

Abstract
We investigated the receptor-mediated regulation of nifedipine-insensitive, high voltage-activated Ca2+ currents in guinea-pig terminal mesenteric arterioles (ImVDCC) using the whole-cell clamp technique. Screening of various vasoactive substances revealed that ATP, histamine and substance P exert modulatory effects on ImVDCC. The effects of ATP on ImVDCC after complete P2X receptor desensitization exhibited a complex concentration dependence. With 5 mm Ba2+, ATP potentiated ImVDCC at low concentrations (∼1–100 μm), but inhibited it at higher concentrations (>100 μm). The potentiating effects of ATP were abolished by suramin (100 μm) and PPADS (10 μm) and by intracellular application of GDPβS (500 μm), whereas a substantial part of ImVDCC inhibition by milimolar concentrations of ATP remained unaffected; due probably to its divalent cation chelating actions. In divalent cation-free solution, ImVDCC was enlarged and underwent biphasic effects by ATPγS and ADP, while 2-methylthio ATP (2MeSATP) exerted only inhibition, and pyrimidines such as UTP and UDP were ineffective. ATP-induced ImVDCC potentiation was selectively inhibited by anti-Gαs antibodies or protein kinase A (PKA) inhibitory peptides and mimicked by dibutyryl cAMP. In contrast, ATP-induced inhibition was selectively inhibited by Gαq/11 antibodies or protein kinase C (PKC) inhibitory peptides and mimicked by PDBu. Pretreatment with pertussis toxin was ineffective. The apparent efficacy for ImVDCC potentiation with PKC inhibitors was: ATPγS > ATP≥ADP and for inhibition with PKA inhibitors was: 2MeSATP > ATPγS > ATP > ADP. Neither ImVDCC potentiation nor inhibition showed voltage dependence. These results suggest that ImVDCC is multi-phasically regulated by external ATP via P2Y11-resembling receptor/Gs/PKA pathway, P2Y1-like receptor/Gq/11/PKC pathway, and metal chelation.
Voltage-dependent Ca2+ channels (VDCCs) serve as a main potential-dependent Ca2+ entry pathway in a wide range of tissues and have been implicated in a variety of cellular processes such as muscle contraction, neurotransmitter release, cell proliferation and development (Bean, 1989a). Several distinct classes of VDCCs have been identified on a biophysical and pharmacological bases (L-, N-, P/Q-, T-, R-type), and subsequently, ten α1-subunit encoding genes responsible for these phenotypes have been cloned (α1A-I and α1S; Davila, 1999; Hofmann et al. 2000). Amongst them, the dihydropyridine-sensitive, L-type VDCC has been found ubiquitously over the whole vasculature and is thought to play a crucial role in the control of blood flow and pressure (Nelson et al. 1990). However, it has recently been reported that, in the peripheral branches of the mesenteric arterial tree (or higher-ordered arterioles), the predominant VDCC is a class of high voltage-activated (HVA) channels, the properties of which do not match up with those of hitherto-known VDCCs (Morita et al. 1999; hereafter designated as mVDCC). In addition to its unique biophysical properties, the mVDCC is totally insensitive to known blockers for L-, N-, P/Q-, T- and R-type VDCCs such as nifedipine, verapamil, diltiazem, ω-conotoxins GVIA and MVIIC, and ω-agatoxin IVA (Morita et al. 1999), thus presumably belonging to a new class of HVA-VDCC that has not yet been characterized at the molecular level.
Detailed electrophysiological analysis of mVDCC has revealed that, despite its rapidly inactivating nature, there is a range of membrane potential in which constant or non-inactivating Ca2+ influx occurs. The physiological significance of non-inactivating Ca2+ influx has been emphasized for L-type VDCC, as the critical determinant of free Ca2+ concentration ([Ca2+]i) in arterial smooth muscle cells and thus of the arterial diameter or tone under pressurized conditions (for review see Nelson et al. 1990). Furthermore, this Ca2+ influx has been thought to be effectively regulated by the modulatory actions of various vasoactive substances such as neurotransmitters (e.g. noradrenaline, neuropeptide Y, acetylcholine, vasointestinal peptide and calcitonin gene-related peptide), vasoactive autacoids which are released from the vascular endothelium or produced during local inflammatory processes (e.g. nitric oxide, endothelium-derived hyperpolarizing factor, endothelin, histamine and bradykinin) and circulating hormones released from distant endocrine organs (e.g. angiotensin II and vasopressin) (Beech, 1998; Kuriyama et al. 1998).
In the present study, we have therefore addressed the question of whether receptor-mediated regulation has a similar physiological significance in modifying the mVDCC activity. To this end, we screened the effects on mVDCC of vasoactive substances known to affect the electrical and contractile properties of vascular smooth muscle. We have found that ATP, a well established fast neurotransmitter of the vascular sympathetic nerves (Burnstock, 1990), exerts the most pronounced dose-dependent modulatory effects on mVDCCs through three distinct mechanisms. The preliminary account of this work has been presented in the 73rd annual meeting of the Japanese Pharmacological Society (Morita et al. 2000).
METHODS
Cell dispersion and electrophysiological measurements
Procedures used for cell dispersion and the system for patch clamp experiments were the same as described previously (Morita et al. 1999) and performed according to the guidelines approved by a local animal ethics committee of Kyushu University. In brief, guinea-pigs of either sex weighing 200–500 g were killed by decapitation after stunning under light anaesthesia with inhalation of diethyl ether. Short segments from the distal half of terminal branches of mesenteric artery measuring 70–100 μm in diameter were mechanically dissected with fine scissors and forceps, and incubated successively in nominally Ca2+-free Krebs solutions without and with 2 mg ml−1 collagenase (Sigma type I) at 35°C for 30 and 60 min, respectively. Single cells, yielded by gently triturating these digested segments using a blunt tipped pipette 20 to 30 times, were stored in 0.5 mm Ca2+-containing Krebs solution at 10°C until use.
A commercial amplifier (Axopatch 1D, Axon Instruments) in conjunction with an A/D, D/A converter was used to generate voltages and sample current signals after low-pass filtering at 1 kHz (digitized at 2 kHz), under the control of an IBM computer (Aptiva) which was driven by a commercial software Clampex v.6.02 (Axon Instruments). The P/4 or P/2 method was used to subtract leak currents, and 50 to 70 % of series resistance (10–15 MΩ) was electronically compensated. Data analyses and illustration were performed using Clampfit v.6.02 (Axon Instruments). All experiments were performed at room temperature (22–25°C).
Solutions
Solutions of the following composition were used (mm): 5 Ba2+-external solution: Na+ 140, K+ 6, Ba2+ 5, Mg2+ 1.2, Cl− 158.4, glucose 10, Hepes 10 (pH 7.4; adjusted by Tris base); divalent cation-free external solution: Na+ 140, K+ 6, Cl− 146, EDTA 0.2, glucose 10, Hepes 10 (pH 7.4; adjusted by Tris base). All external solutions were supplemented with nifedipine 10 μm and were superfused at a rate of 1–2 ml min−1 into the recording chamber (volume ∼0.2 ml), via a gravity-fed perfusion system (time of complete solution change ∼30 s); Cs+-internal solution: Cs+ 140, Mg2+ 2, Cl− 144, phosphocreatine 5, Na2ATP 1, GTP 0.2, EGTA 10, Hepes 10 (pH 7.2; adjusted by Tris base).
Free ATP and Ba2+ concentrations (Fig. 3) were calculated using Fabiato and Fabiato's program with enthalpic and ionic strength corrections (Brooks & Storey, 1992) using association constants for Ba2+ of 103.29 and 105.1, as performed previously (Inoue & Ito, 2000).
Figure 3. Concentration-dependent effects of ATP on ImVDCC with Na+ as the charge carrier (divalent cation-free conditions).
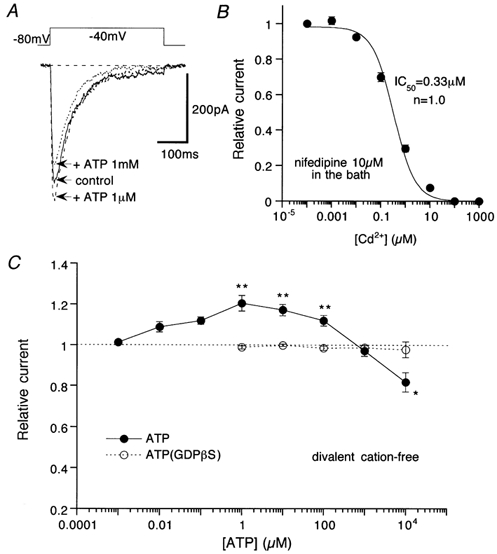
Nifedipine (10 μm) in the bath. A, actual traces of ImVDCC (leak-subtracted). Uppermost trace indicates the waveform of the step pulses used. B, Cd2+ concentration-inhibition curve for ImVDCC. • and bars represent means ±s.e.m. from 5 cells and the smooth continuous curve is the result of Hill fitting. C, relationship between ATP concentration and the ImVDCC amplitude. • and ○, mean of data pooled from 5–20 cells in the absence and presence of 500 μm GDPβS in the pipette, respectively. *P > 0.05 and **P > 0.01 with Student's unpaired t test for the ATP concentration data (• and ○).
Chemicals
The following agents were purchased; angiotensin II, bradykinin, calcitonin gene-related peptide, endothelin-1, histamine, neurokinin A & B, neuropeptide Y, neurotensin, somatostatin, substance P, vasointestinal peptide, vasopressin, ADP, AMP, adenosine, α,β-methylene ATP, ATPγS, AMP-PNP, 2MeSATP, GDPβS, GTPγS, suramin, PPADS (pyridoxalphosphate-6-azophenyl 2′,4′-disulphonic acid), protein kinase A inhibitory peptide (PKI 14–22 amide), protein kinase C inhibitory peptide (PKC 19–31), G-protein α-subunit antibodies (anti-Gαs and anti-Gαq/11) and pertussis toxin from Calbiochem; acetylcholine, noradrenaline, dibutyryl cAMP, SNAP (S-nitroso-N-acetyl penicillamine) from Sigma; ATP, EDTA and EGTA from Dojin (Kumamoto, Japan). Inhibitors were added into the bath at least 5 min, or intracellularly dialysed via the patch pipette for 10–20 min, before application of test agonists.
Statistics
All data are expressed as means ± s.e.m. To evaluate statistical significance of difference between a given set of data, Student's paired and unpaired t tests and one way ANOVA with pooled variance t test were employed.
RESULTS
External ATP exhibits dual actions on mVDCC
We first investigated the effects of various vasoactive agents known to affect the electrical and contractile responses of vascular smooth muscle (Beech, 1998), on the current flowing through mVDCC (ImVDCC) evoked by 100 ms depolarizing pulses to 0 mV at an interval of 20 s from a holding potential of −60 mV, with 5 mm Ba2+ as the charge carrier (10 μm nifedipine present). Under these ionic conditions, the amplitude of ImVDCC was almost tripled compared with that at physiological concentrations of Ca2+ (1–2 mm), without significant changes in the voltage-dependent properties or contamination of other Ca2+-dependent conductances (Morita et al. 1999). The majority of the vasoactive agents tested (5–10 min application; n = 3–6) failed to affect ImVDCC at the sub-maximally or maximally effective concentrations reported to modulate L-type VDCCs (Beech, 1998). These include calcitonin gene-related peptide (100 nm), acetylcholine (1 μm), vasointestinal peptide (1 μm), noradrenaline (10 μm), neuropeptide Y (1 μm), endothelin-1 (1 μm), angiotensin II (1 μm), vasopressin (1 μm), neurokinin A and B (each 1 μm), neurotensin (1 μm), somatostatin (1 μm), bradykinin (10 μm) and the nitric oxide-releasing agent, SNAP (100 μm). In contrast, significant potentiation (10 μm; 119 ± 3 % of control, n = 5) and inhibition (10 mm; 33 ± 2 % of control, n = 5) of ImVDCC occurred with externally applied ATP in a dose-dependent fashion. Modest inhibition was also observed for substance P (1 μm; 77 ± 2 % of control, n = 5) and histamine (10 μm; 90 ± 2 % of control, n = 4). Since ATP is an established major neurotransmitter of vascular sympathetic nerves and is responsible for generating the fast excitatory junction potential (EJP) in the mesenteric arterioles (Starke, 1991; Thapaliya et al. 1999), we concentrated on investigating the effects of ATP in the rest of this study.
Three mechanisms involved in ATP actions
Figure 1A illustrates a typical time course of the effects of ATP on ImVDCC (filled circles) and the holding current under voltage clamp at −60 mV. Immediately after addition of 100 μm ATP in the bath, a rapidly growing and desensitizing inward current was activated, and subsequently, the magnitude of ImVDCC gradually increased. The rapidly desensitizing inward current is likely to be the non-selective cationic current of P2X receptor (Surprenant et al. 1995) and responsible for the fast EJP in the present preparation. This is suggested by the observation that pretreatment with α,β-methylene ATP (10 μm) or total substitution of external cations with a large impermeant cation N-methyl, d-glucamine completely abolished both the inward current and the EJPs (data not shown). In contrast, potentiation of ImVDCC by 100 μm ATP persisted over several minutes and was strongly suppressed by pretreatment with the P2 receptor antagonists, PPADS or suramin (Fig. 1B and C; Fig. 2) but not affected by α,β-methylene ATP (Fig. 1B and C). These results indicate that the non-P2X receptors are involved in the potentiation of ImVDCC.
Figure 1. The effects of ATP and purinergic receptor antagonists on ImVDCC.
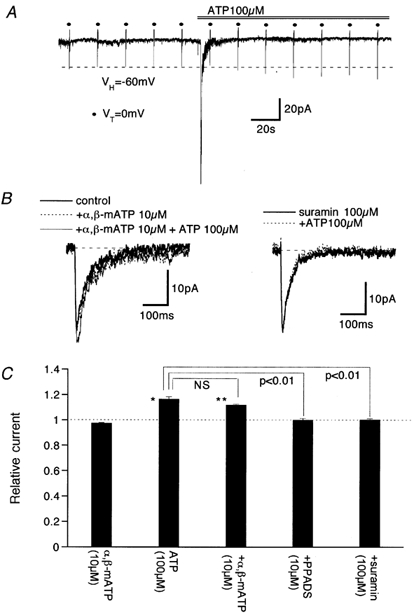
Bath and pipette contained 5 mm Ba2+ external and Cs+ internal solutions, respectively. A, time course of the effects of 100 μm ATP on ImVDCC (•) and holding current. 100 ms depolarizing pulses to 0 mV (VT) from a holding potential of −60 mV (Vh) were applied at an interval of 20 s. B, leak-subtracted traces of ImVDCC in the presence of 10 μmα,β-methylene ATP (α,β-mATP, left) or 100 μm suramin (right) with or without 100 μm ATP. C, summary of the effects of pretreatment with α,β-mATP (10 μm), PPADS (10 μm) and suramin (100 μm) on ATP (100 μm)-induced potentitation of ImVDCC. Columns and bars indicate means ±s.e.m. from 4–5 experiments. *, ** Statistically significant difference (P > 0.05 and 0.01, respectively) from the control value (dotted line). NS (not significant) and P values in the figure indicate the results of one way ANOVA and pooled variance t test.
Figure 2. Concentration-dependent profile of ATP effects on ImVDCC with 5 mm Ba2+ as the charge carrier.
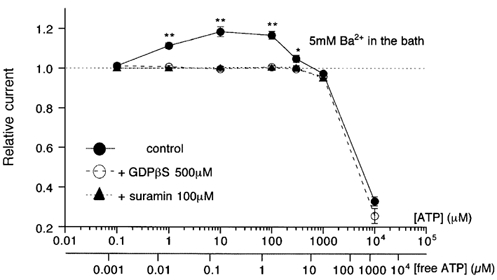
Recording conditions were the same as in Fig. 1. • and ○, and ▴ indicate the amplitude of ImVDCC relative to that before addition of ATP, in the absence and presence of 500 μm GDPβS in the pipette or 100 μm suramin (pretreated for 5 min) in the bath. The effects of ATP were monitored for at least 5 min and the maximum effects were taken. The scale in the lower abscissa (free ATP concentration) was calculated as described in Methods. *, ** Statistically significant difference (P > 0.05 and 0.01, respectively) between • and ○ at each ATP concentration with unpaired t test, respectively.
Significant potentiation of ImVDCC by ATP occurred at a concentration as low as 1 μm, with the maximum response between 10 and 100 μm. However, at higher concentrations of ATP (>100 μm), the potentiating effect declined and was ultimately converted to pronounced inhibition in the milimolar range (filled circles in Fig. 2). ATP-induced ImVDCC potentiation in the micromolar range disappeared when 500 μm GDPβS instead of GTP was included in the pipette (open circles in Fig. 2). However, the inhibition in the milimolar range remained almost unaffected with this procedure (dashed line in Fig. 2). These results strongly suggest that ATP-induced ImVDCC potentiation involves activation of the P2Y receptor/G-protein pathway, whereas inhibition in the milimolar ATP range may result from a mechanism independent of receptor activation, i.e. reduced Ba2+ concentration due to the chelating action of ATP (see below).
In order to eliminate a possible interaction between ATP and divalent cations and also to examine the effects of ATP with a better signal-to-noise ratio, we next recorded ImVDCC using Na+ as the charge carrier under divalent cation-free conditions (200 μm EDTA added in the bath). Under these conditions, the amplitude of ImVDCC was increased 20–40 times and was dependent on the Na+ concentration in the bath, but insensitive to tetrodotoxin (10 μm) and was completely blocked by micromolar concentrations of Cd2+ (Fig. 3A and B). The current-voltage relationship of ImVDCC was shifted negatively by about 20 mV (data not shown).
As summarized in Fig. 3C, the relationship between ATP concentration and the amplitude of the Na+ current through mVDCC channels was shifted to the left by about two logarithmic scales, as compared with that obtained when Ba2+ was the charge carrier (Fig. 2), although marked inhibition of ImVDCC in a very high ATP concentration range disappeared due probably to virtual absence of Na+ chelation by ATP. This can be interpreted as indicating that the free form of ATP is responsible for its effects on ImVDCC. In support of this idea, re-scaling the abscissa of dose-response data with 5 mm Ba2+ solution (Fig. 2) with respect to the free ATP concentration gave a comparable concentration dependence to that observed under divalent cation-free conditions (Fig. 3C). Interestingly, in the micromolar range of ATP (>1 μm), the potentiating effect turned to a clear decline as the concentration of ATP was increased, and substantial inhibition of ImVDCC occurred at concentrations of 1 mm or higher (Fig. 3C). This inhibition could not result from Na+ chelation by ATP (1 mm ATP would cause only ∼0.4 % reduction in Na+ concentration), and seems to correspond to the decline of potentiation observed in the high micromolar range of ATP (100 μm to 1 mm) with Ba2+ as the charge carrier (Fig. 2). The inhibition was more clearly manifested when the potentiating effect was selectively eliminated (see below), and it is likely that a P2Y receptor/G-protein pathway is also involved, since the inhibition was completely abolished when GDPβS (open circles in Fig. 3C) was present in the pipette or P2 antagonists such as suramin and PPADS were added to the bath (not shown).
Two distinct P2Y receptors regulate mVDCC
The P2Y receptors can be pharmacologically distinguished based on the relative potencies of purines and pyrimidines (Kunapuli & Daniel, 1998; King et al. 1998). We therefore compared the potencies of various purines and pyrimidines for potentiating and inhibiting ImVDCC. As summarized in Fig. 4, ADP exhibited concentration-dependent effects on ImVDCC similar to ATP, while AMP and adenosine were ineffective (data not shown). This could suggest that the ATP effects seen were subsequent to its degradation to ADP. However, this possibility is unlikely, since a slowly hydrolysable ATP analogue, ATPγS, also exhibited a comparable efficacy to ATP in both potentiating and inhibiting ImVDCC. We also tested another purine, 2MeSATP, and pyrimidines such as UDP and UTP. While 2MeSATP caused only a dose-dependent inhibition of ImVDCC, UDP or UTP did not exert any discernible effects.
Figure 4. Effects of various nucleotides on ImVDCC evaluated with Na+ as charge carrier (divalent cation-free conditions).
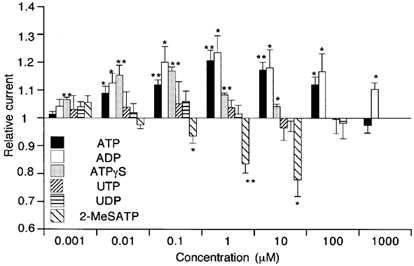
Nifedipine (10 μm) in the bath. Voltage step pulses (100 ms: from −80 to −40 mV) were used to evoke ImVDCC. *P > 0.05 and **P < 0.01 with t test for paired data (n = 5–20) before and after application, respectively, of a given nucleotide at a given concentration.
These pharmacological profiles strongly suggest that there are at least two distinct P2Y receptor/G-protein pathways involved in potentiation and inhibition of ImVDCC (see Discussion).
Involvement of two distinct G-proteins and protein kinases in ATP actions
It has been reported that G-protein-mediated modulation of VDCCs involves both direct interaction with G-protein and phosphorylation/dephosphorylation of VDCC proteins (Bean, 1989b; Dolphin, 1998; Hofmann et al. 1999). However, the former mechanism seems unlikely to account for the observed effects of ATP on ImVDCC. As demonstrated in Fig. 5A, large depolarizing prepulses, which were used to relieve G-protein-mediated inhibition (N- or P/Q-type VDCCs; Dolphin, 1998; Kaneko et al. 1999), merely facilitated the voltage-dependent inactivation of ImVDCC, and did not significantly alter the extent of either ATP-induced potentiation or inhibition of ImVDCC (Fig. 5B). In contrast, inclusion of protein kinase A and C inhibitory peptides in the pipette selectively abolished the ATP-induced potentiation and inhibition of ImVDCC, respectively (open and shaded columns in Fig. 6A), and the simultaneous inclusion of both peptides almost completely abolished the effects of ATP (hatched column in Fig. 6A). The EC50 and IC50 values for ATP-induced ImVDCC potentiation and inhibition evaluated under these conditions are about 10 nm and >10 μm, respectively (Fig. 6A; see also Fig. 7). Consistent with these observations, bath application of dibutyryl cAMP (1 mm), which is a membrane permeable cAMP analogue and directly activates PKA bypassing the receptor, enhanced, whereas that of PKC activator PDBu (250 nm) suppressed, ImVDCC (Fig. 6B).
Figure 5. Large preceding depolarization does not affect the extent of ATP-induced ImVDCC potentiation and inhibition.
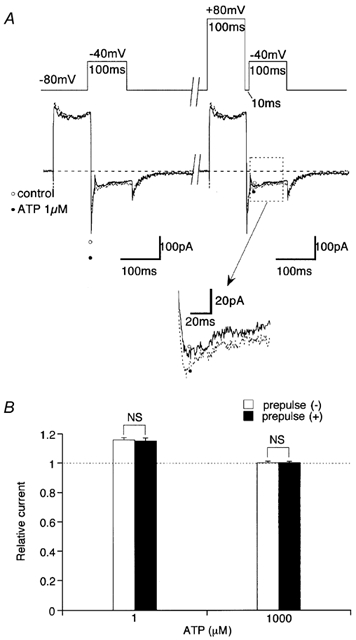
A, voltage protocol (upper trace) and corresponding current traces before (○ and continuous curve) and after (• and dotted curve) addition of 1 μm ATP. Inset indicated by arrow is magnification from a part boxed by dotted line. B, relative amplitude change of ImVDCC after addition of 1 μm (left) or 1 mm (right) ATP with (□) or without (▪) a 100 ms prepulse to 80 mV. Experiments carried out with Na+ as charge carrier (divalent cation-free conditions) in the presence of 10 μm nifedipine in the bath. NS, no statistically significant difference with unpaired t test. n = 4.
Figure 6. Effects of protein kinase inhibitors (A) activators (B), G-protein antibodies (C) and overnight pertussis toxin treatment (D) on ATP-induced modulation of ImVDCC.
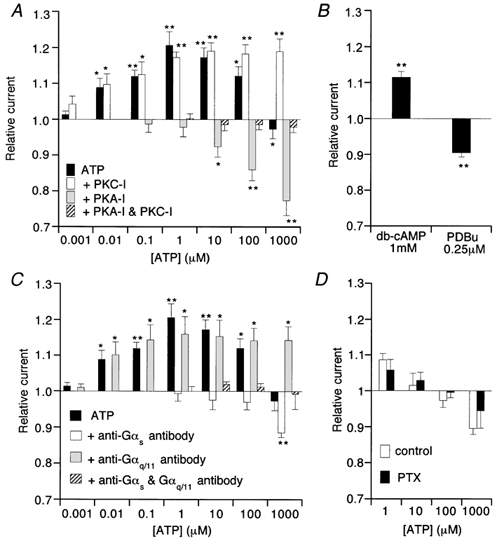
Recording conditions were the same as in Fig. 4. PKC-I, protein kinase C inhibitory peptide (1 μm); PKA-I, protein kinase A inhibitory peptide (41.3 nm); PTX, pertussis toxin. In C, antibodies against Gαs and Gαq/11 were diluted 1:35 in the pipette solution. In D, mesenteric arteriolar myocytes were incubated with (PTX) or without (control) pertussis toxin (500 ng ml−1) at 10°C for 24h. *P > 0.05 and **P < 0.01 with t test for paired data (n = 5) before and after application of a given concentration of ATP, dbcAMP or PDBu.
Figure 7. Efficacies of nucleotides to cause ImVDCC potentiation (A) and inhibition (B) revealed in the presence of protein kinase inhibitors.
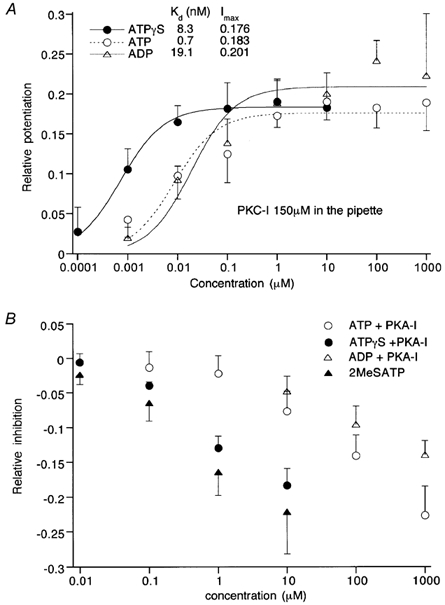
Recording and other experimental conditions were the same as in Fig. 4. A, the extent of ImVDCC potentiation relative to control is plotted against a given nucleotide concentration. Curves are the results of Hill fitting: Imax/(1 +Kd/[nucleotide]), where Imax, Kd and [nucleotide] denote the maximum potentiation, dissociation constant, and given nucleotide concentration, respectively. B, the extent of ImVDCC inhibition relative to control is plotted against a given nucleotide concentration. Symbols and vertical bars represent means ±s.e.m. (n = 4–6).
It is generally thought that receptor-mediated activation of PKA and PKC is mediated through their specific subtypes of G-protein, Gs and Gq/11, respectively. We therefore examined whether antibodies against these G-protein subtypes counteract the ability of ATP to potentiate or inhibit ImVDCC. As summarized in Fig. 6C, 10–20 min intracellular application of Gαs-specific antibody via the patch pipette abolished the potentiating effect but not the inhibitory effect of ATP (open columns in Fig. 6C), and vice versa with Gαq/11- instead of Gαs-specific antibody (shaded columns in Fig. 6C). Simultaneous application of the two antibodies resulted in total abolition of the effects of ATP (hatched columns in Fig. 6C). In addition, overnight pretreatment of mesenteric arteriolar myocytes with pertussis toxin did not significantly alter the effects of ATP, thus excluding the involvement of Gi/Go subtypes in the effects of ATP (Fig. 6D). These results collectively suggest that ATP-induced potentiation and inhibition of ImVDCC primarily involve activation of the P2Y/Gs/PKA and P2Y/Gq/11/PKC pathways, respectively.
The sequence of efficacy of nucleotides to cause ImVDCC potentiation was determined under the conditions in which inhibition via the Gq/11/PKC pathway was selectively eliminated by the PKC inhibitory peptide. As shown in Fig. 7A, Kd values evaluated by Hill fitting suggest that this sequence is ATPγS > ATP ≥ ADP. Similarly, with the PKA inhibitory peptide for eliminating ImVDCC potentiation, the nucleotide sequence to inhibit ImVDCC is 2MeSATP > ATPγS > ATP > ADP (Fig. 7B).
P2Y-mediated modulation of mVDCC shows virtually no alteration of activation and inactivation kinetics
Finally, to gain more insight into the nature of ATP-induced ImVDCC modulation, we investigated what changes would occur in the activation and inactivation kinetics of ImVDCC during application of ATP, using 5 mm Ba2+ as the charge carrier. As illustrated in Fig. 8A, the shape of the current-voltage relationship of ImVDCC remained almost unchanged after potentiation by 100 μm ATP, with a similar extent of increase in ImVDCC amplitude over a wide range of potentials. Correspondingly, there was little discernible shift on application of 100 μm ATP, in either the activation curve for ImVDCC evaluated by the tail current analysis or the quasi steady-state inactivation curve evaluated by 10 s long conditioning prepulses (Fig. 8B). Similar voltage independent properties were also observed when inhibition of ImVDCC by higher concentrations of ATP (1 mm) was separated from potentiation by inclusion of protein kinase A inhibitory peptide in the pipette (Fig. 8C).
Figure 8. ATP-induced ImVDCC potentiation and inhibition is voltage independent.
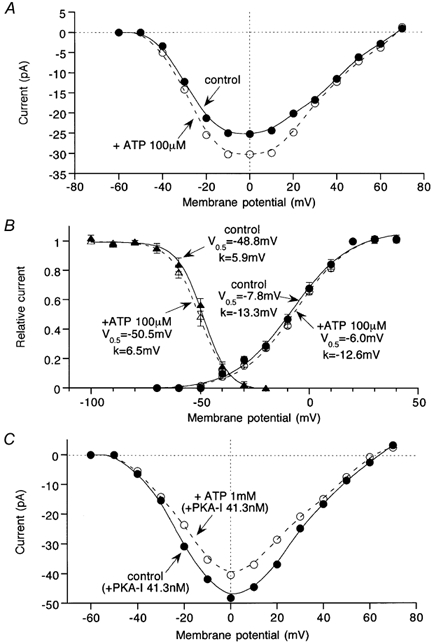
In order to evaluate the voltage-dependent properties of ImVDCC under conditions as similar to the physiological situation as possible, Ba2+ was used as the charge carrier. Bath and pipette contained 5 mm Ba2+-external and Cs+-internal solutions, respectively. A, current-voltage relationships for ImVDCC in the absence and presence of 100 μm ATP (A) in the bath. B, ImVDCC activation curve evaluated by tail current analysis and quasi steady-state inactivation curve evaluated by 10 s preconditioning pulses (see Morita et al. 1999). Smooth curves are the best fit of data points (n = 5) by Boltzmann equation: 1/(1 + exp((Vm - V0.5)/k), where Vm, V0.5 and k denote membrane potential, half activation or inactivation voltage and slope factor, respectively. C, current-voltage relationships for ImVDCC in the absence and presence of 1 mm ATP with 41.3 nm protein kinase A inhibitory peptide in the pipette.
These results, together with the absence of voltage-dependent relief, indicate that P2Y receptor-mediated modulation of ImVDCC occurs through voltage-independent mechanisms which are clearly distinguishable from voltage-dependent receptor-mediated modulation of the other types of dihydropyridine-insensitive, high voltage-activated VDCCs (Dolphin, 1998; Hofmann et al. 1999; Kaneko et al. 1999).
DISCUSSION
The results of the present work clearly show that ATP, but not other potent vasoconstrictors (noradrenaline, angiotensin II and endothelin, etc.), exerts concentration-dependent, triphasic effects on mVDCC activities. In low and high micromolar concentration ranges, ATP caused the potentiation and inhibition of ImVDCC via two distinct G-protein coupled P2Y receptors, while in the milimolar range, it exerted a G-protein-independent inhibition, most likely through divalent cation-trapping actions. These conclusions are supported by the following observations. (1) The first two ATP effects were completely abolished by intracellular application of the G-protein inactivating agent GDPβ and in the presence of P2 antagonists suramin or PPADS but not by the P2X-selective antagonist α,β-methylene ATP. (2) The third effect was not affected by these agents but was almost completely lost under divalent cation-free conditions. Since ATP is well established as a neurotransmitter released from the sympathetic nerves which densely innervate the peripheral resistant arterioles (Burnstock, 1990; Starke, 1991), this novel regulatory mechanism for mVDCCs by ATP might serve as an effective control of blood pressure and local circulation (see below).
Pharmacological investigation using various P2Y agonists/ antagonists and activators/inhibitors for G-proteins and kinases has suggested that the potentiation of ImVDCC by ATP is likely to involve Gs/adenylate cyclase/cAMP/PKA-mediated phosphorylation via a pyrimidine-insensitive P2Y receptor subtype having the agonist sensitivity of ATPγS > ATP ≥ ADP (2MeSATP, UTP and UDP are ineffective) (Table 1; Fig. 4 and Fig. 7). On the other hand, G-protein-dependent inhibition of mVDCCs seems mediated by the Gq/11/PLC β/PKC pathway via a distinct pyrimidine-insensitive P2Y receptor subtype showing an entirely different spectrum of agonist sensitivity, 2MeSATP > ATPγS > ATP > ADP (UTP and UDP are ineffective; Table 1; Fig. 4 and Fig. 7). Compared with the recombinant P2Y receptors so far identified (Kunapuli & Daniel, 1998; King et al. 1998; Jacobson et al. 2000), these overall profiles suggest that the P2Y receptors responsible for inhibition of ImVDCC are most similar to a phosphoinositide turnover-linked receptor, P2Y1 subtype, and those for potentiation have some degree of similarity to an adenylate cyclase-stimulating receptor, P2Y11 subtype (Table 1), although involvement of, as yet, unidentified P2Y isoforms cannot completely be excluded (Boeynaems et al. 2000).
Table 1.
Comparison of nucleotide sensitivity of various P2Y receptors
| Subtype | P2Y1 | P2Y2 | P2Y4 | P2Y6 | P2Y11 | P2Y12 | P2Y(pot) | P2Y(inh) |
| G-protein | Gq/11 | G q/11 | G q/11 | Gq/11 | Gs,G q/11 | Gi/o | Gs | G q/11 |
| Coupling | PLC β/IP3↑ | PLC β/IP3↑ | PLC β/IP3↑ | PLCβ/IP3↑ | PLCb/IP3↑ | AC/cAMP↓ | AC/cAMP↑ | PLC β/IP3↑ |
| AC/cAMP↑ | (DAG/PKC) | |||||||
| Antagonists | ||||||||
| PPADS(10 μm) | + | − | − | +* | na | − | + | + |
| suramin(100 μm) | + | + | − | + | + | + | + | + |
| Agonist | 2MeSATP | UTP≥ATP | UTP=ATP | UDP>UTP | ATPγS>ATP | 2MeSATP | ATPγS> | 2MeSATP> |
| sensitivity | ≫ATP γS | >ATP γS | ≫2MeSATP | >2MeSATP | >ADP | ATP≥ | ATPγS>ATP | |
| >ATP>ADP | ≫ATPγS | ADP | >ADP | |||||
| (MeSATP:NE) |
The agonist sensitivity in the second column is determined based on EC50 values of Jacobson et al. (2000). Other information is based on Boarder & Hourani (1998), King et al. (1998), Kunapuli & Daniel (1998) and Hollopeter et al. (2001). The data in the third column are from the present work. +, effective; + *, effective at 100 μm; −, ineffective. PLCβ, phospholipase Cβ; IP3, inositol 1,4,5-trisphosphate; DAG, diacylglycerol; AC, adenylate cyclase; NE, not effective. Affixes ‘(pot)’ and ‘(inh)’ indicate the potentiation and inhibition of ImVDCC, respectively, na, not available.
Recent contractile studies demonstrated that in several different types of vascular smooth muscle, pyrimidines such as UTP and UDP exert strong vasoconstricting actions via the Gq/11/PLCβ/IP3 pathway (Rubino & Burnstock, 1996; Lagaud et al. 1996; Miyagi et al. 1996; Boarder & Hourani, 1998; Mutafova-Yambolieva et al. 2000; Horiuchi et al. 2001). In support of this, using RT-PCR analysis, mRNA transcripts for pyrimidine-sensitive P2Y2, P2Y4 or P2Y6 receptor subtypes have been amplified from some arterial smooth muscles (Harper et al. 1998; Boarder & Hourani, 1998; Lewis et al. 2000). However, in physiological situations, the contribution of pyrimidine receptors to vasoconstriction is rather uncertain, since it has been shown that with intact endothelium, extraluminally applied UTP and UDP preferentially activate the endothelial P2Y receptors associated with vasodilatation (P2Y1 or P2Y2) and require very high concentrations to produce vasoconstriction (Miyagi et al. 1996; Horiuchi et al. 2001). In this respect, our present results have highlighted a new important target for the vasomotor control via hitherto-unidentified P2Y receptor subtypes in vascular smooth muscle. It would thus be interesting to see to what extent this mechanism contributes to the control of peripheral vascular resistance or circulation.
Possible physiological implications
The degree of ImVDCC potentiation caused by P2Y receptor stimulation was not larger than 20 % (∼10 % with 1 μm ATP, and ∼20 % at 10 and 100 μm ATP), which may raise a question as to the physiological contribution of this mechanism to regulating the small arteriolar tone. However, the observed voltage independence of ImVDCC potentiation (Fig. 8) implies that the magnitude of non-inactivating Ca2+ influx through mVDCCs (several tenths of a pico ampere; Morita et al. 1999), which is indicated by the crossover region of the activation and inactivation curves, would also increase to a similar extent in response to P2Y receptor stimulation in the membrane potential range near the resting level (−60 to −30 mV; Fig. 8B). This change in non-inactivating influx might be significant, albeit small, to elevate the intracellular Ca2+ concentration, since the influx would occur continuously into the cell having an extremely small volume of the order of sub-picolitres (Morita et al. 1999). In fact, a comparable magnitude of non-inactivating Ca2+ entry through dihydropyridine-sensitive L-type VDCC has been shown to cause a significant elevation in the intracellular Ca2+ concentration ([Ca2+]i) and thus arterial smooth muscle tone (see e.g. Nelson et al. 1990). It would therefore be possible to assume a similar role for mVDCCs in the peripheral arterioles. Indeed, our recent preliminary experiments suggest that nifedipine-insensitive but Cd2+-inhibitable [Ca2+]i increases evoked by moderately elevated K+ concentrations (20–40 mm) were enhanced by 100 μm ATP after full P2X receptor desensitization in the same preparation (H. Morita, Y. Ito & R. Inoue, unpublished data). Furthermore, the extent of ImVDCC potentiation by ATP was even larger (50–60 % at 1–10 μm) when recorded with nystatin-perforated technique (H. Morita & R. Inoue, unpublished data), and thus the potentiating mechanism by ATP may have more physiological impact on [Ca2+]i regulation in small arteriolar cells via ImVDCC than expected from the present results. Obviously, further studies will be required to determine the relevance of the above-mentioned speculation more unequivocally.
The observed dose-response relationship for ATP with milimolar concentrations of Ba2+ as a charge carrier (Fig. 2) has indicated that the extent of P2Y-mediated ImVDCC potentiation increases dose dependently from a threshold of submicromolar and reaches the maximum at several tens of micromolars. At higher concentrations, however, this turned to a gradual decrease due to simultaneous activation of a P2Y-mediated inhibitory mechanism (Fig. 3C), and at extremely high concentrations (>1 mm), to a marked inhibition via a G-protein-independent mechanism (Fig. 2). The concentration of ATP in the vicinity of vascular smooth muscle cells (VSMCs) cannot be precisely estimated due to vigorous degradation by the ecto-ATPase tightly bound to the cell membrane (Kennedy & Leff, 1995; Kunapli & Daniel, 1998), but it has been known that considerable overflow of ATP occurs from the sympathetic nerve terminal by electrical stimulation, which lasts for minutes and evokes smooth muscle contractions or relaxations (e.g. Starke, 1991). Provided that the local concentration of ATP around VSMCs derived from the sympathetic nerve parallels nerve activity, the observed concentration dependence enables us to envisage how the sympathetic nervous system regulates the peripheral resistance arteriolar tone. With the basal sympathetic activity of about one half to two impulses per second (Guyton & Hall, 1996), a weak potentiating effect of ATP, and presumably that of its metabolite ADP, on Ca2+ entry through mVDCCs would tend to maintain the basal peripheral vascular tone. With moderately increased sympathetic activities, further increase in Ca2+ influx through mVDCCs via P2Y receptor activation would enhance the vascular tone in proportion to the degree of nerve excitation. However, with excessive sympathetic activity, counteracting mechanisms start to suppress the Ca2+ influx, initially through P2Y receptors (P2Y1-like) and then more potently through the divalent cation chelating action of ATP, leading ultimately to termination of deleterious excessive contractions. Importantly, the apparent ATP concentration range for the latter inhibitory mechanisms (>100 μm) accords with that at which another vasorelaxant mechanism may operate in small resistant arterioles, namely P2Y receptor (P2Y2-like)-mediated endothelium-dependent hyperpolarization (Thapaliya et al. 1999), suggesting synergistic inhibitory actions. Further studies evaluating the arteriolar tension or diameter more directly such as myography and video imaging will be needed to delineate the roles of these several distinct P2Y receptors in the peripheral vasculature.
Acknowledgments
We would like to thank Professor A. F. Brading, University Department of Pharmacology, Oxford, for critical reading of our manuscript. H. M. is a research fellow of the Japanese Society for the Promotion of Sciences. This work is supported by a grant-in-aid for scientific research from the Japan Society for the Promotion of Sciences to Y. I.
REFERENCES
- Bean BP. Class of calcium channels in vertebrate cells. Annual Review of Physiology. 1989a;51:367–384. doi: 10.1146/annurev.ph.51.030189.002055. [DOI] [PubMed] [Google Scholar]
- Bean BP. Neurotransmitter inhibition of neuronal calcium currents by changes in channel voltage dependence. Nature. 1989b;340:153–156. doi: 10.1038/340153a0. [DOI] [PubMed] [Google Scholar]
- Beech DJ. Actions of neurotransmitters and other messengers on Ca2+ channels and K+ channels in smooth muscle cells. Pharmacological Therapeutics. 1998;73:91–119. doi: 10.1016/s0163-7258(97)87271-3. [DOI] [PubMed] [Google Scholar]
- Boarder MR, Hourani SM. The regulation of vascular function by P2 receptors: multiple sites and multiple receptors. Trends in Pharmacological Sciences. 1998;19:99–107. doi: 10.1016/s0165-6147(98)01170-5. [DOI] [PubMed] [Google Scholar]
- Boeymaems J-M, Communi D, Savi P, Herbert J-M. P2Y receptors: in the middle of the road. Trends in Pharmacological Sciences. 2000;21:1–3. doi: 10.1016/s0165-6147(99)01415-7. [DOI] [PubMed] [Google Scholar]
- Brooks SPJ, Storey KB. Bound and determined: a computer program for making buffers of defined ion concentrations. Analytical Biochemistry. 1992;201:119–126. doi: 10.1016/0003-2697(92)90183-8. [DOI] [PubMed] [Google Scholar]
- Burnstock G. Local mechanism of blood flow control by perivascular nerves and endothelium. Journal of Hypertension. 1990;8:95S–106S. [PubMed] [Google Scholar]
- Davila HM. Molecular and functional diversity of voltage-gated calcium channels. Annals of the New York Academy of Sciences. 1999;868:102–117. doi: 10.1111/j.1749-6632.1999.tb11281.x. [DOI] [PubMed] [Google Scholar]
- Dolphin A. Mechanisms of modulation of voltage-dependent calcium channels by G proteins. Journal of Physiology. 1998;506:3–11. doi: 10.1111/j.1469-7793.1998.003bx.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ertel EA, Campbell KP, Harpold MM, Hofmann F, Mori Y, Perez-Reyes E, Schwartz A, Snutch TP, Tanabe T, Birnbaumer L, Tsien RW. Nomenclature of voltage-gated calcium channels. Neuron. 2000;25:533–535. doi: 10.1016/s0896-6273(00)81057-0. [DOI] [PubMed] [Google Scholar]
- Guyton AC, Hall JE. The Textbook of Medical Physiology. 9. Vol. 18. Philadelphia, USA: W. B. Saunders Company; 1996. Chapter. [Google Scholar]
- Harper S, Webb TE, Charlton SJ, Ng LL, Boarder MR. Evidence that P2Y4 nucleotide receptors are involved in the regulation of rat aortic smooth muscle cells by UTP and ATP. British Journal of Pharmacology. 1998;124:703–710. doi: 10.1038/sj.bjp.0701895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann F, Lacinova L, Klugbauer N. Voltage-dependent calcium channels. Reviews in Physiology, Biochemistry and Pharmacology. 1999;139:33–87. doi: 10.1007/BFb0033648. [DOI] [PubMed] [Google Scholar]
- Hollopeter G, Jantzen H-M, Vincent D, Li G, England L, Ramakrishnan V, Yang R-B, Nurden P, Nurden A, Julius D, Conley PB. Identification of the platelet ADP receptor targeted by antithrombotic drugs. Nature. 2001;409:202–207. doi: 10.1038/35051599. [DOI] [PubMed] [Google Scholar]
- Horiuchi T, Dietrich HH, Tsugane S, Dacey RGJr. Analysis of purine- and pyrimidine-induced vascular responses in the isolated rat cerebral arteriole. American Journal of Physiology - Heart and Circulatory Physiology. 2001;280:H767–776. doi: 10.1152/ajpheart.2001.280.2.H767. [DOI] [PubMed] [Google Scholar]
- Inoue R, Ito Y. Intracellular ATP slows time-dependent decline of muscarinic cation current in guinea pig ileal smooth muscle. American Journal of Physiology - Cell Physiology. 2000;279:C1307–1318. doi: 10.1152/ajpcell.2000.279.5.C1307. [DOI] [PubMed] [Google Scholar]
- Jacobson KA, King BF, Burstock G. Pharmacological characterization of P2 (nucleotide) receptors. Cell transmissions. 2000;16:3–16. [Google Scholar]
- Kaneko S, Akaike A, Satoh M. Receptor-mediated modulation of voltage-dependent Ca2+ channels via heterotrimeric G-proteins in neurones. Japanese Journal of Pharmacology. 1999;81:324–331. doi: 10.1254/jjp.81.324. [DOI] [PubMed] [Google Scholar]
- Kennedy C, Leff P. How should P2X pruinoceptors be classified pharmacologically. Trends in Pharmacological Sciences. 1995;16:168–174. doi: 10.1016/s0165-6147(00)89010-0. [DOI] [PubMed] [Google Scholar]
- King BF, Townsend-Nocholson A, Burnstock G. Metabotropic receptors for ATP and UTP: exploring the correspondence between native and recombinant nucleotide receptors. Trends in Pharmacological Sciences. 1998;19:506–514. doi: 10.1016/s0165-6147(98)01271-1. [DOI] [PubMed] [Google Scholar]
- Kunapuli SP, Daniel JL. P2 receptor subtypes in the cardiovascular system. Biochemical Journal. 1998;336:513–523. doi: 10.1042/bj3360513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuriyama H, Kitamura K, Itoh T, Inoue R. Physiological features of visceral smooth muscle cells, with special reference to receptors and ion channels. Physiological Reviews. 1998;87:811–920. doi: 10.1152/physrev.1998.78.3.811. [DOI] [PubMed] [Google Scholar]
- Lagaud GJL, Stoclet JC, Andriantsitohaina R. Calcium handling and purinoceptor subtypes involved in ATP-induced contraction in rat small mesenteric arteries. Journal of Physiology. 1996;492:689–703. doi: 10.1113/jphysiol.1996.sp021338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis CJ, Ennion SJ, Evans RJ. P2 purinoceptor-mediated control of rat cerebral (pial) miscrovasculature; contribution of P2X and P2Y receptors. Journal of Physiology. 2000;527:315–324. doi: 10.1111/j.1469-7793.2000.00315.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyagi Y, Kobayashi S, Nishimura J, Fukui M, Kanaide H. Dual regulation of cerebrovascular tone by UTP: P2U receptor-mediated contraction and endothelium-dependent relaxation. British Journal of Pharmacology. 1996;118:847–856. doi: 10.1111/j.1476-5381.1996.tb15477.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita H, Cousins H, Onoue H, Ito Y, Inoue R. Predominant distribution of nifedipine-insensitive, high voltage-activated Ca2+ channels in the terminal mesenteric artery of guinea pig. Circulation Research. 1999;85:596–605. doi: 10.1161/01.res.85.7.596. [DOI] [PubMed] [Google Scholar]
- Morita H, Inoue R, Ito Y. Dual regulation by ATP through P2Y receptors of a novel nifedipine-insensitive (NI), high voltage-activated (HVA) Ca2+ channel in guinea-pig mesenteric arteriole smooth muscle cells. Japanese Journal of Pharmacology. 2000;82:82P. [Google Scholar]
- Mutafova-Yambolieva VN, Carolan BM, Harden TK, Keef KD. Multiple P2Y receptors mediate contraction in guinea pig mesenteric vein. General Pharmacology. 2000;34:127–136. doi: 10.1016/s0306-3623(00)00054-9. [DOI] [PubMed] [Google Scholar]
- Nelson MT, Patlak JB, Worley JF, Standen NB. Calcium channels, potassium channels, and voltage-dependence of arterial smooth muscle tone. American Journal of Physiology. 1990;259:C3–18. doi: 10.1152/ajpcell.1990.259.1.C3. [DOI] [PubMed] [Google Scholar]
- Rubino A, Burstock G. Evidence for a P2-purinoceptor mediating vasoconstriction by UTP, ATP and related nucleotides in the isolated pulmonary vascular bed of the rat. British Journal of Pharmacology. 1996;118:1415–1420. doi: 10.1111/j.1476-5381.1996.tb15554.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starke K. Noradrenaline-ATP co-transmission in the sympathetic nervous system. Trends in Pharmacological Sciences. 1991;12:319–324. doi: 10.1016/0165-6147(91)90587-i. [DOI] [PubMed] [Google Scholar]
- Surprenant A, Buell G, North RA. P2X receptors bring new structure to ligand-gated ion channels. Trends in Pharmacological Sciences. 1995;18:224–229. doi: 10.1016/0166-2236(95)93907-f. [DOI] [PubMed] [Google Scholar]
- Thapaliya S, Matsuyama H, Takewaki T. ATP released from perivascular nerves hyperpolarizes smooth muscle cells by releasing an endothelium-derived factor in hamster mesenteric arteries. Journal of Physiology. 1999;521:191–199. doi: 10.1111/j.1469-7793.1999.00191.x. [DOI] [PMC free article] [PubMed] [Google Scholar]