

Abstract
In humans, pulmonary oxygen uptake (V̇O2) kinetics may be speeded by prior exercise in the heavy domain. This ‘speeding’ arises potentially as the result of an increased muscle O2 delivery (Q̇O2) and/or a more rapid elevation of oxidative phosphorylation. We adapted phosphorescence quenching techniques to determine the QO2-to-O2 utilization (Q̇O2/V̇O2) characteristics via microvascular O2 pressure (PO2,m) measurements across sequential bouts of contractions in rat spinotrapezius muscle. Spinotrapezius muscles from female Sprague-Dawley rats (n = 6) were electrically stimulated (1 Hz twitch, 3–5 V) for two 3 min bouts (ST1 and ST2) separated by 10 min rest. PO2,m responses were analysed using an exponential + time delay (TD) model. There was no significant difference in baseline and ΔPO2,m between ST1 and ST2 (28.5 ± 2.6 vs. 27.9 ± 2.4 mmHg, and 13.9 ± 1.8 vs. 14.1 ± 1.3 mmHg, respectively). The TD was reduced significantly in the second contraction bout (ST1, 12.2 ± 1.9; ST2, 5.7 ± 2.2 s, P < 0.05), whereas the time constant of the exponential PO2,m decrease was unchanged (ST1, 16.3 ± 2.6; ST2, 17.6 ± 2.7 s, P > 0.1). The shortened TD found in ST2 led to a reduced time to reach 63 % of the final response of ST2 compared to ST1 (ST1, 28.3 ± 3.0; ST2, 20.2 ± 1.8 s, P < 0.05). The speeding of the overall response in the absence of an elevated PO2,m baseline (which had it occurred would indicate an elevated QO2/V̇O2) or muscle blood flow suggests that some intracellular process(es) (e.g. more rapid increase in oxidative phosphorylation) may be responsible for the increased speed of PO2,m kinetics after prior contractions under these conditions.
There is evidence that priming a muscle group with prior exercise can speed oxygen uptake (V̇O2) kinetics in a subsequent bout of exercise. However, the expression of this speeding is controversial and the mechanistic bases for this effect have not been resolved. For instance, Gerbino et al. (1996) have shown a speeding of the pulmonary V̇O2 kinetics (which are thought to be dominated by those of the muscle; Barstow et al. 1990; Grassi et al. 1996) after a prior bout of heavy exercise. Gerbino and colleagues (1996) hypothesized that the accelerated V̇O2 kinetics in the second bout resulted from an improved perfusion and therefore O2 delivery to the exercising muscle consequent to the vasodilatory effects of a residual metabolic acidaemia from the first bout. In a more recent investigation, Burnley et al. (2000) rigorously partitioned their analysis of the V̇O2 kinetics into phase I, phase II and slow component. These authors reported no difference in phase II pulmonary V̇O2 kinetics with prior heavy exercise. Rather, the mean response time (MRT) was significantly reduced due to a decreased amplitude of the V̇O2 slow component. This result has been confirmed independently by Scheuermann and colleagues (2001) and together these studies suggest indirectly that the dominant phase II response is unaffected by prior exercise.
Although the speeding of V̇O2 kinetics by prior exercise has not been measured within intact skeletal muscle, at least two pieces of evidence support the notion that intramyocyte energetics are altered in subsequent contraction bouts. Specifically, Hogan (2001) demonstrated that prior contractions reduce the time delay that occurs prior to the exponential fall of cytosolic O2 pressure (and thus the overall MRT) within single frog lumbrical myocytes at the onset of a second bout of contractions. Moreover, in human calf muscle, [31P] nuclear magnetic resonance spectroscopy measurements indicate that prior exercise reduces the perturbation of phosphocreatine across the on-transient to the second bout (Laurent et al. 1992). Both findings support the conclusion that V̇O2 kinetics are accelerated after priming exercise.
Within humans, the majority of investigations suggest that prior exercise only speeds V̇O2 kinetics if it is performed above the lactate threshold (Gerbino et al. 1996; Burnley et al. 2000; see Koppo & Bouckaert (2000) for one exception). Moreover, it appears important that both exercise bouts are performed by the same muscles because the lactacidaemia and cardiovascular perturbations arising from remote muscle contractions (i.e. arms or legs) either cannot speed V̇O2 kinetics of non-previously exercised legs (Yoshida et al. 1995) or do so to a lesser extent (Bohnert et al. 1998). One common notion is that elevated blood flow within the exercised vascular bed facilitates a more rapid V̇O2 response in the second bout (Bohnert et al. 1998).
The present investigation was designed to address the mechanistic bases by which prior exercise speeds V̇O2 kinetics. Using the technique of phosphorescence quenching (Behnke et al. 2001), we examined the microvascular PO2 (PO2,m), which provides information regarding the dynamic relationship between O2 delivery (Q̇O2) and O2 utilization (V̇O2), across repeated contraction bouts in the rat spinotrapezius muscle. We hypothesized that (1) if blood flow (Q̇O2) is increased out of proportion to V̇O2 following contractions (i.e. immediately preceding the second bout), PO2,m will be higher prior to the second contractile period, and (2) if there is a faster adjustment of oxidative phosphorylation across the second stimulation on-transient, there will be a shorter time delay preceding the decline in PO2,m and thus an overall faster PO2,m response.
METHODS
Female Sprague-Dawley rats (291 ± 7 g, n = 8) were anaesthetized with pentobarbital sodium (40 mg kg−1i.p., to effect). The carotid artery was cannulated using PE-50 tubing (Intra-Medic polyethylene tubing, Clay Adams, Sparks, MD, USA). This provided a route of access for infusion of the phosphorescent probe (see protocol) at 15 mg kg−1, monitoring of arterial blood pressure (Digi-Med BPA model 200, Louisville, KY, USA) and blood sampling. Blood withdrawal for blood gas, pH, and lactate determination (Nova Stat Profile M, Waltham, MA, USA), and haematocrit (Adams Micro-Hematocrit reader, Clay Adams, Parsipanny, NJ, USA) was performed immediately after the ST2 stimulation period. All procedures were approved by the Kansas State University institutional animal care and use committee (IACUC).
Surgical preparation
The left spinotrapezius muscle was exposed with minimal fascial disturbance to facilitate electrical stimulation. The exposed surrounding tissue was protected with Saran Wrap (Dow, Indianapolis, IN, USA) and the spinotrapezius was superfused with a Krebs-Henseleit bicarbonate-buffered solution equilibrated with 5 % CO2-95 % N2 at 38 °C. Body temperature was maintained at 38 °C using a heating pad. Stainless steel plate electrodes (2.5 mm diameter) were attached proximal to the motor point (cathode) and across the caudal region (anode) close to the spinal attachment of the muscle in order to elicit indirect, bipolar muscle contractions.
Protocol
The phosphorescent probe, palladium meso-tetra(4-carboxyphenyl)porphyrin dendrimer (R2), was infused via the arterial cannula approximately 15 min before each experiment. Following a 10–15 min post-surgical preparation stabilization period, twitch muscle contractions (3–5 V, 2 ms pulse duration) were elicited at 1 Hz frequency for 3 min (ST1) using a Grass S88 stimulator (Quincy, MA, USA). After the 3 min stimulation period there was a stimulation-free recovery period of 10 min before a second contraction period (ST2), identical to the first, was elicited. Microvascular PO2 (PO2,m) was determined at 2 s intervals across the rest-contraction period for both stimulation protocols. Mean arterial pressure (MAP) was monitored continuously throughout the protocol. Upon completion of the experiment, the animal was killed with an overdose of pentobarbital sodium (> 80 mg kg−1i.a.)
Principle of O2-dependent phosphorescence quenching
Theory
The O2 dependence of the probe phosphorescence can be described quantitatively through the Stern-Volmer relationship (Rumsey et al. 1988):
thus
where kQ is the quenching constant (in mmHg−1 s−1) and τ and τ° are the phosphorescence lifetimes in an O2-free environment and at the extant PO2, respectively. For R2 bound to albumin at 38 °C and pH 7.4, kQ is 409 mmHg−1 s−1 and τ° is 601 μs (Lo et al. 1997). These values are determined in vitro for the probe bound to albumin. In vivo, the physicochemical environment for the probe is replicated in the blood. The lifetime of the phosphorescence is independent of the excitation light intensity, the probe concentration, or absorbance by other chromophores in the tissue. In the blood, O2 is believed to be the only molecule that quenches phosphorescence, thus facilitating an absolute measurement of PO2 (Rumsey et al. 1988). The palladium porphyrin shows no signs of toxicity or physiological effects on blood gases, blood pressure, or brain electrical activity (Lahiri et al. 1993). The phosphorescence characteristics of the probe allow PO2 measurement from 0–150 mmHg. There is no pH dependence of kQ or τ° between pH 6.8 and pH 7.4 (Lo et al. 1997), but a modest temperature dependence exists (i.e. 3 % °C−1) for kQ and τ°. However, within the normal physiological range this effect is considered to be insignificant (Lo et al. 1997).
Microvascular PO2 measurement
Microvascular PO2 (PO2,m) was determined using a PMOD 1000 Frequency Domain Phosphorometer (Oxygen Enterprises Ltd, Philadelphia, PA, USA) with the common end of the bifurcated light guide placed 2–4 mm above the medial region of the spinotrapezius (i.e. superficial to the dorsal surface). The excitation light (524 nm) is focused on an ∼2 mm diameter circle of exposed muscle surface and samples blood within the microvasculature up to 500 μm deep. The PMOD 1000 uses a 48 kHz, 16-bit Sigma-Delta digitizer to average the phosphorescence signal (700 nm) over 20 ms per scan. Ten scans were performed at each measurement point and the signal was averaged over a 200 ms interval for each PO2,m measurement, with measurements being repeated at 2 s intervals.
The R2 phosphorescent probe is bound to albumin in the blood, and therefore it is assumed to be uniformly distributed in the blood plasma and provide a signal corresponding to the volume average O2 pressure in the vascular compartment (i.e. principally the capillary bed). The phosphorescence lifetime was obtained by taking the logarithm of the intensity values at each time point and fitting the linearized decay to a straight line by the least-squares method (Bevington, 1969). PO2,m values were then curve-fitted to a monoexponential plus delay model (Behnke et al. 2001) using an iterative least-squares technique by means of a commercial graphing/analysis package (KaleidaGraph 3.5, Synergy Software, Reading, PA, USA). For the KaleidaGraph analysis program a user-defined function to the data was fitted using the following equation:
where PO2,m(t) is the change in PO2,m at time t, ΔPO2,m is the change in PO2,m from baseline to steady-state during contractions, TD is the time delay, and τ is the time constant of the response. The same model was applied to PO2,m responses for both stimulation periods. In addition, the overall time taken to reach 63 % of the final response (t63) was measured directly from the response to provide an indication of the time course of PO2,m change independent of any modelling procedure.
Blood flow measurements
Blood flow was determined in a subset of four animals (female Sprague-Dawley rats, 363 ± 49 g) using the radionuclide-tagged microsphere technique (Musch & Terrell 1992). Initially, rats were anaesthetized with sodium pentobarbital (40 mg kg−1i.p., to effect). Polyethylene catheters (PE-10 connected to PE-50) were placed in the right carotid and caudal (tail) arteries. The carotid artery catheter was advanced 2–3 mm rostral to the aortic valve and secured. The tail artery catheter was advanced toward the bifurcation of the descending aorta and secured. The carotid artery catheter was connected to a pressure transducer. Arterial blood pressure and heart rate were measured (Digi-Med BPA model 200, Louisville, KY, USA). The tail artery catheter was connected to a 1 ml plastic syringe, which was attached to a Harvard Withdrawal Pump (model 907, Cambridge, MA, USA).
Blood flow measurements were taken at rest and at both 5 and 10 min after electrical stimulation (as detailed above). Three different microspheres (46Sc, 85Sr, 113Sn) with a diameter of 15 μm (New England Nuclear, Boston, MA, USA) were injected in random order. Prior to infusion, the microspheres were agitated by sonication to suspend the beads and prevent clumping. Thirty seconds prior to initiating infusion, blood withdrawal from the caudal artery at 0.25 ml min−1 was begun. The right carotid artery catheter was disconnected from the pressure transducer and a specified microsphere (∼2.5 × 105 in number) was injected into the ascending aorta and flushed with saline to assure clearance of the beads. Blood withdrawal from the caudal artery continued for 45 s after microsphere infusion.
Following the final microsphere injection, the rats were killed with an overdose of sodium pentobarbital (> 80 mg kg−1) via the right carotid artery catheter. After verifying correct placement of the carotid catheter, the following muscles and organs were removed: left and right spinotrapezius, solei and kidneys. The radioactivity levels of the tissues were determined by a two-channel γ scintillation counter (Packard Auto Gamma Spectrometer, model 5230) set to record the peak energy activity of each isotope for 5 min. Total blood flow to each tissue was calculated by the reference sample method (Ishise et al. 1980; Musch & Terrell 1992) and expressed in millilitres per minute per 100 g of tissue. Adequate mixing of the microspheres was verified by demonstrating a < 15 % difference in blood flow to the right and left kidneys, and/or the right and left solei.
Statistical analysis
Microvascular PO2 and resultant model parameters from rest to electrical stimulation, as well as differences between first and second stimulation periods and blood flow, were analysed by means of a paired t test. Data are presented as mean ± standard error of the mean (s.e.m.). Significance was accepted at P ≤ 0.05.
RESULTS
Responses from two animals were discarded due to movement of the PO2,m measurement plane during stimulation. Therefore, data were collected and presented from the remaining animals (n = 6). Individual data on blood gases, pH, lactate, and haematocrit are presented in Table 1.
Table 1.
Haematocrit, oxygen pressure, and acid-base status of arterial blood
| Rat | Hct (%) | PO2 (mmHg) | [La] (mmol l−1) | pH |
|---|---|---|---|---|
| 1 | 49 | 95 | 1.2 | 7.34 |
| 2 | 52 | 89 | 1.3 | 7.38 |
| 3 | 51 | 80 | 1.6 | 7.34 |
| 4 | 50 | 84 | 1.3 | 7.39 |
| 5 | 51 | 92 | 1.1 | 7.37 |
| 6 | 53 | 86 | 0.9 | 7.39 |
| Mean ±s.e.m. | 51 ± 0.5 | 87. 4 ± 2.3 | 1.2 ± 0.1 | 7.36 ± 0.01 |
Hct, haematocrit; PO2, pressure of O2 in arterial blood; [La], blood lactate concentration.
The criterion established for adequate mixing (i.e. ≤15 % difference in flow between left and right solei and kidneys) was met in all four animals used for spinotrapezius blood flow measurements. No difference was observed in blood flow between pre-stimulation (19.6 ± 4.6 ml min−1 (100 g)−1) and post-5 min (16.8 ± 3.8 ml min−1 (100 g)−1) or 10 min (corresponding to pre-ST2; 14.0 ± 1.3 ml min−1 (100 g)−1, both P > 0.1 with respect to control) after cessation of ST1 stimulation. In addition, there were no significant differences in conductance between the three conditions (pre-ST1, 0.24 ± 0.07; post-5 min, 0.21 ± 0.04; post-10 min, 0.19 ± 0.02 ml min−1 (100 g)−1 mmHg−1, P > 0.10).
There was no difference in mean arterial pressure between ST1 and ST2 (104.7 ± 8.3 vs. 105.7 ± 8.3 mmHg, respectively). Neither the pre-stimulation baseline values for PO2,m (ST1, 28.5 ± 2.6; ST2, 27.9 ± 2.4 mmHg) nor the change (Δ) in PO2,m (pre-stimulation minus steady-state contracting, ΔST1, 13.9 ± 1.8; ΔST2, 14.1 ± 1.3 mmHg) were different between the two stimulation periods (both P > 0.05, Fig. 1).
Figure 1. Comparison of pre-contractions baseline PO2,m (left) and change in PO2,m from rest to contractions (right) for the first (ST1) and second (ST2) contraction bouts.

No change in pre-contracting baseline PO2,m between first and second bouts reflects a similar QO2/V̇O2 prior to the two contraction bouts.
Figure 2 shows representative responses for PO2,m and resultant model fits for ST1 and ST2. The mono-exponential + delay curve fitted the data qualitatively well as evidenced from visual inspection of Fig. 2, and this observation was supported by the high correlation coefficients (ST1, r = 0.976 ± 0.014; ST2, r = 0.961 ± 0.014) and low chi-square values (ST1, χ2 = 37.7 ± 11.9; ST2, χ2 = 91.1 ± 37.9) found. ST2 consistently exhibited a significantly (P < 0.05) reduced time delay (TD) compared to ST1 (TD1, 12.2 ± 1.9 vs. TD2, 5.7 ± 2.2 s, Fig. 3). However, the prior contraction period did not affect the time constant of the response (τ) significantly (τST1, 16.3 ± 2.6; τST2, 17.6 ± 2.7 s, P > 0.1, Fig. 3). As expected with a reduced TD2, the time to reach 63 % of the final response (t63) was significantly (P < 0.05) shorter for ST2 (20.2 ± 1.8 s) compared to ST1 (28.3 ± 3.0 s).
Figure 2. Comparison of PO2,m dynamics in first (ST1) and second (ST2) contraction bouts for an individual muscle from onset of stimulation (time 0).
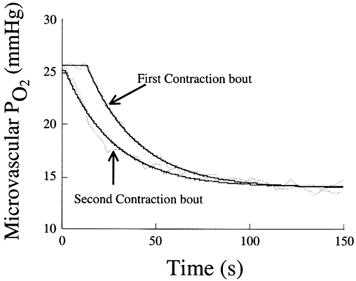
Note the shorter time delay observed across the second contractions transient with relatively no change in the primary (exponential) component of the response. Smoothed lines represent model fits.
Figure 3. Mean response data for first (ST1) and second (ST2) contraction bouts.
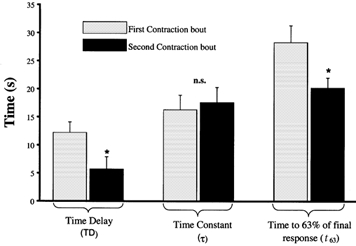
Note the significantly reduced time delay in the second contraction bout leading to a reduced time to 63 % of the final response (t63). *P < 0.05.
DISCUSSION
The present investigation demonstrates that the dynamics of microvascular PO2 (PO2,m) are speeded by a bout of prior or priming contractions as demonstrated by the reduced time to 63 % (t63) of the final response. This speeding seen during the second contraction bout (ST2) was caused by a shortened time delay, which accelerated the onset of PO2,m fall, the time constant of which were essentially unchanged. Our results indicate that this speeding occurred in the absence of a pre-existing arterial lactic acidaemia or an elevated PO2,m. We believe the latter observation provides the first evidence in a mammalian muscle preparation with intact blood supply that an increased muscle blood flow and/or PO2,m is not requisite for accelerating the dynamics of microvascular O2 exchange in a subsequent bout of contractions. Thus, the present data support the notion that priming exercise speeds the dynamics of PO2,m at the onset of contractions via an intracellular mechanism.
Effect of priming exercise on V̇O2 kinetics
It is well known that pulmonary V̇O2 kinetics can be accelerated by prior exercise in the human (moderate domain (Koppo & Bouckaert, 2000); heavy-domain (Gerbino et al. 1996; MacDonald et al. 1997; Bohnert et al. 1998; Burnley et al. 2000)) as well as the horse (moderate-domain (Tyler et al. 1996); moderate & heavy-domains (Geor et al. 2000)). Gerbino et al. (1996) demonstrated that, after a heavy exercise warm-up, there is a speeding of muscle V̇O2 kinetics in the second bout of exercise that reduces the ‘effective’ time constant. Subsequently, Burnley et al. (2000), using a more complex three component model, demonstrated that the faster V̇O2 kinetics in the second bout resulted from a decreased amplitude of the slow component with no change in phase II kinetics. However, the precise mechanisms underlying the accelerated V̇O2 kinetics remain obscure. Recognizing that the pulmonary V̇O2 response reflects closely that occurring across the contracting muscle(s) for both the primary fast and slow components (Barstow et al. 1990; Poole et al. 1991; Grassi et al. 1996;), the obvious next step was to examine O2 exchange across (or within) the active muscle across the transition to sequential contraction bouts. However, prior to the present investigation, measurement of O2 exchange within mammalian muscle (blood supply intact) across the transition to sequential bouts of contractions has not, to our knowledge, been made.
Mechanistic hypothesis
The rate of oxygen delivery (Q̇O2) to the exercising muscle has been suggested as a possible mechanism limiting the speed of the V̇O2 dynamics at exercise onset for moderate, heavy, and severe domain exercise (Hughson et al. 1993). Indeed, Gerbino et al. (1996) argue for an increased O2 delivery to the exercising muscle as a potential mechanism for the speeding of V̇O2 kinetics during the second heavy exercise bout. They attributed this speeding of V̇O2 kinetics to a residual vasodilation resulting from the metabolic acidaemia caused by the first exercise bout. Using the electrically stimulated gastrocnemius model, Grassi et al. (1998a,b) have demonstrated that neither increasing bulk O2 delivery (via adenosine infusion) nor enhancing peripheral O2 diffusion (via RSR-13 with hyperoxia) to the muscle at exercise onset speeds the t63 in the moderate exercise intensity domain. However, increased QO2 and also arterial O2 content (and PO2) both speed V̇O2 kinetics in the heavy/severe domains (MacDonald et al. 1997; Grassi et al. 2000). These findings in the moderate domain are consistent with the speed of V̇O2 kinetics being determined by a metabolic inertia intrinsic to the exercising muscle, whereas in the heavy/severe domains muscle QO2 (and possibly arterial PO2) may limit V̇O2 kinetics. It is also pertinent that, within single amphibian muscle fibres that are not subject to vascular O2 delivery limitations, cytosolic PO2 dynamics are speeded (via reduced time delay) by prior contractions (Hogan 2001). Thus, there is compelling evidence that prior exercise speeds muscle O2 dynamics via an intracellular mechanism in both isolated single fibres (Hogan 2001) and whole muscle (blood supply intact, present results).
Phosphorescence quenching (Rumsey et al. 1988) is a powerful tool that allows observation of the relationship between the local QO2 and V̇O2 through the measurement PO2,m. From the model used herein, if there was an improved vascular perfusion (i.e. increased QO2) above that of the metabolic demand (V̇O2), prior to the start of the second bout of contractions, the presence of an elevated baseline PO2,m would be expected. Pertinent to this, Yoshida & Whipp (1994) demonstrated that cardiac output (and presumably QO2) and pulmonary V̇O2 at the off-transient from whole body exercise in the moderate-intensity domain both return to baseline values within ∼4 min post-exercise. Therefore, with the 10 min recovery period used in the present investigation, no significant differences in V̇O2 or QO2in separatum or QO2/V̇O2 prior to the second contraction period would be expected, and indeed none were found. Pre-stimulation baseline PO2,m values were almost exactly the same prior to ST2 as found earlier for ST1 (i.e. ST1, 28.5 ± 2.6; ST2, 27.9 ± 2.4 mmHg, P > 0.05). In conjunction with unchanged baseline blood flow (ST1, 19.6 ± 4.6; ST2, 14.0 ± 1.3 ml min−1 100g−1, P > 0.05), these findings demonstrate that, after 10 min of recovery and prior to ST2, both V̇O2 and QO2 had returned to values close to those present before ST1. This finding is in agreement with the work of McDonough et al. (2001) who demonstrated recently in the same model used herein that PO2,m, and thus the local QO2/V̇O2 ratio, reaches its prior contraction baseline value at approximately 4 min of recovery. The data from the present study therefore provide strong evidence that an increased muscle QO2, or an increased QO2/V̇O2 ratio, is not necessary to accelerate the dynamics of O2 exchange of the contracting muscle.
Intracellular energetics
The results from the present study, particularly the reduced time delay for ST2, suggest an earlier onset in the increase of V̇O2 (or oxidative phosphorylation) compared to that of QO2 across the second contraction transient. There are many mechanisms postulated for an earlier activation of oxidative phosphorylation and one is an increased rate in, or driving force for, the delivery of reducing equivalents (NADH, FADH2) to the mitochondria at exercise onset. This process is controlled, in part, by the pyruvate dehydrogenase complex (PDC), of which an earlier or more rapid activation (via dichloroacetate) has been demonstrated to reduce phosphocreatine (PCr) degradation during dynamic exercise in some (Timmons et al. 1998) but not all (Gladden et al. 2001) investigations. If a reduced energetic contribution of PCr breakdown reflects a decreased participation of non-oxidative energy sources across the second exercise on-transient, an accelerated V̇O2 kinetics and consequent reduction of the O2 deficit would be expected.
In vivo, PDC is a highly regulated multienzyme complex (Wieland, 1983, for review), of which there are multiple enhancers (e.g. Ca2+, ADP, pyruvate) and inhibitors (e.g. ATP, increased NADH/NAD+ ratio) that control the flux of pyruvate-derived acetyl-CoA through the tricarboxylic acid (TCA) cycle. As stated earlier, two key papers addressed PDC activation: Timmons et al. (1998) demonstrated that early PDC activation has the potential to speed muscle V̇O2 kinetics (inferred from measurements of [PCr] breakdown), whereas in contrast Gladden et al. (2001) found no evidence of speeding of V̇O2 kinetics with pre-exercise PDC activation. Thus, this issue remains controversial.
Theoretical considerations
At the onset of a single contraction or the first bout of sequential muscle contractions, two PO2,m profiles have been measured. In the majority of instances (∼70 %), there is a delay of 10–20 s prior to the onset of an exponential fall of PO2,m as seen in Fig. 4, profile ‘b’ (Behnke et al. 2001; Fig. 2). In the remaining ∼30 % of instances, PO2,m becomes elevated transiently before its exponential descent to the steady-state (Fig. 4, profile ‘a’). Profile ‘b’ is consistent with the precise matching of QO2-to-V̇O2 across the first 20 s of the transition such that PO2,m is unchanged for that period, whereas profile ‘a’ corresponds to an over-perfusion (increased QO2-to-V̇O2 ratio) resulting in an elevated PO2,m. Profile ‘a’ (elevation of PO2,m for 10–20 s after onset of contractions) would be predicted from the human studies of De Cort et al. (1991) and Grassi et al. (1996) where a more rapid QO2 than V̇O2 (i.e. τ QO2< τV̇O2) response at exercise onset led to a transient elevation of effluent muscle venous PO2. As discussed by Behnke et al. (2001), it is possible that the anaesthesia requisite for the muscle preparation used herein may blunt the QO2 response preventing the PO2,m overshoot (response ‘a’) from being manifested in more instances.
Figure 4. Theoretical responses in PO2,m across the rest-to-contraction transition for three muscles with differentQO2-to-V̇O2 dynamics.
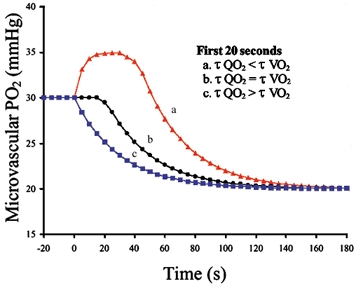
Responses ‘a’ and ‘b’ reflect data from Behnke et al. (2001). Responses close to ‘b’ and ‘c’ were observed in the present investigation for ST1 and ST2, respectively. τ denotes the time constant of the response.
At the onset of the second bout of muscle contractions a response closer to profile ‘c’ in Fig. 4 was observed. Specifically, PO2,m began to decrease after a foreshortened delay, which suggests that muscle V̇O2 increased more rapidly than QO2 after only ∼6 s in ST2vs. ∼12 s in ST1. At present, we have no reason to suspect that QO2 dynamics were slowed on the second bout and an accelerated V̇O2 response is in agreement with the findings of Hogan (2001) that intramyocyte PO2 falls after a reduced time delay in single amphibian myocytes.
Although a reduced time delay was apparent across the second contractile transient, the time constant (τ) of the response was unaltered. This would suggest that, after the delay phase, a proportionality exists in the V̇O2 and QO2 responses (i.e. accelerated QO2 in concert with V̇O2 response) between ST1 and ST2 such that the τ of the PO2,m responses was unaltered. This is supported by data from human studies where no change in the primary component of the pulmonary V̇O2 response (Burnley et al. 2000) was observed after a bout of priming exercise. It should be noted, however, that a reduced time delay for muscle V̇O2 might theoretically shorten phase I of the pulmonary V̇O2 response if the resultant reduction in venous PO2 was of greater amplitude than the cardiodynamic phase (phase I). It is pertinent, however, that the PO2,m measurements made herein are not confined to the temporal resolution of breathing frequency or subject to breath-to-breath variation. Rather, the measurement system used herein has a high fidelity and a high frequency of measurement that permits detection of small changes in the QO2/V̇O2 (i.e. corresponding to < 1 mmHg PO2,m). With pulmonary breath-to-breath measurements, an earlier increase in pulmonary V̇O2 may possibly be masked by noise or lost in the averaging functions commonly employed for data analysis. It is pertinent that a reduced time delay of muscle V̇O2 on-kinetics would result in an attenuated oxygen deficit. However, a mechanistic link between faster V̇O2 dynamics at exercise onset following priming exercise and the subsequent reduction of the V̇O2 slow component (e.g. Burnley et al. 2000) remains to be resolved.
Preparation considerations
In vivo, during voluntary contractions there is a heterogeneity of motor unit recruitment (Gollnick et al. 1974), whereas when a muscle is electrically stimulated, there will be a more homogeneous motor unit activation which is likely to change blood flow distribution due to the concurrent activation of all fibres. In addition, there would not be the rapid increase of cardiac output and elevated MAP that attends voluntary muscle activation (De Cort et al. 1991).
Conclusions
In conclusion, prior contractions in the spinotrapezius muscle elicited faster PO2,m dynamics in a subsequent bout of contractions as evidenced from the reduced time delay and accelerated t63, but there was no change in the τ of the response. Moreover, this speeding was evident in the absence of an elevated PO2,m, muscle blood flow or QO2/V̇O2, or arterial lactacidaemia prior to the onset of the second bout of contractions. Therefore, the results of the present investigation provide strong evidence that the speeding of V̇O2 kinetics by prior contractions is not dependent on a residual vasodilation or local lactacidaemia, at least within the experimental model utilized herein. This supports the notion that the mechanism(s) by which prior exercise speeds V̇O2 kinetics resides within the oxidative machinery of the contracting musculature.
Acknowledgments
The authors gratefully acknowledge the technical assistance of K. Sue Hageman, Troy E. Richardson and Janet K. Bailey, and also the insightful discussions with Drs Thomas J. Barstow and Paul McDonough. This work was supported, in part, by grants from The National Institutes of Health HL-50306, HL-57226, R21AG19228, and AHA (Heartland Affiliate) 51321Z.
REFERENCES
- Barstow TJ, Lamarra N, Whipp BJ. Modulation of muscle and pulmonary O2 uptakes by circulatory dynamics during exercise. Journal of Applied Physiology. 1990;68:979–989. doi: 10.1152/jappl.1990.68.3.979. [DOI] [PubMed] [Google Scholar]
- Behnke BJ, Kindig CA, Musch TI, Koga S, Poole DC. Dynamics of muscle microvascular oxygen pressure across the rest-exercise transition. Respiration Physiology. 2001;126:53–63. doi: 10.1016/s0034-5687(01)00195-5. [DOI] [PubMed] [Google Scholar]
- Bevington PR. Data Reduction and Error Analysis for Physical Sciences. New York: McGraw-Hill; 1969. chap. 1–4. [Google Scholar]
- Bohnert B, Ward SA, Whipp BJ. Effects of prior arm exercise on pulmonary gas exchange kinetics during high-intensity leg exercise in humans. Experimental Physiology. 1998;83:557–570. doi: 10.1113/expphysiol.1998.sp004138. [DOI] [PubMed] [Google Scholar]
- Burnley M, Jones AM, Carter H, Doust JH. Effects of prior exercise on phase II pulmonary oxygen uptake kinetics during heavy exercise. Journal of Applied Physiology. 2000;89:1387–1396. doi: 10.1152/jappl.2000.89.4.1387. [DOI] [PubMed] [Google Scholar]
- De Cort SC, Innes JA, Barstow TJ, Guz A. Cardiac output, oxygen consumption and arteriovenous oxygen difference following a sudden rise in exercise level in humans. Journal of Physiology. 1991;441:501–512. doi: 10.1113/jphysiol.1991.sp018764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geor RJ, McCutcheon J, Hinchcliff KW. Effects of warm-up intensity on kinetics of oxygen consumption and carbon dioxide production during high-intensity exercise in horses. American Journal of Veterinary Research. 2000;61:638–645.C. doi: 10.2460/ajvr.2000.61.638. [DOI] [PubMed] [Google Scholar]
- Gerbino A, Ward SA, Whipp BJ. Effects of prior exercise on pulmonary gas-exchange kinetics during high-intensity exercise in humans. Journal of Applied Physiology. 1996;80:99–107. doi: 10.1152/jappl.1996.80.1.99. [DOI] [PubMed] [Google Scholar]
- Gladden LB, Hogan MC, Greenhaff PL, Hamann JJ, Kelley KM, Aschenbach WG, Constantin-Teodosiu D, Grassi B. Metabolic effects of PDC activation in dog gastrocnemius. Medicine and Sience in Sports and Exercise (abstract) 2001;33:S328. [Google Scholar]
- Gollnick PD, Peihl K, Saltin B. Selective glycogen depletion pattern in human muscle fibres after exercise of varying intensity and at varying pedalling rates. Journal of Physiology. 1974;241:45–57. doi: 10.1113/jphysiol.1974.sp010639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grassi B, Gladden LB, Samaja M, Stary CM, Hogan MC. Faster adjustment of O2 delivery does not affect V̇O2 on-kinetics in isolated in situ canine muscle. Journal of Applied Physiology. 1998b;84:1398–1403. doi: 10.1152/jappl.1998.85.4.1394. [DOI] [PubMed] [Google Scholar]
- Grassi B, Gladden LB, Stary CM, Wagner PD, Hogan MC. Peripheral O2 diffusion does not affect V̇O2 on-kinetics in isolated in situ canine muscle. Journal of Applied Physiology. 1998a;85:1404–1412. doi: 10.1152/jappl.1998.85.4.1404. [DOI] [PubMed] [Google Scholar]
- Grassi B, Hogan MC, Kelley KM, Aschenbach WG, Hamann JJ, Evans RK, Patillo RE, Gladden LB. Role of convective O2 delivery in determining V̇O2 on-kinetics in canine muscle contracting at peak V̇O2. Journal of Applied Physiology. 2000;89:1293–1301. doi: 10.1152/jappl.2000.89.4.1293. [DOI] [PubMed] [Google Scholar]
- Grassi B, Poole DC, Richardson RS, Knight DR, Erickson BK, Wagner PD. Muscle O2 uptake kinetics in humans: implications for metabolic control. Journal of Applied Physiology. 1996;80:988–998. doi: 10.1152/jappl.1996.80.3.988. [DOI] [PubMed] [Google Scholar]
- Hogan MC. Fall in intracellular PO2 at the onset of contractions in Xenopus single skeletal muscle fibers. Journal of Applied Physiology. 2001;90:1871–1876. doi: 10.1152/jappl.2001.90.5.1871. [DOI] [PubMed] [Google Scholar]
- Hughson RL, Cochrane JE, Butler CG. Faster O2 uptake kinetics at onset of supine exercise with than without lower body negative pressure. Journal of Applied Physiology. 1993;75:1962–1967. doi: 10.1152/jappl.1993.75.5.1962. [DOI] [PubMed] [Google Scholar]
- Ishise S, Pegram BL, Yamamoto J, Kitamura Y, Frolich ED. Reference sample microsphere method: cardiac output and blood flows in conscious rat. American Journal of Physiology. 1980;239:H433–H449. doi: 10.1152/ajpheart.1980.239.4.H443. [DOI] [PubMed] [Google Scholar]
- Koppo K, Bouckaert J. In humans the oxygen uptake slow component is reduced by prior exercise of high as well as low intensity. European Journal of Applied Physiology. 2000;83:559–565. doi: 10.1007/s004210000295. [DOI] [PubMed] [Google Scholar]
- Lahiri S, Rumsey WL, Wilson DF, Iturriaga R. Contribution of in vivo microvascular PO2 in the cat carotid body chemotransduction. Journal of Applied Physiology. 1993;75:1035–1043. doi: 10.1152/jappl.1993.75.3.1035. [DOI] [PubMed] [Google Scholar]
- Laurent D, Arthier B, Lebas JF, Rossi A. Effect of prior exercise in Pi/PC ratio and intracellular pH during standardized exercise. A study on human muscle using [31P]NMR. Acta Physiologica Scandinavica. 1992;144:31–38. doi: 10.1111/j.1748-1716.1992.tb09264.x. [DOI] [PubMed] [Google Scholar]
- Lo L-W, Vinogradov SA, Koch CJ, Wilson DF. A new, water soluble, phosphor for oxygen measurement in vivo. Advances in Experimental Medicine and Biology. 1997;411:577–583. doi: 10.1007/978-1-4615-5399-1_91. [DOI] [PubMed] [Google Scholar]
- MacDonald M, Pederson PK, Hughson RL. Acceleration of V̇O2 kinetics in heavy submaximal exercise by hyperoxia and prior high-intensity exercise. Journal of Applied Physiology. 1997;83:1318–1325. doi: 10.1152/jappl.1997.83.4.1318. [DOI] [PubMed] [Google Scholar]
- McDonough P, Behnke BJ, Kindig CA, Poole DC. Muscle microvascular PO2 kinetics during the exercise off-transient. Experimental Physiology. 2001;86:349–356. doi: 10.1113/eph8602192. [DOI] [PubMed] [Google Scholar]
- Musch TI, Terrell JA. Skeletal muscle blood flow abnormalities in rats with chronic myocardial infarction: rest and exercise. American Journal of Physiology. 1992;262:H411–H419. doi: 10.1152/ajpheart.1992.262.2.H411. [DOI] [PubMed] [Google Scholar]
- Poole DC, Schaffartzik W, Knight DR, Derion T, Kennedy B, Guy HJ, Prediletto R, Wagner PD. Contributions of exercising legs to the slow component of oxygen uptake kinetics in humans. Journal of Applied Physiology. 1991;71:1245–1260. doi: 10.1152/jappl.1991.71.4.1245. [DOI] [PubMed] [Google Scholar]
- Rumsey WL, Vanderkooi JM, Wilson DF. Imaging of phosphorescence: a novel method for measuring oxygen distribution in perfused tissue. Science. 1988;241:1649–1651. doi: 10.1126/science.241.4873.1649. [DOI] [PubMed] [Google Scholar]
- Scheuermann BW, Hoelting BD, Noble ML, Barstow TJ. The slow component of O2 uptake is not accompanied by changes in muscle EMG during repeated bouts of heavy exercise in humans. Journal of Physiology. 2001;531:245–256. doi: 10.1111/j.1469-7793.2001.0245j.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmons JA, Gustafsson T, Sundberg CJ, Jansson E, Greenhaff PL. Muscle acetyl group availability is a major determinant of oxygen deficit in humans during submaximal exercise. American Journal of Physiology. 1998;274:E377–E380. doi: 10.1152/ajpendo.1998.274.2.E377. [DOI] [PubMed] [Google Scholar]
- Tyler CM, Hodgson DR, Rose RJ. Effect of a warm-up on energy supply during high intensity exercise in horses. Equine Veterinary Journal. 1996;28:117–120. doi: 10.1111/j.2042-3306.1996.tb01602.x. [DOI] [PubMed] [Google Scholar]
- Wieland OH. The mammalian pyruvate dehydrogenase complex: structure and regulation. Reviews of Physiology, Biochemistry and Pharmacology. 1983;96:123–170. doi: 10.1007/BFb0031008. [DOI] [PubMed] [Google Scholar]
- Yoshida T, Kamiya J, Hishimoto K. Are oxygen uptake kinetics at the onset of exercise speeded up by local metabolic status in active muscles. European Journal of Applied Physiology and Occupational Physiology. 1995;70:482–486. doi: 10.1007/BF00634376. [DOI] [PubMed] [Google Scholar]
- Yoshida T, Kamiya J, Hishimoto K. Are oxygen uptake kinetics at the onset of exercise speeded up by local metabolic status in active muscles? European Journal of Applied Physiology and Occupational Physiology. 1994;70:482–486. doi: 10.1007/BF00634376. [DOI] [PubMed] [Google Scholar]