or inactive cSrc (□, n = 9) applied intracellularly are shown. C, IGly from α1βY413F receptors. D, mean value of IGly (1 mm) from α1β and α1βY413F GlyRs. Currents were measured at 25 min and were normalized to the response measured 1 min following patch breakthrough (n values are listed above). E, traces of IGly at 1 and 25 min after breakthrough in control solution, followed by PP2 applied extracellularly to α1β and α1βY413F GlyRs. F, PP2 (▪, P < 0.05, n = 6) but not PP3 (○, n = 4) inhibited α1β GlyRs whereas PP2 did not inhibit α1βY413F GlyRs (□, n = 6).
The run-down of IGly in α1β and α1βY413F GlyRs, with active or inactive cSrc in the pipette solution, was examined (Fig. 4B). For α1β GlyRs, the current ran down to 61 ± 5 and 39 ± 4 % of the control values measured at 1 min with active and inactive cSrc in the pipette solution, respectively (P < 0.01, n = 6, Fig. 4D, at 25 min). However, for α1βY413F GlyRs, the current ran down to 37 ± 7 % (n = 10) and 34 ± 6 % (n = 9) in the presence of active and inactive cSrc, respectively (Fig. 4C and D, at 25 min). Hence, cSrc reduced the run-down of α1β GlyR-mediated currents by 22 ± 7 % (P < 0.01) but did not affect α1βY413F GlyRs. Next, the effect of a selective inhibitor of the Src family of PTKs, PP2, was examined in GlyRs containing the β or βY413F subunit. PP2 (0.5 μm) depressed α1β-mediated currents (P < 0.05, n = 6, at 13 min) but had no effect on α1βY413F-mediated currents (n = 6). A control analogue for PP2, PP3, failed to inhibit α1β currents (n = 4, Fig. 4E and F), consistent with the hypothesis that phosphorylation of βY413 enhances GlyR function.
DISCUSSION
This study provides evidence that PTKs play a role in maintaining and upregulating the function of GlyRs in neurons as well as recombinant heteromeric GlyRs. Lavendustin A, an inhibitor of PTKs, reduced IGly while intracellularly applied cSrc increased IGly. The enhancement by cSrc required the β subunit and point substitution of tyrosine-413 abolished both the enhancement by cSrc and the inhibition by PP2. Furthermore, cSrc increased the potency of glycine and accelerated GlyR desensitization.
The simplest explanation for our findings is that tyrosine phosphorylation of the β subunit at position 413 increases GlyR-mediated conductance by at least two possible mechanisms: (1) enhanced glycine potency and (2) alterations in intrinsic receptor properties, including channel conductance or gating. However, our results do not exclude interactions of PTKs with other intermediary cytosolic proteins that modulate GlyR function. The β subunit regulates interactions with the anchoring protein, gephyrin (Meyer et al. 1995) or scaffolding proteins that can be modified by phosphorylation. Also, GlyR activity is modulated by divalent cations, including Ca2+ (Xu et al. 1999, 2000) and Zn2+ (for review see Legendre, 2001). Src specifically interacts with the intracellular domains of the GABAA receptor β and γ2 subunits (Brandon et al. 2001) and potentiates the function of recombinant NMDA receptors by reducing voltage-independent inhibition by Zn2+ (Zheng et al. 1998). Furthermore, PTKs may be involved in cross-modulation of GlyR function with other intracellular kinases, including PKA and PKC (for review see Moss & Smart, 1996). PKA increased glycine potency in neurons from the ventral tegmental area (Ren et al. 1998), although neither PKA- nor PKC-dependent mechanisms influenced the EC50 of glycine in trigeminal neurons (Song & Huang, 1990; Gu & Huang, 1998). Biochemical experiments to examine whether the GlyR β subunit is tyrosine phosphorylated will address protein phosphorylation directly although such studies have been hampered by unsuccessful attempts to develop antibodies selective for the GlyR β subunit (correspondence with C. F. Valenzuela, University of New Mexico, Health Science Center).
Potentiation of GlyRs by tyrosine phosphorylation may have therapeutic implications. For example, GlyRs have been implicated in the regulation of nociceptive input to the spinal cord. Inhibition of GlyRs by intrathecal administration of strychnine yields touch-evoked allodynia (Yaksh, 1989) and contributes to hyperalgesia (Beyer et al. 1988) and neuropathic pain (Simpson et al. 1996). Thus, the manipulation of GlyR function by second messenger regulation may represent a novel therapeutic strategy, particularly for patients with narcotic-resistant pain states.
Acknowledgments
We thank Dr C. Fernando Valenzuela (Department of Neuroscience, University of New Mexico Health Sciences Centre, Albuquerque, New Mexico, USA) for providing the cDNAs, Dr Michael W. Salter (Programmes in Brain and Behaviour & Cell Biology, Hospital for Sick Children) for reviewing the manuscript, and Lidia Brandes and Elzbieta Czerwinska for technical assistance. Grants from CIHR to B.A.O. and J.F.M. and the International Anesthesia Research Society to B.A.O. supported these studies.
REFERENCES
- Ali DW, Salter MW. NMDA receptor regulation by Src kinase signaling in excitatory synaptic transmission and plasticity. Current Opinion in Neurobiology. 2001;11:336–342. doi: 10.1016/s0959-4388(00)00216-6. [DOI] [PubMed] [Google Scholar]
- Beyer C, Banas C, Gomora P, Komisaruk BR. Prevention of the convulsant and hyperalgesic action of strychnine by intrathecal glycine and related amino acids. Pharmacology, Biochemistry, and Behavior. 1988;29:73–78. doi: 10.1016/0091-3057(88)90276-6. [DOI] [PubMed] [Google Scholar]
- Bormann J, Rundstrom N, Betz H, Langosch D. Residues within transmembrane segment M2 determine chloride conductance of glycine receptor homo- and hetero-oligomers. EMBO Journal. 1993;12:3729–3737. doi: 10.1002/j.1460-2075.1993.tb06050.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandon NJ, Delmas P, Hill J, Smart TG, Moss SJ. Constitutive tyrosine phosphorylation of the GABAA receptor γ2 subunit in rat brain. Neuropharmacology. 2001;41:745–752. doi: 10.1016/s0028-3908(01)00121-6. [DOI] [PubMed] [Google Scholar]
- Dunne EL, Moss SJ, Smart TG. Inhibition of GABAA receptor function by tyrosine kinase inhibitors and their inactive analogues. Molecular and Cellular Neuroscience. 1998;12:300–310. doi: 10.1006/mcne.1998.0717. [DOI] [PubMed] [Google Scholar]
- Grenningloh G, Pribilla I, Prior P, Multhaup G, Beyreuther K, Taleb O, Betz H. Cloning and expression of the 58 kD beta subunit of the inhibitory glycine receptor. Neuron. 1990;4:963–970. doi: 10.1016/0896-6273(90)90149-a. [DOI] [PubMed] [Google Scholar]
- Gu Y, Huang LYM. Cross-modulation of glycine-activated Cl- channels by protein kinase C and cAMP-dependent protein kinase in the rat. Journal of Physiology. 1998;506:331–339. doi: 10.1111/j.1469-7793.1998.331bw.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handford CA, Lynch JW, Baker E, Webb GC, Ford JH, Sutherland GR, Schofield PR. The human glycine receptor beta subunit: primary structure, functional characterization and chromosomal localization of the human and murine genes. Molecular Brain Research. 1996;35:211–219. [PubMed] [Google Scholar]
- Huang R, Dillon GH. Direct inhibition of glycine receptors by genistein, a tyrosine kinase inhibitor. Neuropharmacology. 2000;39:2195–2204. doi: 10.1016/s0028-3908(00)00046-0. [DOI] [PubMed] [Google Scholar]
- Huang RQ, Fang MJ, Dillon GH. The tyrosine kinase inhibitor genistein directly inhibits GABAA receptors. Molecular Brain Research. 1999;67:177–183. doi: 10.1016/s0169-328x(99)00061-3. [DOI] [PubMed] [Google Scholar]
- Legendre P. A reluctant gating mode of glycine receptor channels determines the time course of inhibitory miniature synaptic events in zebrafish hindbrain neurons. Journal of Neuroscience. 1998;18:2856–2870. doi: 10.1523/JNEUROSCI.18-08-02856.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legendre P. The glycinergic inhibitory synapse. Cellular and Molecular Life Sciences. 2001;58:760–793. doi: 10.1007/PL00000899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald JF, Mody I, Salter MW. Regulation of N-methyl-d-aspartate receptors revealed by intracellular dialysis of murine neurones in culture. Journal of Physiology. 1989;414:17–34. doi: 10.1113/jphysiol.1989.sp017674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer G, Kirsch J, Betz H, Langosch D. Identification of a gephyrin binding motif on the glycine receptor beta subunit. Neuron. 1995;15:563–572. doi: 10.1016/0896-6273(95)90145-0. [DOI] [PubMed] [Google Scholar]
- Moss SJ, Gorrie GH, Amato A, Smart TG. Modulation of GABAA receptors by tyrosine phosphorylation. Nature. 1995;377:344–348. doi: 10.1038/377344a0. [DOI] [PubMed] [Google Scholar]
- Moss SJ, Smart TG. Modulation of amino acid-gated ion channels by protein phosphorylation. International Review of Neurobiology. 1996;39:1–52. doi: 10.1016/s0074-7742(08)60662-5. [DOI] [PubMed] [Google Scholar]
- Pribilla I, Takagi T, Langosch D, Bormann J, Betz H. The atypical M2 segment of the beta subunit confers picrotoxinin resistance to inhibitory glycine receptor channels. EMBO Journal. 1992;11:4305–4311. doi: 10.1002/j.1460-2075.1992.tb05529.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren J, Ye JH, McArdle JJ. cAMP-dependent protein kinase modulation of glycine-activated chloride current in neurons freshly isolated from rat ventral tegmental area. Brain Research. 1998;811:71–78. doi: 10.1016/s0006-8993(98)00959-7. [DOI] [PubMed] [Google Scholar]
- Simpson RK, Jr, Gondo M, Robertson CS, Goodman JC. Reduction in the mechanonociceptive response by intrathecal administration of glycine and related compounds. Neurochemical Research. 1996;21:1221–1226. doi: 10.1007/BF02532399. [DOI] [PubMed] [Google Scholar]
- Song Y, Huang LYM. Modulation of glycine receptor chloride channels by cAMP-dependent protein kinase in spinal trigeminal neurons. Nature. 1990;348:242–245. doi: 10.1038/348242a0. [DOI] [PubMed] [Google Scholar]
- Valenzuela CF, Cardoso RA, Wick MJ, Weiner JL, Dunwiddie TV, Harris RA. Effects of ethanol on recombinant glycine receptors expressed in mammalian cell lines. Alcoholism, Clinical and Experimental Research. 1998;22:1132–1136. [PubMed] [Google Scholar]
- Wan Q, Man HY, Braunton J, Wang W, Salter MW, Becker L, Wang YT. Modulation of GABAA receptor function by tyrosine phosphorylation of beta subunits. Journal of Neuroscience. 1997;17:5062–5069. doi: 10.1523/JNEUROSCI.17-13-05062.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong Z, Pelkey KA, Lu WY, Lu YM, Roder JC, MacDonald JF, Salter MW. Src potentiation of NMDA receptors in hippocampal and spinal neurons is not mediated by reducing zinc inhibition. Journal of Neuroscience. 1999;19:RC37. doi: 10.1523/JNEUROSCI.19-21-j0003.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu TL, Dong XP, Wang DS. N-methyl-D-aspartate enhancement of the glycine response in the rat sacral dorsal commissural neurons. European Journal of Neuroscience. 2000;12:1647–1653. doi: 10.1046/j.1460-9568.2000.00065.x. [DOI] [PubMed] [Google Scholar]
- Xu TL, Li JS, Jin YH, Akaike N. Modulation of the glycine response by Ca2+-permeable AMPA receptors in rat spinal neurones. Journal of Physiology. 1999;514:701–711. doi: 10.1111/j.1469-7793.1999.701ad.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaksh TL. Behavioral and autonomic correlates of the tactile evoked allodynia produced by spinal glycine inhibition: effects of modulatory receptor systems and excitatory amino acid antagonists. Pain. 1989;37:111–123. doi: 10.1016/0304-3959(89)90160-7. [DOI] [PubMed] [Google Scholar]
- Zheng F, Gingrich MB, Traynelis SF, Conn PJ. Tyrosine kinase potentiates NMDA receptor currents by reducing tonic zinc inhibition. Nature Neuroscience. 1998;1:185–191. doi: 10.1038/634. [DOI] [PubMed] [Google Scholar]