

Abstract
Microtubule disassembly by colchicine increases spontaneous beating of neonatal cardiac myocytes by an unknown mechanism. Here, we measure drug effects on spontaneous calcium transients and whole cell ionic currents to define the route between microtubule depolymerization and the increase in the rate of contraction. Colchicine treatment disassembles microtubules resulting in free tubulin dimers, thereby increasing the spontaneous beating frequency and changing both the rates of rise and decay of calcium transients. In addition, colchicine treatment produces an increase of the sodium current (INa) while ICa is not modified. The colchicine-enhanced INa was blocked by the addition of 10 μm TTX. In addition, the colchicine-induced increase of INa was prevented when GTP was omitted from the patch pipette. Vinblastine also depolymerizes microtubules but re-aggregates tubulin into paracrystalline structures. Free tubulin dimers are not increased with vinblastine treatment. We found no modification in calcium transients or INa in the presence of vinblastine. Action potential durations measured at 50 % and 90 % repolarization were shorter, and the dV/dt was larger, in colchicine-treated cells compared to untreated cells. The resting membrane potential and overshoot of the action potentials were comparable in both kinds of cells. Our data suggest that release of free tubulin dimers may activate G proteins, which in turn modulate the sodium channel. An increase in whole cell INa changes the spontaneous firing rate and this may be the underlying cause of the increase in the frequency of contraction in neonatal cardiac myocytes. We suggest a new role for dimeric tubulin in regulating membrane excitability.
The contractile cycle of cardiac myocytes is controlled by the activity of membrane channels and regulation of cell calcium. Membrane excitability can be modulated by a wide variety of ligands (agonists or antagonists) and second messengers. While the major function of the microtubular component of the cytoskeleton is transporting subcellular components (Perhonen et al. 1998), it may also be indirectly involved in contractile output (Zile et al. 1999). Microtubules are constantly remodelled in cells by their depolymerization into the constituent tubulin dimers. Hydrolysis of GTP bound to β-tubulin is the energy source for rebuilding new microtubules. This offers the possibility that a G protein-coupled process related to microtubule assembly has other cellular roles indirectly related to contractility in the heart. This paper attempts to establish these links.
It is well established that the number of microtubules increases in some forms of cardiac hypertrophy (Zile et al. 1999; Hein et al. 2000). Cooper and collaborators initially described the role of microtubules on cardiac cell contraction and changes in number using a feline model of pressure or volume overload hypertrophy (Tsutsui et al. 1994). Depolymerization of microtubules in adult myocytes isolated from pressure-overloaded hearts resulted in an increase in contractile function with a considerable improvement of shortening velocity. Their explanation was predominantly mechanical and relied on decreased stiffness when these rigid microtubules are removed.
Neonatal myocytes in culture display spontaneous beating but adult cells do not. Furthermore, the rate of spontaneous beating is altered by colchicine (Klein, 1983; Lampidis et al. 1986; Lampidis et al. 1992) and, clearly, these alterations in chronicity in neonatal cells have nothing to do with changes in stiffness seen in the adult (Tsutsui et al. 1994). Furthermore, existing explanations do not separate the role of free tubulin dimers from the mechanical properties of an intact microtubule cytoskeleton (Tagawa et al. 1998). Thus, in this paper we report a series of experiments to answer questions about the mechanism of free tubulin dimer action on chronicity.
We found that there is an increase in the spontaneous calcium transients, and both the rising and falling phases of the transients are faster when microtubules are depolymerized by colchicine in neonatal rat cardiac myocytes. Colchicine depolymerizes microtubules and increases the free tubulin in cardiac tissue (Takahashi et al. 1998). Whole cell ionic currents were recorded with the patch clamp technique to detect possible changes in membrane excitability. Depolymerization of microtubules with vinblastine, which leaves the tubulin dimer concentration unchanged (Jordan et al. 1991), did not alter calcium transients or ionic currents. Colchicine treatment resulted in an increase in INa, mediated by a GTP-dependent mechanism since it was reversed when GTP was omitted from the patch pipette. We hypothesize that an increase in free GTP-bound α,β-tubulin heterodimers potentiates the sodium channel either through a G protein or by a direct pathway. An increase in INa would change the spontaneous rate of firing of cardiac myocytes. This would be a novel regulatory mechanism of sodium channels by the microtubular component of the cytoskeleton.
METHODS
Cell isolation
Experiments were performed according to Institutional Animal Care and Use Committee and NIH guidelines. Primary cardiac cell cultures were performed as previously described (Goldspink et al. 1996). Briefly, hearts were removed from 1- to 2-day-old neonatal Sprague-Dawley rats after decapitation and kept in cold Moscona's saline (136.8 mm NaCl, 28.6 mm KCl, 11.9 mm NaHCO3, 9.4 mm glucose, 0.08 mm NaH2PO4, pH 7.4) on ice. Ventricles were trimmed free and minced with dissecting scissors. Cells were dissociated in a 37 °C shaker bath with collagenase type 2 at 0.42 mg ml−1 (Worthington Biochemical Corporation) in Krebs buffered Ringer (118.4 mm NaCl, 2.4 mm MgSO4, 4.7 mm KCl, 23.8 mm NaHCO3, 1.5 mm KH2PO4, 11.1 mm glucose, 20 mg ml−1 BSA Fraction V, 10 μl ml−1 penicillin G/ streptomycin solution (Sigma), phenol red, pH 7.4, gassed to top with 5 % CO2 in air to prevent pH change). Cells were pelleted by centrifugation at 5000 r.p.m. for 6 min at room temperature. The supernatant was discarded and cells were resuspended in complete medium (Dulbecco's modified Eagle's medium/ Nutrient Mixture F-12 HAM without l-glutamine (Sigma), standard amino acid concentrations plus palmitic (2.56 mg l−1) and linoleic (0.84 mg l−1) fatty acids, penicillin G/streptomycin solution (10 μl ml−1) and gentamicin (50 mg l−1)), then filtered through a metal sieve to filter out large material. The resulting mixture was pre-plated for 1 h in 37 °C, CO2 incubator. Cells were plated on collagen-coated dishes at high density (1000–2000 cells mm−2) and maintained in DMEM with 5 % fetal bovine serum for 48 h.
Calcium transient measurement
Calcium transients were measured in a Nikon Diaphot 300 inverted microscope using an excitation filter centred at 470 nm (half bandwith 20 nm), a dichroic long-pass mirror centred at 510 nm, and a long-pass emission filter centred at 520 nm. The fluorescence signal was detected with a photomultiplier tube, filtered at 1.0 kHz, and sampled at 400–500 μs intervals. For recording of calcium transients, cells were switched to serum-free medium for 30 min in order to load the cells with 5 μm Fluo-3 AM (Molecular Probes, Inc.). The medium was then changed to complete medium with serum lacking phenol red (Sigma) to prevent interference with the fluorescent beam. Pharmacological agents were added to dissociate microtubules (colchicine (15 μm) or vinblastine (0.5 μm) for 1.5–2 h) prior to measuring calcium transients. Transients were measured in each plate prior to the addition of such agents so that each dish would serve as its own control. Calcium transients were assessed for frequency, rate of rise and rate of decay.
Resting (diastolic) and systolic calcium was measured from cells loaded with 5 μm Fura-2 AM (Molecular Probes, Inc.). For these measurements, cells were plated on collagen-coated glass coverslips. The rest of the procedure for loading cells was identical to that followed when using Fluo-3. Two high temperature filters, centred at 355 and 380 nm, were used to select the wavelength of excitation light; the light was reflected with a long-pass dichroic mirror centred at 430 nm. Fluorescence emission was recorded with a filter centred at 510 nm (wide band of 40 nm) and a photomultiplier tube.
Ionic currents and action potentials measurements
Ionic currents were recorded in the whole cell configuration of the patch clamp technique (Hamill et al. 1981). The membrane linear components were subtracted with negative control pulses delivered from the holding potential. Series resistance compensation was used in all recordings. ICa was recorded with the following solutions: extracellular (mm): TEACl 145, CaCl2 10, tetrodotoxin 0.001, Hepes 10 (pH 7.4 adjusted with CsOH); intracellular (mm): caesium aspartate 140, MgCl2 5, Mg-ATP 2.5, Tris-GTP 0.5, Cs2-EGTA 10, Hepes 10 (pH 7.2 adjusted with CsOH). ICa was elicited with 160 ms pulses delivered every 10 s, from a holding potential of −80 mV to test potentials between −60 and +60 mV. The sampling interval for ICa measurements was 200 μs per point and the traces were filtered at 2 kHz. The solutions for INa recording were the following: extracellular (mm): NaCl 20, CsCl 115, MgCl2 3, CdCl2 0.3, glucose 5, and Hepes 10 (pH 7.4 adjusted with CsOH); intracellular (mm): NaCl 20, CsCl 115, Mg-ATP 2.5, Tris-GTP 0.5, Cs2-EGTA 5, Hepes 10 (pH 7.2 adjusted with CsOH). INa was elicited every 10 s with 35 ms pulses, from a holding potential of −80 mV; test potentials ranged between −70 and +60 mV. The sampling interval for INa recording was 50 μs per point and the traces were filtered at 5 kHz. Junction potentials produced with the different combinations of extracellular and intracellular recording solutions were measured and used to correct the estimated reversal potentials. Membrane capacitance was measured by integrating the area under the capacity transient elicited by a 20 ms pulse to −90 mV, and was used to normalize ionic currents obtained from different myocytes. Currents were recorded with an Axopatch 200B amplifier. Pulse sequences, data acquisition and analysis were controlled and performed with pCLAMP suite software (version 7.0, Axon Instruments, Foster City, CA, USA).
The experimental current-voltage relationships were fitted to the equation:
where Gmax is the maximum conductance calculated from the linear part of the curves, Vrev is the reversal potential, V1/2 is the mid-point potential of activation, and k is the steepness of the curve.
Control and test currents were recorded from cells in the same culture and on the same day to avoid possible changes due to time and culture-to-culture variation. Control currents were first recorded from a dish while cells in another dish were incubated with either colchicine or vinblastine using the same concentrations as for calcium transient measurements. Because measurement of ionic currents requires longer than measurement of transients, cells were exposed to the drugs for a minimum of 4 h.
Action potentials were also recorded in the whole cell configuration of the patch clamp technique. The external solution consisted of (mm): NaCl 145, KCl 5, CaCl2 2, MgCl2 1, Hepes 10 (pH 7.4 adjusted with NaOH). The internal solution contained (mm): KCl 140, MgCl2 5, K2-EGTA 10, Na-ATP 2.5, Tris-GTP 0.5, Hepes 10 (pH 7.2 adjusted with KOH). The sampling rate was 50 μs per point and the signals were filtered at 5 kHz.
Immunolabelling
Cells were washed in phosphate-buffered saline (pH 7.4) and then fixed in 4 % paraformaldehyde/PBS for 10 min. Microtubules were detected with a monoclonal antibody specific for β-tubulin (Sigma, St Louis, MO, USA) at a working dilution of 1:200. A FITC-labelled goat anti-mouse antibody was used at a dilution of 1:200 in 1 % BSA. Coverslips were mounted in Vectashield mounting medium for fluorescence (Vector Laboratories) to prevent fading.
Statistics
To determine the statistical significance, data were analysed with GraphPad Prism software (version 3.0, San Diego, CA, USA) using a paired t test when comparing calcium transients. Ionic currents were analysed with Statistica 5.1 software (StatSoft, Inc., Tulsa, OK, USA) using analysis of variance (ANOVA) with repeated measures. Data are presented as means ± s.e.m. A level of significance was set at a value of P < 0.05.
RESULTS
Effect of colchicine on calcium transients
Microtubule disruption was examined by immunolabelling of cultured neonatal rat cardiac myocytes. The majority of the microtubules disappeared after treatment with either colchicine or vinblastine (Fig. 1). Tubulin labelling appears diffuse throughout cells treated with colchicine to depolymerize the microtubules (Tagawa et al. 1998). In contrast, labelling tubulin in vinblastine-treated cells shows paracrystals of re-aggregated tubulin dimers (Bhattacharyya & Wolff, 1976; Na & Timasheff, 1982; Jordan et al. 1991). These results indicate that microtubular disruption was complete at the time we performed our experiments.
Figure 1. Effects of colchicine or vinblastine on microtubule depolymerization and spontaneous calcium transients.
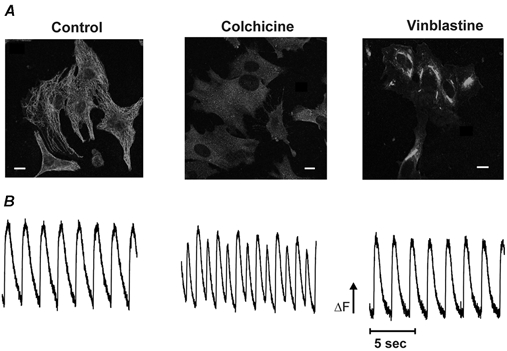
A, immunohistochemical staining of neonatal rat cardiac myocytes stained for β-tubulin and visualized by a FITC-labelled goat anti-mouse secondary antibody with a confocal microscope. The microtubules are intact in control cardiac myocytes (left); disassembled by colchicine treatment into a diffuse cloud of tubulin dimers (middle); or disassembled by vinblastine treatment and reaggregated into paracrystalline structures (right). Scale bar = 10 μm. B, myocytes were loaded with Fluo-3-AM and a representative 15 s recording of spontaneous calcium transients is shown before treatment (left), or after 1.5–2 h incubation in colchicine (middle) or vinblastine (right). Compared to the control, the calcium transients in colchicine-treated cells had a higher frequency and increased rates of rise and decay than in control; they also show the presence of alternans. Note the lack of effect with vinblastine compared to control.
We studied the effect of colchicine on spontaneous calcium transients because of the positive chronotropic and inotropic effects of microtubule depolymerization. Figure 1B shows calcium transients recorded from neonatal cardiac myocytes before and after the addition of 15 μm colchicine. Colchicine treatment (middle) had several effects on the calcium transients. We observed a 190.0 ± 19.0 % (n = 70 cells from seven different dishes) increase in the frequency of calcium transients compared to untreated cells (13.7 ± 4.1 transients per min, ranging from 7 to 35, in control cells; 26.9 ± 7.9 transients per min, ranging from 13 to 55, in colchicine-treated cells). The increase in frequency agrees with the study by Klein (1983) in that changes in spontaneous beating started taking place only after the cells had been incubated in colchicine for more than 20 min and reached a steady level at about 1–1.5 h. In addition, the rates of rise and decay of the transients were faster in colchicine-treated cells compared to untreated cells. As shown in Fig. 2A, the rising phase of the transients was best fitted to a two-exponential function while the decay was fitted to a one-exponential. In the presence of colchicine (Fig. 2B), the fast and slow time constants of the rising phase decreased to 70.3 ± 11.0 % and 58.8 ± 8.9 % of the control values, respectively. The absolute values of the time constants in control cells were: τfast = 33.5 ± 2.7 ms and τslow = 370.5 ± 48.3 ms, and in colchicine-treated cells the values were: τfast = 23.4 ± 4.0 ms and τslow = 220.1 ± 44.2 ms. The rate of decay in control cells was 1375 ± 176 ms and in colchicine-treated cells the τdecay was 883 ± 101 ms. The value of τdecay in treated cells represents 65.8 ± 3.3 % of the control value. All these changes were significantly different from control cells (see legend to Fig. 2 for P values).
Figure 2. Effects of colchicine or vinblastine on the properties of spontaneous calcium transients.
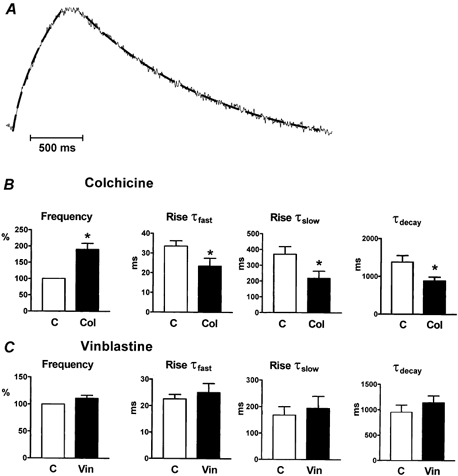
The bar graphs show the average of the calcium transient parameters obtained from all control or treated cells. For each parameter, measurements from treated cells in one dish were averaged and compared to previous control measurements from the same dish. A shows an example of the exponential fitting to calcium transients used to obtain the rates of rise and decay. The interrupted line on the transient corresponds to the fitted curve. B, colchicine-treated cells showed significantly higher frequency and faster rates of rise and decay compared to control (n = 70 cells from seven dishes). * Statistically significant difference between colchicine-treated and control parameters. P values were: frequency, P = 0.012; rise τfast, P = 0.032; rise τslow, P = 0.004; decay τ, P = 0.002. C, vinblastine did not cause a significant modification of calcium transient parameters (n = 90 cells from nine dishes). For all bar graphs, open bars represent controls and filled bars correspond to treated cells. P values were: frequency, P = 0.111; rise τfast, P = 0.228; rise τslow, P = 0.538; decay τ, P = 0.063.
Interestingly, the increase in frequency caused by colchicine was accentuated in cells with a slow rate of contraction in the untreated state. An extreme measurement resulted in a 760 % increase in frequency because the basal rate of contraction of control cells was on average about 5 beats min−1. This set of data was not included in the average given above since it fell beyond the second standard deviation.
Another interesting effect produced by colchicine was the development of alternans (a decrease in amplitude of every other event) observed in the calcium transient recordings (see Fig. 1B middle). Alternans occurred in about half the cells treated with colchicine. Although the increase in frequency was intimately associated with alternans, we did not see the same phenomenon in control cells even when those cells had a high basal rate of contraction. This suggests that colchicine induced alternans.
We studied the effect of vinblastine on spontaneous calcium transients to test whether the effect of colchicine was due to an increase in free tubulin dimers or simply a result of microtubule disruption. Representative transients are shown in Fig. 1B (right). Exposure of the cells to 0.5 μm vinblastine for 1.5–2 h did not have any significant effect on calcium transients. In the presence of vinblastine, the frequency and the rates of rise and decay of the calcium transients were: 111.1 ± 5.6 %, τfast = 94.9 ± 14.8 %, τslow = 136.7 ± 32.5 %, and 127 ± 15.4 % of those transients in control conditions, respectively. For this group of experiments, the control values were: 27.8 ± 3.7 transients per min, τfast = 22.6 ± 1.7 ms, τslow = 167.8 ± 32.2 ms, and τdecay = 951 ± 142 ms. The absolute values in the presence of vinblastine were: 30.9 ± 4.3 transients per min, τfast = 25.0 ± 3.4 ms, τslow = 194.1 ± 45.5 ms, and τdecay = 1139 ± 137 ms. The differences between the values obtained from control and vinblastine-treated cells were not statistically significant (see legend to Fig. 2). Figure 2C shows the means of 90 cells from nine dishes in control or vinblastine. Examination of Fig. 2 shows that colchicine, but not vinblastine, modifies the properties of calcium transients in neonatal cardiac myocytes. Thus, our results suggest that an increase in free tubulin dimers is associated with the changes observed in the calcium transient in the presence of colchicine.
Effect of colchicine on membrane ionic currents
Ionic currents tightly regulate calcium transients in cardiac myocytes, either directly or through changes in the action potential. In cardiac myocytes isolated from adult rats, it has been recently reported that colchicine treatment increases the L-type ICa (Gómez et al. 2000). Thus, we wanted to investigate whether the same phenomenon was present in neonatal cells and if that could explain the increase in frequency of contraction observed upon exposure to colchicine.
ICa was initially recorded from neonatal cardiac myocytes. To prevent rundown of the calcium channel, we included ATP and GTP in the patch pipette. With this recording solution, the amplitude of the ICa remained stable for a period of over 20 min. Figure 3A shows ICa recorded from a control cell (top) and from a cell that was exposed to 15 μm colchicine for 4 h (bottom). The current-voltage relationships for the control group and the colchicine-treated group are shown in Fig. 3B. It is evident from the data in this figure that microtubule disruption by colchicine did not cause any modifications in either the kinetics or amplitude of the L-type ICa. This is in contrast to the recent study by Gómez et al. (2000) who reported that colchicine increased the amplitude of ICa from adult rat cardiac myocytes. In our experiments with neonatal rat cardiac cells, the average peak ICa amplitude was −3.79 ± 0.74 pA pF−1 (n = 9) in control cells and −3.73 ± 0.35 pA pF−1 (n = 9) in colchicine-treated cells. Thus, no significant difference was found (P = 0.88). The extrapolated reversal potential, steepness of activation and mid-point potential of activation of ICa were not modified in the presence of colchicine either. The average values of these parameters in control cells (n = 9) were Vrev = 82.92 ± 3.87 mV, k = 13.54 ± 1.38 mV, and V1/2 = 14.88 ± 2.99 mV; while in colchicine-treated cells (n = 9) the average values were Vrev = 77.83 ± 2.29 mV, k = 16.34 ± 1.49 mV, and V1/2 = 23.52 ± 3.96 mV. The difference between these values was not statistically significant (P = 0.067).
Figure 3. ICa is not modified by colchicine in neonatal cardiac myocytes.
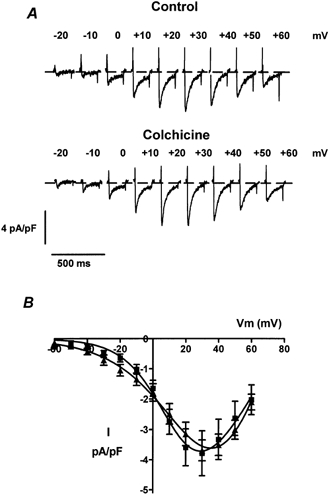
A, ICa traces from representative control (top) or colchicine-treated (bottom) cells. Cells were exposed to 15 μm colchicine at 37 °C for at least 4 h before recording. Colchicine was also present in the recording solution. ICa was recorded from control and colchicine-treated cells the same day to avoid culture variability. Only ICa elicited at membrane potentials between −20 and +60 mV is shown. B, current-voltage relationships of the averaged ICa in control (▪, n = 9 cells) or in the presence of colchicine (▴, n = 9 cells). Currents were normalized to cell size and expressed as pA pF−1. Colchicine did not cause any significant modifications of the ICa.
The INa is another possible candidate that might indirectly affect the observed changes in the calcium transient produced by colchicine treatment. The INa was recorded with 20 mm sodium in both the extracellular and pipette solutions to minimize errors due to loss of voltage clamp. The patch pipette also contained ATP and GTP. Figure 4A shows examples of INa recorded from a control cell in the absence of colchicine (top) and a cell that had been exposed to colchicine (bottom). In contrast to ICa, microtubule disruption by colchicine caused a significant increase in the amplitude of INa. The average peak INa amplitude in control cells was −2.01 ± 0.58 pA pF−1 (n = 10) and the amplitude in colchicine-treated cells was −5.15 ± 1.06 pA pF−1 (n = 10, P = 0.02). The current-voltage relationships for control and colchicine-treated cells are shown in Fig. 4B. As expected, INa reversed around 0 mV in control cells (−0.5 ± 1.5 mV, n = 10). The reversal potential in colchicine-treated cells was on average 2.5 ± 1.1 mV (n = 10); however, this difference was not significant (P = 0.06). Additionally, neither the mid-point potential of activation nor the slope of the curves were changed in the presence of colchicine (V1/2 = −28.2 ± 3.3 mV, k = 9.47 ± 1.3 mV for control cells; V1/2 = −35.4 ± 2.01 mV, k = 7.8 ± 0.7 mV for colchicine-treated cells; P = 0.074).
Figure 4. Increase of INa by colchicine treatment in neonatal cardiac myocytes.
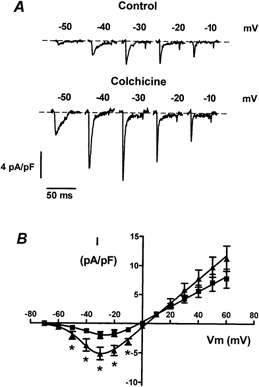
A, INa traces from representative control (top) or colchicine-treated (bottom) cells. Cells were exposed to 15 μm colchicine. INa was recorded from control and colchicine-treated cells the same day to avoid culture to culture variability. INa elicited at potentials between −50 and −10 mV is shown. B, current-voltage relationships of the averaged INa density in control (▪, n = 10 cells) or in the presence of colchicine (▴, n = 10 cells). Colchicine induced a significant increase of the INa. Asterisks indicate a P value < 0.05.
We used tetrodotoxin to verify that the sodium channel mediated the colchicine-enhanced inward current. The inset in Fig. 5A illustrates the blockade of the INa in a colchicine-treated cell before and after the addition of 5 or 10 μm tetrodotoxin. In agreement with previous reports (Satin et al. 1992; Nuss & Marban, 1994), 5 μm tetrodotoxin caused a partial blockade of the INa and 10 μm almost completely blocked the current. The amplitude of the INa recorded from different colchicine-treated cells averaged −1.84 ± 0.29 pA pF−1 (n = 6) in the presence of 5 μm tetrodotoxin and −0.17 ± 0.08 pA pF−1 (n = 3) in 10 μm tetrodotoxin. Figure 5A shows the current-voltage relationships of the INa obtained after colchicine treatment and in the absence (triangles) or presence of 5 (inverted triangles) or 10 μm (diamonds) tetrodotoxin.
Figure 5. INa recorded from neonatal cardiac myocytes under different conditions.
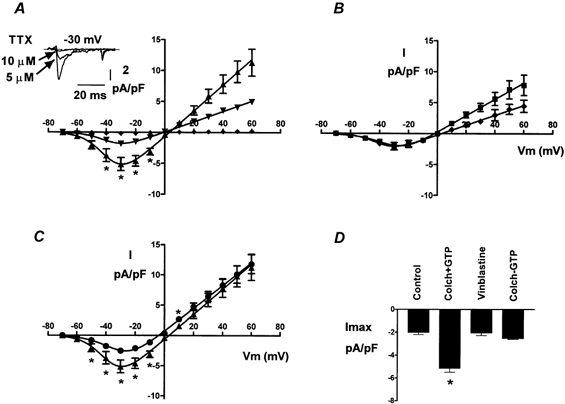
A, the colchicine-enhanced INa was reduced with 5 μm tetrodotoxin and almost completely blocked with 10 μm. The inset shows INa recorded from the same cell before and after the addition of TTX. * Statistically significant difference (P < 0.05) between colchicine-treated cells with 5 μm (▾, n = 6 cells), 10 μm (♦, n = 3 cells), or no tetrodotoxin added (▴, n = 10 cells). No statistical analysis was performed for membrane potentials ≥+20 mV and no error bars were added since we recorded from only two cells in this range. B, INa recorded from vinblastine-treated cells. Vinblastine (♦, n = 7 cells) did not increase INa at any membrane potential and the current-voltage relationship practically overlaps with that of control cells (▪, n = 10). Although the INa was somewhat smaller at positive potentials in the presence of vinblastine, the difference was not significant. C, INa recorded in the absence of intracellular GTP. When GTP was omitted from the patch pipette, the enhancement of the INa by colchicine was prevented. * Significant difference (P < 0.05) between colchicine-treated cells in the presence (▴, n = 10) or the absence (•, n = 11 cells) of GTP. INa recorded in the absence of GTP was not different from control currents. D, bar graph of the maximum INa amplitude under different conditions. Note that only the bar corresponding to INa recorded in the presence of colchicine and GTP is significantly different from control.
We also tested the effect of vinblastine on INa. Exposure of the cells to 0.5 μm vinblastine for more than 4 h did not cause significant modifications of INa (Fig. 5B). The amplitude of the INa after vinblastine treatment was −2.06 ± 0.6 pA pF−1 (n = 7), similar to the amplitude of control cells (P = 0.96). Vinblastine has no effect on the inward INa or on calcium transients. Therefore, it is possible that INa is the underlying cause for the changes observed with colchicine.
How does microtubule depolymerization alter INa?
Our results demonstrate that microtubule depolymerization with colchicine increases an inward current that is mediated by the sodium channel in some way. In other systems, it has been shown that tubulin dimers can activate G proteins directly through transfer of GTP (Popova et al. 1994). To determine whether a similar mechanism was present in neonatal cardiac cells, we removed GTP from the patch pipette when recording INa in another set of experiments. The rest of the recording conditions were identical. INa recorded in the absence of GTP was similar to the currents recorded under control conditions (i.e. in the presence of GTP but without colchicine). The maximum INa in the absence of GTP averaged −2.53 ± 0.25 pA pF−1 (n = 11). This value was not statistically different from control cells (P = 0.40), but it was different from currents recorded in the presence of GTP (P = 0.02). Figure 5C shows the current-voltage relationships of colchicine-treated cells studied with GTP (triangles) and without GTP (circles) in the patch pipette. The curve corresponding to the currents in the absence of GTP is similar to the curve from control cells shown in Fig. 4B. These results are consistent with previous observations, which show that cardiac INa is enhanced by the Gs protein in cardiac muscle (Lu et al. 1999). Furthermore, in rat cardiac muscle, the α subunit of Gs can directly activate INa (Lu et al. 1999).
Figure 5D summarizes the average maximum INa recorded under the different conditions presented in this study. It is evident from the figure that the only bar that was significantly different from control corresponds to the current recorded in the presence of colchicine and GTP. INa recorded in the presence of vinblastine or in the absence of internal GTP had a similar amplitude to control INa.
The role of an increased INa on membrane depolarization
We recorded action potentials from control and colchicine-treated cells to explore the functional significance of the increase in INa induced by colchicine. We found no significant differences in the resting membrane potential (control, −73.59 ± 0.96 mV (n = 10); colchicine, −76.93 ± 1.55 mV (n = 10); P = 0.152) or the overshoot of action potentials (control, +47.7 ± 3.6 mV; colchicine, +51.9 ± 2.80 mV; P = 0.370). However, the durations of the action potentials at 50 % and 90 % repolarization were significantly reduced in colchicine-treated cells compared to control. The values of these parameters were AP50 = 82.1 ± 15.3 ms and AP90 = 114.7 ± 19.5 ms in the presence of colchicine and AP50 = 127.3 ± 15.1 ms and AP90 = 172.1 ± 19.1 ms in control cells (P = 0.048). Figure 6 shows examples of action potentials recorded from a control (A) or a colchicine-treated cell (B). The overall shape and duration of the action potentials (top) were maintained in successive depolarizations in the control cell, while they changed from depolarization to depolarization in the colchicine-treated cell. Furthermore, the dV/dt in control cells remained unchanged from one action potential to the next (Fig. 6, bottom) and in four out of ten cells treated with colchicine, we found that this parameter was smaller in every other action potential. The smaller dV/dt in colchicine-treated cells corresponded to the shorter and smaller action potentials. The average dV/dt in the colchicine group, measured from the wider and taller action potentials, was significantly larger than in the control group (colchicine, 104.4 ± 3.41 mV ms−1; control, 95.1 ± 2.65 mV ms−1; P = 0.046). The changes in every other action potential in the presence of colchicine is reminiscent of the alternans found in calcium transients recorded with Fluo-3 and suggests that the increase in INa may be responsible for the increase in frequency of calcium transient.
Figure 6. Effect of colchicine on action potentials.
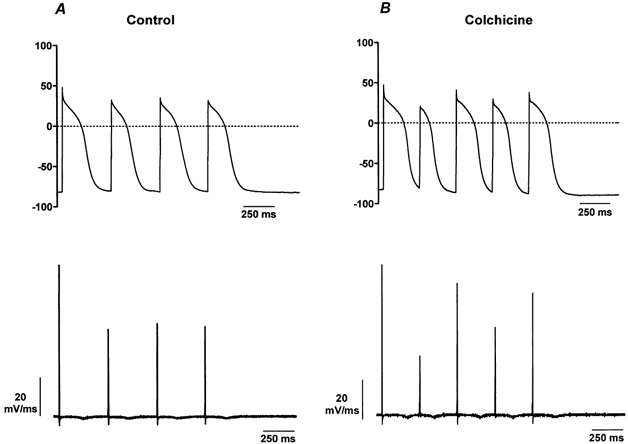
A, action potentials recorded from a representative control cell (top) and the corresponding dV/dt (bottom). The duration and morphology of the action potentials remain constant throughout the recording. The amplitude of the dV/dt also remains constant for the second to fourth action potentials. B, action potentials from a colchicine-treated cell (top) and its dV/dt (bottom). Action potentials changed in shape and duration after colchicine treatment. For this particular cell, the dV/dt showed smaller amplitudes on every other action potential. Records were taken from different dishes on the same day of the culture.
To test further this hypothesis, we quantified the frequency of contraction of control and colchicine-treated cells in a separate set of experiments before and after the addition of tetrodotoxin. Control cells treated with 5 μm tetrodotoxin showed a dramatic reduction of the frequency of contraction, from 35–39 beats min−1 to 3 beats min−1 (n = 30 cells from three dishes). Similarly, the frequency of contraction of colchicine-treated cells decreased from 63 beats min−1 to 3–7 beats min−1 after the addition of tetrodotoxin (n = 30 cells from three dishes). We also tested whether tetrodotoxin was able to revert the alternans induced by colchicine. We measured calcium transients with Fura-2 and found that the frequency of the transient decreased and the alternans disappeared after the addition of tetrodotoxin. Diastolic and systolic calcium levels were not significantly different in control and colchicine-treated cells, as estimated with Fura-2. Diastolic calcium in control cells was 107 ± 5.6 nm (n = 6) and in colchicine-treated cells was 108 ± 3.3 nm (n = 7) (P = 0.67). Systolic calcium in control cells was 403 ± 58.5 nm (n = 6) and in colchicine-treated cells was 369 ± 17.0 nm (n = 7) (P = 0.67). Thus, it is unlikely that changes in calcium concentrations made a significant contribution to the changes reported in this paper. Instead, our results indicate that microtubule depolymerization by colchicine may increase the INa, which modulates the rate of contraction and membrane excitability in neonatal cardiac myocytes.
DISCUSSION
In this paper we confirm that microtubule depolymerization with colchicine produces a positive chronotropic effect in neonatal cardiac myocytes (Klein 1983; Lampidis et al. 1986; Lampidis et al. 1992). Colchicine treatment leads to an increase in both the frequency and the rates of rise and decay of the calcium transients. These effects can be explained by an increase in the INa mediated by a G protein-dependent process since removal of GTP prevented INa potentiation. Vinblastine did not alter any of the parameters studied. Therefore, the changes in the calcium transient appear to be caused by modulation of INa and to be specific to the free tubulin dimer concentration. A similar mechanism has also been reported recently in adult cardiac myocytes (Kerfant et al. 2001).
Our results for neonatal cells seem to contradict results from a recent study performed in adult myocytes (Gómez et al. 2000) where microtubule depolymerization by colchicine caused an increase in ICa and sarcoplasmic reticulum calcium release through an adenylyl cyclase-mediated mechanism. Unfortunately, currents other than the ICa were not included in that study and the possible effect of tubulin dimers on the adult rat INa is still unknown. Perhaps age of the rat may, alone, explain the observed discrepancies. It has been shown that in neonatal cells, colchicine treatment does not increase cAMP (Klein 1983) and that the increase in the rate of contraction is independent of the β-adrenergic system (Lampidis et al. 1986). Another difference may lie in the regulation of the ICa at early stages of development and in newborn animals by β-adrenergic receptors and G proteins. Addition of isoproterenol has been shown to either increase the ICa (Liu et al. 1999) or have no effect at all on this current (Xiao et al. 1999). The concurrent coupling of Gs and Gi to β-adrenergic receptors may explain the null response of isoproterenol on the ICa in young animals (Xiao et al. 1999). Thus the calcium channel is coupled to more than one G protein whereas the sodium channel may be coupled only to Gs. To the best of our knowledge, there is no report describing coupling of the sodium channel to a Gi protein in cardiac myocytes of the neonatal rat. Furthermore, the rat sodium channel can be directly activated by the α subunit of Gs, independently of protein kinase A (Lu et al. 1999).
We propose a mechanism of action of free tubulin on the activation of the sodium channel that results in an increase in membrane excitability (Fig. 7). This mechanism is based on a previous study by Wang et al. (1990) and the results presented here. Depolymerization of the microtubules with colchicine, but not vinblastine, releases GTP-bound α,β-tubulin dimers. The tubulin dimers transfer the GTP to a G protein, presumably Gs, releasing the α and β/γ subunits. The Gs α directly activates the sodium channel, causing an increase in the amplitude of the current. The increase in the INa increases membrane excitability, resulting in a higher frequency of contraction. We have also included the possibility that the GTP-bound α,β-tubulin dimers can activate the sodium channel directly since this mechanism is possible and deserves further examination.
Figure 7. Model of the mechanism of action of colchicine.
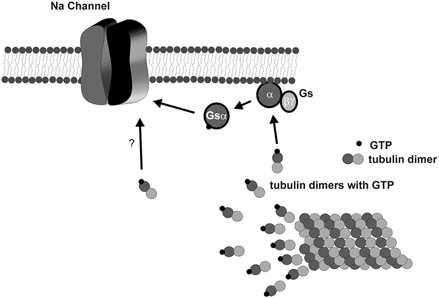
When microtubules are depolymerized with colchicine, GTP-bound α,β-tubulin dimers are free in the cytoplasm of neonatal cardiac myocytes. The GTP from the tubulin dimers is transferred to the stimulatory G protein, Gs, which causes a dissociation of the α and β,γ subunits. The activated Gs α subunit interacts with the sodium channel causing an increase in the whole cell INa. Also shown in this model is a possible direct activation of the sodium channel by GTP-bound α,β-tubulin dimers. Note that we did not include here the activation of protein kinase A since it has been shown that colchicine treatment of neonatal cardiac myocytes does not increase cAMP. The overall increase in INa caused by the activated Gs α and/or GTP-bound α,β-tubulin dimers results in an increase of membrane excitability.
The changes in duration and shape of the action potentials in the presence of colchicine directly implicate the INa as the underlying cause for these effects. An increase in INa would cause a faster depolarization, which would speed up activation and inactivation of other ion channels; the result is that ion channels respond earlier to subsequent depolarizations. The overall effect of these changes is that the action potentials become shorter and more frequent, explaining the faster decay and the increase in frequency of the transients, respectively. These results also agree with the idea that a simple alteration in the magnitude of an ionic current has a profound effect on other ionic currents, as shown by Campbell et al. (1991).
An increase in the INa also may explain, to some extent, the presence of alternans seen in some of the cells after colchicine treatment. Evidence supporting this hypothesis is provided by the reduction of calcium transient frequency and disappearance of the alternans after a colchicine-treated cell was exposed to tetrodotoxin. Furthermore, although a change in sarcoplasmic reticulum calcium loading is not ruled out with our experiments, alterations of cytoplasmic calcium as the cause of alternans was excluded because control and colchicine-treated cells had similar levels, as measured with Fura-2.
The similarity of the calcium levels between control and colchicine-treated cells is intriguing since the frequency of the transients was higher in the latter group of cells. The maintenance of the peak calcium transients measured with Fura-2 could indicate that other changes in calcium handling might occur as well. In fact, the faster decay of the calcium transient in the presence of colchicine may be explained by a faster uptake of calcium by the sarcoplasmic reticulum (Langer, 1997).
Several experimental observations argue against a direct effect of colchicine on the processes that affect calcium handling, and instead favour the idea that the effect of colchicine is mediated by depolymerization of the microtubules. Our own experiments and previous work (Klein, 1983; Gómez et al. 2000) show that the effect of colchicine is observed only after several minutes of exposure to the drug and not with acute application. Kerfant et al. (2001) also have found that the faster decay of calcium transients is related to activation of Gs proteins and not to a direct effect of colchicine. Furthermore, it has been shown recently that nocodaloze, another agent that also promotes depolymerization of the microtubules in a way similar to colchicine, increases the beating frequency of neonatal cardiac myocytes (Webster & Patrick, 2000).
In summary, our results demonstrate that microtubules serve more than their intracellular transport function in cardiac myocytes. Our new results suggest that disassembly of the microtubules by colchicine is able to modulate membrane excitability via the sodium channel of neonatal cardiac cells by changing the concentration of tubulin dimers in the cytoplasm.
Acknowledgments
This work was supported by grants from NSF (IBN-9733570 to J.G.), and NIH (HL40880 and P01-HL62426 to B.R.) and NIH training grants T32-DK07739 and HL07692 (K.J.A. and D.M.).
REFERENCES
- Bhattacharyya B, Wolff J. Tubulin aggregation and disaggregation: mediation by two distinct vinblastine-binding sites. Proceedings of the National Academy of Sciences of the USA. 1976;73:2375–2378. doi: 10.1073/pnas.73.7.2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell DL, Rasmusson RL, Strauss HC. Electrophysiology of the sinus node. Ionic and cellular mechanisms underlying primary cardiac pacemaker activity. In: Dangman KH, Miura DS, editors. Electrophysiology and Pharmacology of the Heart, Clinical Guide. New York: M. Dekker; 1991. pp. 59–108. [Google Scholar]
- Goldspink PH, Thomason DB, Russell B. Beating affects the posttranscriptional regulation of alpha-myosin mRNA in cardiac cultures. American Journal of Physiology. 1996;271:H2584–2590. doi: 10.1152/ajpheart.1996.271.6.H2584. [DOI] [PubMed] [Google Scholar]
- Gómez AM, Kerfant BG, Vassort G. Microtubule disruption modulates Ca2+ signaling in rat cardiac myocytes. Circulation Research. 2000;86:30–36. doi: 10.1161/01.res.86.1.30. [DOI] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Archiv. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Hein S, Kostin S, Heling A, Maeno Y, Schaper J. The role of the cytoskeleton in heart failure. Cardiovascular Research. 2000;45:273–278. doi: 10.1016/s0008-6363(99)00268-0. [DOI] [PubMed] [Google Scholar]
- Jordan MA, Thrower D, Wilson L. Mechanism of inhibition of cell proliferation by Vinca alkaloids. Cancer Research. 1991;51:2212–2222. [PubMed] [Google Scholar]
- Kerfant BG, Vassort G, Gomez AM. Microtubule disruption by colchicine reversibly enhances calcium signaling in intact rat cardiac myocytes. Circulation Research. 2001;88:E59–65. doi: 10.1161/hh0701.090462. [DOI] [PubMed] [Google Scholar]
- Klein I. Colchicine stimulates the rate of contraction of heart cells in culture. Cardiovascular Research. 1983;17:459–465. doi: 10.1093/cvr/17.8.459. [DOI] [PubMed] [Google Scholar]
- Lampidis TJ, Kolonias D, Savaraj N, Rubin RW. Cardiostimulatory and antiarrhytmic activity of tubulin-binding agents. Proceedings of the National Academy of Sciences of the USA. 1992;89:1256–1260. doi: 10.1073/pnas.89.4.1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampidis TJ, Trevorrow KW, Rubin RW. Effects of colchicine on cardiac cell function indicate possible role for membrane surface tubulin. Experimental Cell Research. 1986;164:463–470. doi: 10.1016/0014-4827(86)90044-3. [DOI] [PubMed] [Google Scholar]
- Langer GA. The Myocardium. San Diego, CA, USA: Academic Press; 1997. [Google Scholar]
- Liu W, Yasui K, Arai A, Kamiya K, Cheng J, Kodama I, Toyama J. β-adrenergic modulation of L-type Ca2+-channel currents in early-stage embryonic mouse heart. American Journal of Physiology. 1999;276:H608–613. doi: 10.1152/ajpheart.1999.276.2.H608. [DOI] [PubMed] [Google Scholar]
- Lu T, Lee HC, Kabat JA, Shibata EF. Modulation of rat cardiac sodium channel by the stimulatory G protein α subunit. Journal of Physiology. 1999;518:371–384. doi: 10.1111/j.1469-7793.1999.0371p.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Na GC, Timasheff SN. In vitro vinblastine-induced tubulin paracrystals. Journal of Biological Chemistry. 1982;257:10387–10391. [PubMed] [Google Scholar]
- Nuss HB, Marban E. Electrophysiological properties of neonatal mouse cardiac myocytes in primary culture. Journal of Physiology. 1994;479:265–279. doi: 10.1113/jphysiol.1994.sp020294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perhonen M, Sharp WW, Russell B. Microtubules are needed for dispersal of α-myosin heavy chain mRNA in rat neonatal cardiac myocytes. Journal of Molecular and Cellular Cardiology. 1998;30:1713–1722. doi: 10.1006/jmcc.1998.0734. [DOI] [PubMed] [Google Scholar]
- Popova JS, Johnson GL, Rasenick MM. Chimeric Gαs/Gαi2 proteins define domains on Gαs that interact with tubulin for β-adrenergic activation of adenylyl cyclase. Journal of Biological Chemistry. 1994;269:21748–21754. [PubMed] [Google Scholar]
- Satin J, Kyle JW, Chen C, Bell P, Cribbs LL, Fozzard HA, Rogart RB. A mutant of TTX-resistant cardiac sodium channels with TTX-sensitive properties. Science. 1992;256:1202–1205. doi: 10.1126/science.256.5060.1202. [DOI] [PubMed] [Google Scholar]
- Tagawa H, Koide M, Sato H, Zile MR, Carabello BA, Cooper G. Cytoskeletal role in the transition from compensated to decompensated hypertrophy during adult canine left ventricular pressure overloading. Circulation Research. 1998;82:751–761. doi: 10.1161/01.res.82.7.751. [DOI] [PubMed] [Google Scholar]
- Takahashi M, Tsutsui H, Tagawa H, Igarashi-Saito K, Imanka-Yoshida K, Takeshita A. Microtubules are involved in early hypertophic responses of myocardium during pressure overload. American Journal of Physiology. 1998;44:H341–348. doi: 10.1152/ajpheart.1998.275.2.H341. [DOI] [PubMed] [Google Scholar]
- Tsutsui H, Tagawa H, Kent RL, Mccollam PL, Ishihara K, Nagatsu M, Cooper G. Role of microtubules in contractile dysfunction of hypertrophied cardiocytes. Circulation. 1994;90:533–555. doi: 10.1161/01.cir.90.1.533. [DOI] [PubMed] [Google Scholar]
- Wang N, Yan K, Rasenick MM. Tubulin binds specifically to the signal-transducing proteins, Gs alpha and Gi alpha 1. Journal of Biological Chemistry. 1990;25:1239–1242. [PubMed] [Google Scholar]
- Webster DR, Patrick DL. Beating rate of isolated neonatal cardiomyocytes is regulated by the stable microtubule subset. American Journal of Physiology - Heart and Circulatory Physiology. 2000;278:H1653–1661. doi: 10.1152/ajpheart.2000.278.5.H1653. [DOI] [PubMed] [Google Scholar]
- Xiao RP, Avdonin P, Zhou YY, Cheng H, Akhter SA, Eschenhagen T, Lefkowitz RJ, Koch WJ, Lakatta EG. Coupling of β2-adrenoceptor to Gi proteins and its physiological relevance in murine cardiac myocytes. Circulation Research. 1999;84:43–52. doi: 10.1161/01.res.84.1.43. [DOI] [PubMed] [Google Scholar]
- Zile MR, Koide M, Sato H, Ishiguro Y, Conrad CH, Buckley JM, Morgan JP, Cooper G. Role of microtubules in the contractile dysfunction of hypertrophied myocardium. Journal of the American College of Cardiology. 1999;33:250–260. doi: 10.1016/s0735-1097(98)00550-6. [DOI] [PubMed] [Google Scholar]