

Abstract
The effects of nitric oxide (NO) donors on the L-type Ca2+ current (ICa,L) and the muscarinic activated K+ current (IK,ACh) were studied in isolated rat cardiac myocytes. The nitrosothiol S-nitroso-N-acetyl-d,d-penicillamine (SNAP, 1 pm-1 μm) strongly potentiated the stimulation of the ICa,L elicited by subthreshold concentrations of isoprenaline (Iso, 0.1–0.5 nm) in ventricular myocytes. The effect of SNAP was mimicked by 2-(N,N-diethylamino)-diazenolate-2-oxide (DEANO, 1 pm-1 nm), a NONOate that spontaneously releases NO in a pH-controlled manner, and was blunted by 2-(4-carboxyphenyl)-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide (100 μm), a NO trap. 1H-[1,2,4]Oxadiazolo[4,3-a]quinoxaline-1-one (10 μm), a guanylyl cyclase inhibitor, did not alter the effect of SNAP. SNAP (1 pm-1 μm) did not modify the effect of L858051 (0.1–0.3 μm), a forskolin analogue that activates adenylyl cyclase, on ICa,L and did not enhance the basal ICa,L in the presence of rolipram (1 μm), a phosphodiesterase type 4 inhibitor. Superfusion with Rp-CPT-cAMPS (500 μm), or internal dialysis with cAMP-dependent protein kinase (cA-PK) inhibitory peptide (PKI; 20 μm), inhibitors of the cA-PK, blunted the effect of SNAP (1 nm and 1 μm) on the Iso-stimulated (1‐100 pm) ICa,L. SNAP (1 nm and 1 μm) potentiated the threshold stimulation of ICa,L elicited by internal GTP-γS (10 μm), a non-hydrolysable analogue of GTP. SNAP (1 pm-1 μm) and DEANO (1 μm) potentiated the stimulation of IK,ACh elicited by low concentrations of ACh (1–2 nm) in rat atrial myocytes. The threshold stimulation of IK,ACh elicited by internal 5′-guanylylimidodiphosphate (10 μm) was also potentiated by NO donors. SNAP (1 μm) did not modify IK,ACh reconstituted in human embryonic kidney 293 cells, in the absence or in the presence of ACh (1 or 10 nm). Taken together, these data suggest that NO is a cGMP-independent modulator of G-protein-coupled muscarinic and β-adrenergic receptor actions on cardiac ion channels. Although this action of NO seemed to occur at the level of G proteins, it appeared to require a component distinct from receptors, G proteins or their effectors.
Biochemical data has established the presence of a nitric oxide (NO) pathway in the heart, including sources of NO and several effectors. Yet the role of NO synthesis in cardiac physiology remains rather controversial, since it has been reported that endogenous or exogenous NO has either no effect or multiple effects on cardiac contraction, excitability and respiration (reviewed in Méry et al. 1997; Kojda & Kottenberg, 1999; Shah & MacCarthy, 2000). The heterodimeric guanylyl cyclase, which produces cGMP, was recognised as an important target of NO in the heart, as well as in purified cardiac myocytes (see Kojda & Kottenberg, 1999). In turn, cGMP co-ordinates the activity of various proteins, such as the cGMP-stimulated phosphodiesterase (PDE2), the cGMP-inhibited-PDE (PDE3), and the cGMP-dependent protein kinase (cG-PK). Accordingly, cGMP was often found to mediate the effects of NO donors on myocyte shortening, ionic currents and contractile proteins.
The heterodimeric guanylyl cyclase is no longer viewed as the only target of NO in the heart. Indeed, cGMP-independent effects of NO donors modulate the activity of cardiac proteins, including the ryanodine receptor-Ca2+ channel (Hu et al. 1997; Xu et al. 1998), the β2-adrenergic receptor (Adam et al. 1999), adenylyl cyclase (McVey et al. 1999; Hill et al. 2000), creatine kinase (Gross et al. 1996) and cytochrome C (Wolin et al. 1997). In addition, cGMP is not involved in the effects of NO donors on the cardiac L-type Ca2+ channels expressed in human embryonic kidney (HEK)293 cells (Hu et al. 1997; Poteser et al. 2001), on the basal L-type Ca2+ current (ICa,L) in ferret ventricular myocytes (Campbell et al. 1996), and on the β-adrenergic stimulation of cardiac contractility and Ca2+ transients in rat ventricular myocytes (Vila-Petroff et al. 1999).
NO donors exert multiple effects on the activity of L-type Ca2+ channels, which regulate cardiac contraction. Interestingly, the effects of NO donors on ICa,L seem to exhibit tissue specificity. In nodal myocytes, NO donors inhibit the β-adrenergic stimulation of ICa,L, and this effect involves the cGMP-dependent activation of PDE2 (Han et al. 1994, 1996; see also Martynyuk et al. 1997). In cat and human atrial myocytes, NO donors potentiate the cAMP-dependent stimulation of ICa,L, and this effect involves the cGMP-dependent inhibition of PDE3 (Kirstein et al. 1995; Wang et al. 1998). In rabbit atrial myocytes, NO donors enhance ICa,L in a cAMP-independent manner through the activation of cG-PK (Wang et al. 2000). In ventricular myocytes, NO donors were generally found to exert opposite effects on ICa,L. The inhibitory effect (a reduction in cAMP-dependent stimulation) was attributed to cG-PK activation in mammalian myocytes (Levi et al. 1994; Whaler & Dollinger, 1995; Campbell et al. 1996; Abi-Gerges et al. 2001a) and to PDE2 activation in frog myocytes (Méry et al. 1993). The stimulatory effect was attributed to PDE3 inhibition in frog ventricular myocytes (Méry et al. 1993) and to a cGMP-independent and, possibly, a direct effect of NO in ferret ventricular myocytes (Campbell et al. 1996). In rat ventricular myocytes, where contractility instead of ICa,L was examined, the potentiating effect of NO donors was suggested to involve PDE3 (Kojda et al. 1996), or to alter a step located upstream from cAMP in the adenylyl cyclase pathway (Vila-Petroff et al. 1999).
In the present study, we investigated the mechanism underlying the potentiating effect of the NO donors S-nitroso-N-acetyl-d,l-penicillamine (SNAP) and 2-(N,N-diethylamino)-diazenolate-2-oxide (DEANO) on ICa,L in rat ventricular myocytes. We found that NO donors modulated strongly the adenylyl cyclase pathway, in a cGMP-independent manner. Our data support the hypothesis that NO might interfere with the activity of G proteins. Moreover, NO donors potentiated the muscarinic activation of the inwardly rectifying potassium channel, IK,ACh. Importantly, while the effect of NO donors on IK,ACh appeared to occur at the level of the G protein in rat atrial myocytes, further experiments conducted in HEK293 cells suggested that G proteins per se are not the target of NO. Part of this work has been presented in abstract form (Abi-Gerges et al. 2001b).
METHODS
The investigation conforms to our institutional guidelines, which are defined by the European Community guiding principles in the care and use of animals and the French decree no. 87/848 of 19th October 1987. Authorisations to perform animal experiments according to this decree were obtained from the French Ministère de l'Agriculture, de la Pêche et de l'Alimentation (no. 7475, 27th May 1997).
Cell isolation and storage
Male Wistar rats (160–250 g) were anaesthetised by intraperitoneal injection of urethane (2 g kg−1) and heparin (2.5 mg kg−1). After all reflex activity had ceased, the animal was killed by opening the chest and rapidly removing the heart. Ventricular myocytes were dispersed using collagenase A (0.255 mg ml−1, Boehringer-Mannheim Biochemica, Mannheim, Germany), as described previously (Abi-Gerges et al. 1997). An additional digestion with 0.5 mg ml−1 protease (Sigma Chemical, St Louis, MO, USA) and 0.255 mg ml−1 collagenase A (Boehringer-Mannheim) allowed the collection of atrial myocytes (Abi-Gerges et al. 1997). Ventricular myocytes (stored at 37 °C) and atrial myocytes (kept at 4 °C) were used within 1–12 h following isolation.
Cell culture and transfection
HEK293 cells stably expressing the GIRK4 (Kir3.4 or CIR) subunit (Xu et al. 1996) were grown on 12 well plates in Dulbecco's modified Eagle's medium (DMEM, Invitrogen, Carlsbad, CA, USA), supplemented with 10 % fetal bovine serum (Invitrogen) and 1 % penicillin-streptomycin (Invitrogen), under 5 % CO2 at 37 °C. Lipofectamine reagent (Invitrogen) was used for the transient transfection of HEK293 cells according to the manufacturer's instructions. The following transfection vectors were used: GIRK1 (Kir3.1, gift of Dr L. Jan) inserted in the pCDNA3.1/Zeo(+)-vector (Invitrogen, 0.745 μg); M2-receptor (in pCMV, gift of Dr E. Peralta, see Peralta et al. 1987; 0.745 μg) and the pGreen Lantern plasmid (Life Technologies; 0.19 μg). Typically, the total amount of plasmid used for transfection was 1.68 μg per well. All experiments were performed 24–48 h post-transfection on green-fluorescent-protein-expressing cells.
Electrophysiology
The whole-cell configuration of the patch-clamp technique was used to record ICa,L and K+ currents in cardiac myocytes (Abi-Gerges et al. 1997, 2001a). For ICa,L measurements, the routine protocol consisted of a test pulse to 0 mV (400 ms duration) elicited from a holding potential of −50 mV, every 8 s. The maximal amplitude of ICa,L, the steady-state currents, as well as current-voltage relationships and inactivation curves were recorded as described previously (Abi-Gerges et al. 2001a). In the present study, the density of basal ICa,L was 5.51 ± 0.30 pA pF−1 and the density of the current at the end of the 400 ms pulse was 0.39 ± 0.02 pA pF−1 (n = 94). For atrial IK,ACh measurements, 200 ms pulses to 0 mV and −120 mV were successively applied every 5 s from a holding potential of −50 mV (Abi-Gerges et al. 1997). IK,ACh was quantified as the change in steady-state current at the end of the pulse at 0 mV. In the present study, the density of this current was 2.4 ± 0.2 pA pF−1 under basal conditions (n = 75). Currents were not compensated for capacitive and leak currents. On-line analysis of the recordings was made possible by programming a PC-compatible 486/50 microcomputer in Assembly language (Borland) to determine, for each membrane depolarisation, peak and steady-state current values.
The muscarinic potassium current Ik,ACh reconstituted in HEK293 cells was measured as an inward (at −110 mV for 300 ms) and outward (at −5 mV for 300 ms) current using a holding potential of −40 mV for 200 ms. Currents were recorded using an Axopatch 200A current-to-voltage converter (Axon Instruments, Union City, CA, USA), filtered at 5 kHz and recorded using a VCR-based converter (Instrutech VR-10B, Port Washington, NY, USA). Tracings are from data filtered at 1 kHz and digitised at 500 μs intervals. All experiments were carried at room temperature.
Solutions for patch-clamp recordings
Solutions were superfused onto floating myocytes as described previously (Abi-Gerges et al. 1997). The external caesium-containing solution contained in (mm): 107 NaCl, 10 Hepes, 20 CsCl, 4 NaHCO3, 0.8 NaH2PO4, 1.8 MgCl2, 1.8 CaCl2, 5 d-glucose, 5 sodium pyruvate and 6 × 10−4 tetrodotoxin, pH 7.4 adjusted with CsOH. In the external potassium-containing solution, 2.5 mm KCl was substituted for CsCl, and the pH was adjusted to 7.4 with NaOH. The patch pipettes (0.5–1.0 MΩ resistance for ventricular cells, 1.0–2.5 MΩ for atrial cells) used to record ICa,L were filled with an internal caesium-containing solution that contained (mm): 119.8 CsCl, 5 EGTA (acid form), 4 MgCl2, 5 disodium phosphocreatine, 3.1 Na2ATP, 0.42 Na2GTP, 0.062 CaCl2 (pCa 8.5) and 10 Hepes, pH 7.3 adjusted with CsOH. For the internal potassium-containing solution, 102 mm KCl was substituted for CsCl and the pH was adjusted to 7.3 with KOH. Na2GTP was omitted in the internal solution containing 400 μm GTP-γS.
For the measurement of IK,ACh in HEK293 cells, the external solution contained (mm): 150 NaCl, 5.4 KCl, 5 Hepes, 1 MgCl2, 2 CaCl2 and 1.1 glucose, pH 7.4 adjusted with KOH. Patch pipettes (2.5–5 MΩ) were filled with an internal potassium-containing solution, which contained (mm): 20 KCl, 130 potassium aspartate, 1 KH2PO4, 5 Hepes, 5 EGTA (acid form), 3 MgCl2, 3 MgATP and 0.2 GTP, pH 7.3 adjusted with KOH.
Drugs
2-(4-carboxyphenyl)-4,4,5,5-tetramethylimidazoline-1-oxyl 3-oxide (cPTIO), S-nitroso-N-acetyl-d,l-penicillamine (SNAP), 2-(N,N-diethylamino)-diazenolate-2-oxide (DEANO) and 1H-[1, 2,4]oxadiazolo[4,3-a]quinoxaline-1-one (ODQ) were from Alexis (San Diego, CA, USA). 8,13-Epoxy-7b-(N-methyl-piperazino-g-butyryloxy)-1a,6b-trihydroxy-labd-14-en-11-one (L858051), cilostamide and adenosine-3′,5′-cyclic monophosphothioate, Rp-isomer (Rp-cAMPS) were from Calbiochem-France Biochem (Meudon, France). Tetrodotoxin was from Latoxan (Rosans, France). All others drugs were from Sigma Aldrich (L'Isle d'Abeau Chesnes, France). Drugs were prepared and used according to the manufacturer's instructions. Solutions were prepared by dilution to the desired concentration in the physiological solution at the beginning of each experiment.
Statistical analysis and fitting equations
Results are expressed as means ±s.e.m. Differences between mean values were tested for statistical significance using the paired Student's t test, as indicated. In the text, the ‘basal’ condition for ICa,L refers to the absence of isoprenaline (Iso) or cAMP-elevating agents. The ‘basal’ condition for IK,ACh refers to the absence of ACh or non-hydrolysable analogues of GTP.
The Michaelis-Menten equation was used as follows:
where Emax is the maximal effect of ACh and EC50, the concentration of ACh ([ACh]) that induces a half-maximal effect.
RESULTS
Effect of SNAP on the stimulation of ICa,L by threshold concentrations of Iso
ICa,L, which is enhanced in rat ventricular myocytes by commonly used concentrations (1 nm to 1 μm) of the β-adrenergic agonist Iso, was found to be inhibited by NO donors (Abi-Gerges et al. 2001a). In this report, we examined an unexpected stimulatory effect of NO donors on ICa,L observed when threshold or sub-threshold concentrations of Iso were used to stimulate ICa,L. In the experiment shown in Fig. 1A, a rat ventricular myocyte was first exposed to internal and external solutions. Superfusion of the myocyte with Iso (100 pm) elicited a small stimulatory effect on ICa,L. In the continuing presence of Iso, addition of the nitrosothiol SNAP (1 pm and 1 nm) induced a dose-dependent potentiation of the Iso-stimulated ICa,L. As expected from our previous study (Abi-Gerges et al. 2001a), a further increase in SNAP concentration to 1 μm induced a reversible inhibitory effect on ICa,L that counteracted the stimulatory effect seen at lower doses. However, under our current experimental conditions, this secondary inhibitory effect of SNAP was observed in only 4 out of 11 experiments, while the stimulatory effect of SNAP was maintained or even increased at a concentration of 1 μm in the remaining 7 experiments.
Figure 1. The nitric oxide (NO) donor S-nitroso-N-acetyl-d,l-penicillamine (SNAP) modulates the threshold β-adrenergic stimulation of the L-type Ca2+ current (ICa,L) in rat ventricular myocytes.
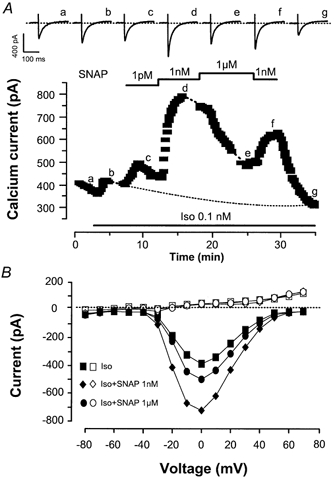
A, a myocyte was first exposed to external and internal control solutions. ICa,L (▪) was elicited at 0 mV from a holding potential of −50 mV. Superfusion of the myocyte with 0.1 nm isoprenaline (Iso) alone or in combination with SNAP (1 pm, 1 nm, or 1 μm) is indicated by the lines. Traces were recorded at the times indicated by the corresponding letters on the main graph (a–g). The dotted line indicates the zero-current level. B, (same experiment as in A) current-voltage relationships of ICa,L (filled symbols) and of the steady-state current at the end of the pulse (open symbols) obtained in the presence of Iso 0.1 nm alone (squares) or in combination with SNAP (1 nm, triangles; 1 μm, circles).
Current-voltage relationships of ICa,L (filled symbols) and the steady-state current measured at the end (400 ms) of the test pulse (open symbols) recorded during this experiment are shown in Fig. 1B. The effects of SNAP on the Iso-stimulated ICa,L were smooth along the voltage range, and SNAP did not change the steady-state current (similar findings were obtained in four other experiments). Moreover, SNAP did not change the inactivation curve of the Iso-stimulated ICa,L (data not shown). Thus, the potentiating effect of SNAP on the Iso-stimulated ICa,L occurred in a voltage-independent manner.
Interestingly, SNAP alone had no effect on ICa,L. Indeed, at SNAP concentrations of 1 pm, 1 nm and 1 μm, ICa,L was 96.5 ± 1.4 % (n = 3), 101.2 ± 1.9 % (n = 3) and 97.3 ± 1.0 % (n = 6) of the control value, respectively. Thus, the stimulatory effect of SNAP required the presence of Iso. A summary of the effect of 1 nm SNAP on the Iso-stimulated ICa,L is shown in Fig. 2A. Iso was used at low concentrations (0.01–500 pm) which, on average, induced slight but non-significant stimulations of ICa,L. The potentiating effect of this nitrosothiol was observed over the entire range of concentrations of Iso. Surprisingly, the mean effect of 1 nm SNAP appeared to be similar at the different concentrations of Iso examined. Figure 2B summarises the effects of different concentrations of SNAP (1 pm, 1 nm, 1 μm) in the presence of Iso (0.1–0.5 nm). For a comparison, the effects of DEANO, a NONOate, were studied in similar conditions. At low concentration (1 pm), both NO donors induced non-significant increases in the Iso-stimulated ICa,L, but consistently potentiated the β-adrenergic stimulation of ICa,L at higher concentrations (1 nm and 1 μm). On average, the effect of DEANO was weaker than that of SNAP.
Figure 2. Summary of the effects of NO donors on the threshold β-adrenergic stimulation of ICa,L in rat ventricular myocytes.
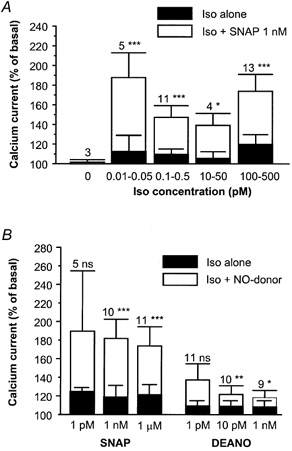
A, mean stimulatory effects of various low concentrations of Iso on ICa,L, in the absence (filled bars) and in the presence of 1 nm SNAP (open bars). B, mean stimulatory effects of Iso (0.1–0.5 nm) in the absence (filled bars) and in the presence of different concentrations of SNAP or 2-(N,N-diethylamino)-diazenolate-2-oxide (DEANO; open bars). The amplitude of ICa,L in the presence of agonists was normalised to the amplitude of the basal ICa,L in the absence of agonists. Bars are the means and vertical lines are the s.e.m. of the number of experiments indicated near the bars. Significant differences from the Iso level: *P≤ 0.05; **P≤ 0.01; ***P≤ 0.005.
The involvement of NO was examined using cPTIO, a NO scavenger (Akaike et al. 1993). In these experiments, Iso (0.01–2 pm) induced a slight increase in ICa,L (+21.3 ± 6.9 % over basal, n = 12, P≤ 0.01), and this effect was not affected by 100 μm cPTIO (+26.8 ± 6.4 % over basal, n = 4). The Iso-stimulated ICa,L was potentiated by 1 nm SNAP (+76.8 ± 14.9 % over basal, n = 5, P≤ 0.01) and the addition of 100 μm cPTIO totally antagonised this stimulatory effect (to +25.2 ± 10.6 % over basal, n = 5; P≤ 0.01versus Iso plus SNAP). Similarly, the potentiating effect of 1 μm SNAP on the Iso-stimulated ICa,L (+60.7 ± 13.5 % over basal, n = 3, P≤ 0.05) was abolished by the further addition of cPTIO 100 μm (to +35.6 ± 9.3 % over basal, n = 3, P≤ 0.05versus Iso plus SNAP). Therefore, the stimulatory effect of SNAP must be due to NO release.
Role of guanylyl cyclase in the potentiating effect of SNAP on the Iso-stimulated ICa,L
The effects of NO on ICa,L in cardiac myocytes have generally been attributed to the production of cGMP. However, in two studies, nitrosothiols were found to enhance the basal ICa,L or to potentiate the cardiac contractility of isolated myocytes in a cGMP-independent manner (Campell et al. 1996; Vila-Petroff et al. 1999). The selective guanylyl cyclase inhibitor, ODQ (Sandirasegarane & Diamond, 1999; Stojanovic et al. 2001), was used here to examine the possible involvement of heterodimeric guanylyl cyclase in the potentiating effect of SNAP.
In the typical experiment shown in Fig. 3A, the basal ICa,L was slightly enhanced by a low concentration of Iso (0.2 nm), and addition of 1 nm SNAP induced a further increase in ICa,L. The effect of SNAP was not changed by treatment with ODQ (10 μm). After wash-out of ODQ, the concentration of SNAP was raised to 1 μm, inducing a decrease in ICa,L. This inhibitory effect of SNAP was antagonised by ODQ, as reported previously (Abi-Gerges et al. 2001a).
Figure 3. The potentiating effect of NO donors on the Iso-stimulated ICa,L is cGMP-independent in rat ventricular myocytes.
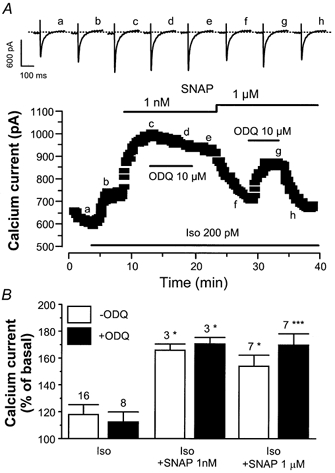
A, a myocyte was first exposed to control solutions. Superfusion of the myocyte with 0.2 nm Iso alone or in combination with SNAP (1 nm or 1 μm) or 1H-[1, 2, 4]oxadiazolo[4, 3-a]quinoxaline-1-one (ODQ) (10 μm) is indicated by the lines. Traces were recorded at the times indicated by the corresponding letters on the main graph (a-h). The dotted line indicates the zero-current level. B, mean stimulatory effects of Iso (0.1 and 0.2 nm) on ICa,L, in the presence (filled bars) and in the absence of 10 μm ODQ (open bars), with or without SNAP (1 nm or 1 μm), as indicated. The amplitude of ICa,L in the presence of agonists was normalised to the amplitude of the basal ICa,L in the absence of agonists. Bars are the means and vertical lines are the s.e.m. of the number of experiments indicated near the bars. Significant differences from the Iso level: *P≤ 0.05; ***P≤ 0.005.
As summarised in Fig. 3B, the effect of low concentrations of Iso (0.1 and 0.2 nm) was not altered by ODQ (10 μm). On average, the guanylyl cyclase inhibitor did not modify the stimulatory effects of SNAP (1 nm and 1 μm). In this set of experiments, the inhibitory effect of 1 μm SNAP was observed only twice, and was then antagonised upon the addition of ODQ. In the remaining five experiments where 1 μm SNAP induced a stimulatory effect similar or larger than that of a lower concentration (1 nm), addition of ODQ had no further effect. Thus, unlike the inhibitory effect of NO donors on ICa,L, which is clearly due to cGMP production (see also Abi-Gerges et al. 2001a), these data suggest that the potentiating effect of SNAP does not require cGMP.
PDE3 is not involved in the potentiating effect of SNAP on the Iso-stimulated ICa,L
In a study by Vila-Petroff et al. (1999), it was shown that in rat ventricular myocytes, SNAP increases cardiac contractility and intracellular Ca2+ in the presence of Iso. The effect of SNAP, independent of cGMP, was suggested to occur at the level of cAMP production. We investigated whether this mechanism could account for the effect of SNAP by using L858051, a forskolin analogue that activates directly adenylyl cyclase (Hartzell & Budnitz, 1992). In the typical experiment shown in Fig. 4A, a myocyte was exposed to 0.2 μm L858051, which induced a slow and non-maximal increase in ICa,L. Unlike what was observed with Iso, addition of SNAP (1 nm) with L858051 induced a slight decrease in ICa,L. For a comparison, addition of cilostamide (0.3 μm), a specific PDE3 inhibitor, elicited a strong potentiation of the L858051 stimulated ICa,L. In other experiments where L858051 (0.1–0.3 μm) had a slight but non-significant stimulatory effect on ICa,L, or those using different concentrations of SNAP (1 pm, 1 nm, 1 μm), ICa,L remained unaltered in the presence of the forskolin analogue (Fig. 4B). Under the same conditions, cilostamide (0.3 μm) potentiated markedly the effect of L858051 on ICa,L.
Figure 4. Effect of SNAP on the L858051-stimulated ICa,L in rat ventricular myocytes.
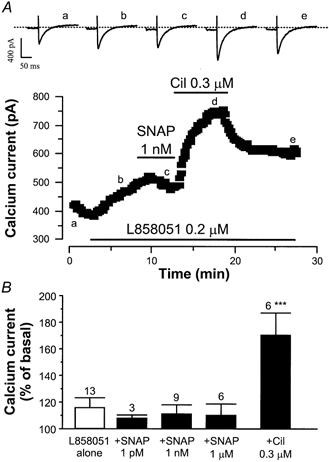
A, a myocyte was first exposed to control solutions. Applications of L858051 (0.2 μm), SNAP (1 nm) and cilostamide (Cil, 0.3 μm) are indicated by the lines. Traces were recorded at the times indicated by the corresponding letters on the main graph (a-e). The dotted line indicates the zero-current level. B, summary of the effects of L858051 alone (open bar) or in the presence (filled bars) of SNAP (1 pm, 1 nm, 1 μm), or cilostamide (0.3 μm). The amplitude of ICa,L in the presence of agonists was normalised to the amplitude of the basal ICa,L in the absence of agonists. Bars are the means and lines are the s.e.m. of the number of experiments indicated near the bars. Significant differences from L858051 level: ***P≤ 0.005.
The results of these experiments suggest that the effect of SNAP does not require a cGMP-dependent inhibition of PDE3. To further support this conclusion, we examined the effect of SNAP in the presence of a PDE4 inhibitor, rolipram, since dual inhibition of PDE3 and PDE4 was found to stimulate basal ICa,L in rat myocytes (Verde et al. 1999). As reported previously (Verde et al. 1999), basal ICa,L was not modified in the presence of rolipram alone (1 μm; 94.4 ± 2.7 % of basal, n = 4). Moreover, addition of SNAP (at 1 nm or 1 μm) in the presence of the PDE4 inhibitor had no effect on basal ICa,L (which was 94.7 ± 5.2 and 98.6 ± 3.4 % of basal, respectively, n = 4). Taken together, these experiments rule out the participation of a cGMP-inhibited PDE3 in the stimulatory effect of SNAP. Furthermore, they demonstrate that the effect occurs neither at the level nor downstream of adenylyl cyclase, but rather takes place at a step located upstream (i.e. at the level of the receptor or the G protein).
Role of cAMP-dependent protein kinase (cA-PK) in the potentiating effect of SNAP on the Iso-stimulated ICa,L
In ferret ventricular myocytes, NO donors were found to potentiate the basal ICa,L, and this effect was not prevented by inhibition of cA-PK (Campbell et al. 1996). Since cA-PK is ultimately responsible for the β-adrenergic stimulation of ICa,L (see e.g. Striessnig, 1999, for a review), we investigated its involvement in the potentiating effect of SNAP on the Iso-stimulated ICa,L. In the experiment shown in Fig. 5A, a myocyte was dialysed with the cA-PK inhibitory peptide (PKI, 20 μm) and superfused with control solution. While an initial brief exposure of the myocyte to Iso (10 nm) enhanced ICa,L, dialysis of the myocyte with PKI for ≥ 20 min totally suppressed this effect, as evidenced by the lack of response of the cell to subsequent applications of 10 nm or 100 pm Iso. However, PKI also suppressed the stimulatory effect of SNAP since, when applied in the continuing presence of Iso (100 pm), SNAP (1 nm and 1 μm) had no effect on ICa,L.
Figure 5. cAMP-dependent protein kinase (cA-PK) inhibitors antagonise the effects of NO donors on the Iso-stimulated ICa,L in rat ventricular myocytes.
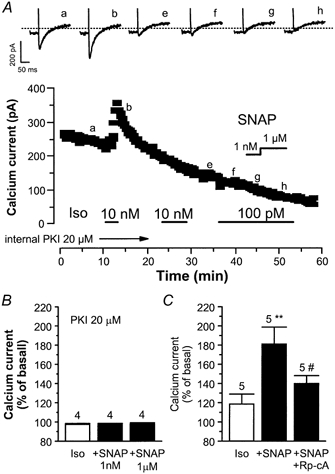
A, a myocyte was first dialysed with cA-PK inhibitory peptide (PKI; 20 μm) and exposed to external control solution. Applications of Iso (100 pM, 10 nm) and SNAP (1 nm, 1 μm) are indicated by the lines. Traces were recorded at the times indicated by the corresponding letters on the main graph (a-h). The dotted line indicates the zero-current level. B, summary of the effects of Iso (100 pm), SNAP (1 nm, 1 μm), in myocytes dialysed with PKI (20 μm). C, overview of the effects of Iso (1–100 pm), SNAP (1 nm), in the absence or in the presence of external adenosine-3′,5′-cyclic monophosphothioate, Rp-isomer (Rp-cA 300 μm). The amplitude of ICa,L in the presence of agonists was normalised to the amplitude of the basal ICa,L in the absence of agonists. Bars are the means and vertical lines are the s.e.m. of the number of experiments indicated near the bars. Significant differences from the Iso (*) or the Iso plus SNAP (#) levels: *P≤ 0.05; #P≤ 0.01.
On average, Iso (100 pm) had no effect in four similar experiments with PKI-dialysed myocytes (Fig. 5B). In addition, SNAP did not stimulate ICa,L in these cells. The effect of an extracellular perfusion with Rp-cAMPS, another cA-PK inhibitor, was also examined (Fig. 5C). In these experiments, Iso (1–100 pm) induced a small stimulatory effect, which as expected, was potentiated by the application of SNAP (1 nm). However, this effect was antagonised by Rp-cAMPS (500 μm). Thus, like the stimulatory effect of Iso, the potentiating effect of SNAP on Iso-stimulated ICa,L requires the activation of cA-PK.
In rat cardiomyocytes, adrenergic agonists can induce NOS2 expression, and this effect involves (at least, in part) the mitogen-activated protein kinase pathway (Kan et al. 1999). However, intracellular dialysis of rat ventricular myocytes with PD98059 (50 μm), an MEK inhibitor, did not modify the effects of SNAP (1 pm to 1 μM) on either basal or Iso-stimulated (1 pm) ICa,L (data not shown).
Role of G proteins in the potentiating effect of SNAP on Iso-stimulated ICa,L
Since the NO donors tested here potentiated the effect of Iso on ICa,L, but not that of L858051, one possibility is that the NO donors enhance the binding of Iso to its receptor. Alternatively, the NO donors may enhance the activation of Gs proteins via the activated β-adrenergic receptor and/or strengthen the effect of activated Gs proteins on adenylyl cyclase. To discriminate between these two hypotheses, we investigated the effect of SNAP in the presence of GTP-γS, a non-hydrolysable GTP analogue that can activate Gs proteins and hence adenylyl cyclase, in the absence of β-adrenergic agonists. In the experiment shown in Fig. 6A, GTP-γS (10 μm) was included in the patch pipette solution, and dialysis of the GTP analogue started when the membrane patch was ruptured. GTP-γS induced a spontaneous run-up of the basal ICa,L, consistent with the activation of Gs proteins. SNAP (1 nm) was superfused during the run-up of ICa,L, inducing a strong stimulatory effect. Increasing the concentration of SNAP to 1 μm induced no further effect on ICa,L. On average, when the myocytes were dialysed with GTP-γS (10 μm) for 18.6 ± 2.2 min before the addition of SNAP (n = 4), the GTP-γS-stimulated ICa,L was consistently enhanced by 1 nm and 1 μm SNAP (Fig. 6B). Thus, the potentiating effect of the NO donors does not require the binding of the β-adrenergic agonist to its receptor, but requires some degree of activation of Gs proteins.
Figure 6. Effect of SNAP on ICa,L in the presence of GTP-γS in rat ventricular myocytes.
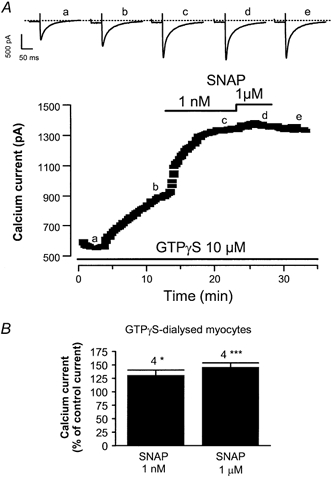
A, a myocyte was first dialysed with 10 μm GTP-γS, a non-hydrolysable analogue of GTP, and exposed to control external solution. The lines indicate applications of SNAP (1 nm, 1 μm). Traces were recorded at the times indicated by the corresponding letters on the main graph (a-e). The dotted line indicates the zero-current level. B, summary of the effects of SNAP (1 nm, 1 μm) in 10 μm GTP-γS-dialysed myocytes. The amplitude of ICa,L in the presence of SNAP was normalised to the amplitude of the basal ICa,L in the absence of SNAP. Bars are the means and vertical lines are the s.e.m. of the number of experiments indicated near the bars. Significant differences from the GTP-γS level are: *P≤ 0.05; ***P≤ 0.005.
Potentiating effect of SNAP on IK,ACh
The results of our study so far support the view that the NO released by NO donors modifies the activation of adenylyl cyclase by G proteins. In the heart, the muscarinic activation of IK,ACh is a simple membrane-delimited pathway involving a single G protein (Gi/o). Therefore, we next studied the effects of NO donors on IK,ACh in rat atrial myocytes. In the experiment shown in Fig. 7A, where currents are shown at two membrane potentials (−120 and 0 mV), a rat atrial myocyte was first exposed to control potassium-containing solutions. Superfusion of the myocyte with SNAP (1 nm and 1 μm) did not modify the basal current, although after wash-out of SNAP, ACh (3 μm) strongly stimulated the inwardly rectifying IK,ACh (see also Abi-Gerges et al. 1997). Individual recordings also demonstrate that SNAP did not modify the kinetics of the basal current (top panel in Fig. 7A). In similar experiments, the steady-state current at 0 mV, which averaged 2.4 ± 0.5 pA pF−1 (n = 13), was not modified by SNAP (+11.7 ± 12.0 % over basal at 1 nm SNAP, n = 5, and +9.0 ± 11.4 % at 1 μm SNAP, n = 4). The basal steady-state current also remained unaltered by the application of either 1 μm DEANO (+18.9 ± 17.1 % over basal, n = 4) or 100 μm cPTIO (−1.8 ± 6.9 % over basal, n = 4).
Figure 7. SNAP potentiates the muscarinic activation of the muscarinic-activated K+ current, IK,ACh, in rat atrial myocytes.
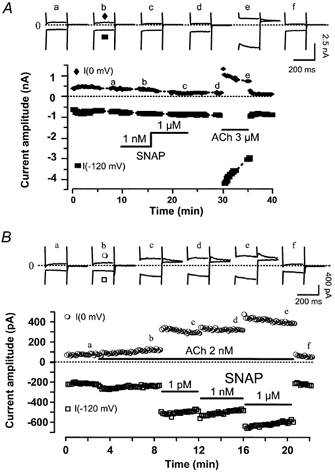
A, an atrial myocyte was first exposed to control potassium-rich solutions, and superfusion with SNAP (1 nm, 1 μm) or ACh (3 μm) was performed as indicated by the lines. IK,ACh was measured at 0 mV (•) or −120 mV (▪) from a holding potential of −50 mV. B, same as in A, except that SNAP (1 pm, 1 nm, 1 μm) was applied in the presence of ACh (2 nm). Traces were recorded at the times indicated by the corresponding letters on the main graphs. The dotted lines indicate the zero-current level.
In the experiment shown in Fig. 7B, the myocyte was first exposed to ACh (2 nm), which induced a slow stimulation of IK,ACh. Surprisingly, this effect was potentiated dramatically in a dose-dependent manner upon further superfusion with increasing concentrations of SNAP (1 pm, 1 nm and 1 μm). The effect of SNAP was rapidly reversible. The effects of SNAP (1 μm) on IK,ACh in the presence of different concentrations of ACh (10 pm to 3 μm) are summarised in Fig. 8A. SNAP modified the effect of low concentrations of ACh (below 30 nm) and did not change the maximal effect of the muscarinic agonist on IK,ACh. In addition, SNAP did not modify the kinetics of the desensitisation of IK,ACh to saturating concentrations of ACh (data not shown). Further analysis (inset in Fig. 8A) suggests that the effects of ACh alone are well fitted to a Michaelis-Menten equation (EC50 = 17.5 nm; Emax = 404 %). However, the data points obtained for the effect of ACh in the presence of SNAP show clearly that a single Michaelis-Menten equation would be inappropriate to fit the data. Although a quantitative analysis was not permitted due to the lack of data in the lowest range of ACh concentrations (between 0.03 and 0.3 nm), the results obtained at 1 and 2 nm ACh suggest that SNAP converts a fraction of the effective binding sites to an apparent high-affinity state.
Figure 8. Summary of the effects of NO donors on IK,ACh in rat atrial myocytes.
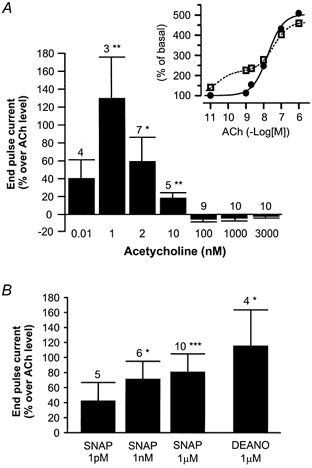
A, mean stimulatory effects of SNAP (1 μm) on IK,ACh, in the presence of various concentrations of ACh. Inset (same experiments as in the main graph), the effects of ACh, in the absence (•) and in the presence of SNAP (□), were normalised to the amplitude of the end pulse current at 0 mV in the absence of agonist. The dose-response curve to ACh alone was fitted to the Michaelis-Menten equation (continuous line), as described in the text. The dose-response curve to ACh in the presence of SNAP was fitted by eye (dotted line). B, mean stimulatory effects of SNAP (1 pm, 1 nm, 1 μm) or DEANO (1 μm) on IK,ACh, in the presence of ACh (1 and 2 nm). In A and B, the amplitude of IK,ACh (the end pulse current at 0 mV) in the presence of ACh + NO donors was normalised to the amplitude of IK,ACh in the presence of ACh alone. Bars are the means and vertical lines are the s.e.m. of the number of experiments indicated near the bars. Significant differences from the ACh level: *P≤ 0.05; **P≤ 0.01: ***P≤ 0.005.
The effects of three different concentrations of SNAP (1 pm, 1 nm and 1 μm) and of DEANO (1 μm) were also studied in the presence of threshold concentrations of ACh (1 or 2 nm, Fig. 8B), which alone induced only a slight stimulation of IK,ACh (+41.7 ± 13.1 % over basal, n = 10, P≤ 0.01). While 1 pm SNAP had no significant effect on the ACh-stimulated IK,ACh, 1 nm or 1 μm SNAP or 1 μm DEANO potentiated the effect of the muscarinic agonist. In addition, the stimulation of IK,ACh elicited by a saturating concentration of ACh (3 μm) was unchanged in the presence of DEANO, even when used at 100 μm (−5.7 ± 3.4 % over ACh level, n = 6).
NO is involved in the effect of NO donors on IK,ACh
The involvement of NO in the potentiating effects of the NO donors on the ACh-stimulated IK,ACh was examined using cPTIO, which antagonised the effects of the NO donors on the Iso-stimulated ICa,L (as shown above). In Fig. 9A, a rat atrial myocyte was exposed to a low concentration of ACh (10 pm), which had a small stimulatory effect on IK,ACh. The addition of DEANO (1 μm) potentiated the effect of ACh on IK,ACh, and the effect of the NO donor was totally antagonised by superfusion with cPTIO (100 μm). The effects of the compounds were reversible upon wash-out. A saturating concentration of ACh (3 μm) was then superfused to elicit the maximal activation of IK,ACh.
Figure 9. The NO scavenger 2-(4-carboxyphenyl)-4,4,5,5-tetramethylimidazoline-1-oxyl 3-oxide (cPTIO) antagonises the effects of DEANO on the ACh-activated IK,ACh in rat atrial myocytes.
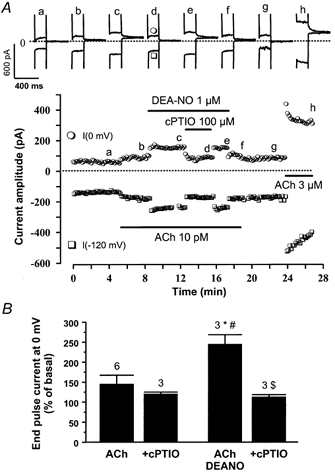
A, an atrial myocyte was first exposed to control potassium-rich solutions, and superfusion with DEANO (1 μm), cPTIO (100 μm) and ACh (10 pm, or 3 μm) was performed as indicated by the lines. Traces were recorded at the times indicated by the corresponding letters on the main graphs (a-h). The dotted line indicates the zero-current level. B, mean stimulatory effects of ACh (1 pm, 1 nm, 1 μm) on IK,ACh, with or without DEANO (1 μm) in the absence or presence of cPTIO (100 μm). The amplitude of IK,ACh (at 0 mV) in the presence of compounds was normalised to the amplitude of the end pulse current (at 0 mV) in the absence of compounds. Bars are the means and lines are the s.e.m. of the number of experiments indicated near the bars. Significant differences (P≤ 0.05) from basal level (*), or the Iso level (#), or the Iso plus DEANO level ($) are indicated near the bars.
On average, cPTIO (100 μm) alone exerted no significant effect on the small stimulatory effect of ACh (10 pm and 1 nm) on IK,ACh, but fully antagonised the potentiating effect of DEANO (1 μm, Fig. 9B). Thus, NO generation most likely contributed to the effect of DEANO on the ACh-stimulated IK,ACh. In contrast, guanylyl cyclase inhibition with ODQ did not modify the effects of NO donors on IK,ACh (data not shown), indicating that similar to ICa,L regulation, cGMP does not contribute to the effect of DEANO on IK,ACh.
Effect of NO donors on the direct activation of IK,ACh by GTP analogues
Since we found that NO donors potentiate ICa,L in the presence of a non-hydrolysable GTP analogue and in the absence of β-adrenergic agonists, we studied the corresponding effects of NO donors on IK,ACh in the presence of the non-hydrolysable GTP analogue 5′-guanylylimidodiphosphate (GppNHp). In the experiment shown in Fig. 10A, a low concentration of GppNHp (10 μm) was included in the patch-pipette solution, and the dialysis of this analogue started as the membrane patch was ruptured, inducing a small and slow rise in IK,ACh in the absence of ACh. Fifteen minutes after the beginning of the dialysis, superfusion of the myocyte with SNAP (1 nm and 1 μm) induced a dose-dependent increase in IK,ACh. Interestingly, the effect of SNAP was reversible, and could be repeated when SNAP (1 μm) was applied a second time. For a comparison, a maximal concentration of ACh (3 μm) was then superfused, inducing a large (approximately three-fold) increase in IK,ACh.
Figure 10. Effects of SNAP on IK,ACh in the presence of 5′-guanylylimidodiphosphate (GppNHp) in rat atrial myocytes.
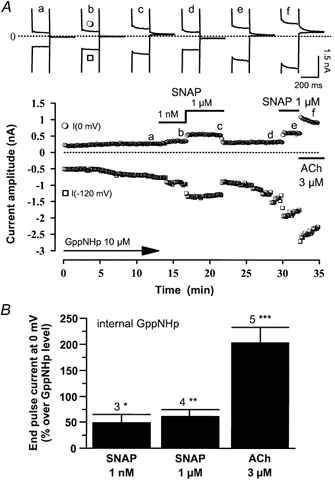
A, an atrial myocyte was dialysed with GppNHp (10 μm) and exposed to external control K+-solutions. Superfusion with SNAP (1 nm, 1 μm) and ACh (3 μm) was performed as indicated by the lines. Traces were recorded at the times indicated by the corresponding letters on the main graph (a-f). The dotted line indicates the zero-current level. B, mean stimulatory effects of SNAP (1 nm, 1 μm) and ACh (3 μm) on IK,ACh in GppNHp-dialysed (10 μm) rat atrial myocytes. The amplitude of IK,ACh (at 0 mV) in the presence of agonists was normalised to the amplitude of the end pulse current (at 0 mV) in the presence of GppNHp alone. Bars are the means and vertical lines are the s.e.m. of the number of experiments indicated near the bars. Significant differences from the GppNHp level: *P≤ 0.05; **P≤ 0.01; ***P≤ 0.005.
On average, SNAP (1 nm and 1 μm) significantly enhanced IK,ACh in GppNHp-dialysed (10 μm) atrial myocytes (Fig. 10B). The effect of SNAP was weaker than the effect of ACh (3 μm) tested in the same experiments. However, the potentiating effect of SNAP was not additive with the stimulatory effect of a saturating concentration of non-hydrolysable GTP analogue. Indeed, in four atrial myocytes dialysed with 400 μm GTP-γS, which induced a large and saturating increase in IK,ACh (to 10.4 ± 2.7 pA pF−1 at 0 mV), superfusion with 10 μm DEANO did not further modify IK,ACh (+2.6 ± 0.6 % over GTP-γS-stimulated current at 0 mV). Thus, NO donors can potentiate IK,ACh in the presence of a low, threshold concentration of GTP analogue, and this effect is blunted by a higher, maximal concentration of GTP analogue. These results indicate that the stimulatory effect of NO donors on IK,ACh does not require agonist binding and activation of the muscarinic receptor.
Lack of effect of NO donors on IK,ACh in HEK293 cells
Our results suggest that NO donors potentiate the effect of ACh on IK,ACh by acting at a step located between the muscarinic receptor and the K+ channel. To examine whether NO was acting directly on the muscarinic receptor, on the G protein, or on the muscarinic K+ channel itself, the effects of NO donors were re-examined on IK,ACh reconstituted in HEK293 cells. As shown in Fig. 11A, Ba2+ ions (0.5–1 mm) inhibited a basal background current in these cells, identified previously as IK,ACh (Bunemann & Hosey, 1998). Application of ACh (10 nm) produced an additional current exhibiting kinetics and voltage-dependence indistinguishable from the native IK,ACh in atrial myocytes (data not shown, Xu et al. 1996; Bunemann & Hosey, 1998). On average (Fig. 11B), Ba2+ ions inhibited a significant fraction of the basal current, suggesting that IK,ACh was spontaneously active in the HEK293 cells. Nevertheless, IK,ACh was enhanced by ACh (1 nm to 1 μm) in a dose-dependent manner.
Figure 11. Effects of SNAP on IK,ACh in human embryonic kidney (HEK)293 cells.
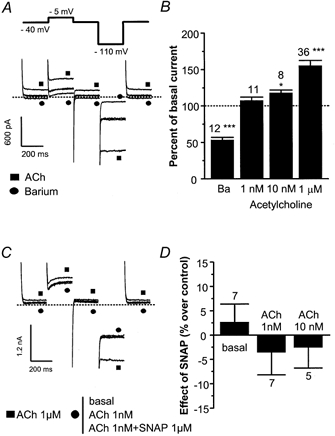
A, representative traces of the effects of Ba2+ ions (1 mm, •) or ACh (1 μm, ▪) in HEK293 cells. Control traces are unlabelled. The dotted line indicates the zero-current level. The voltage steps are indicated on the top of the panel. B, mean effects of Ba2+ ions (1 mm) and ACh (1 nm, 10 nm, 1 μm) on IK,ACh in HEK293 cells. The amplitude of IK,ACh (at −110 mV) in the presence of compounds was normalised to the amplitude of the end pulse current in the absence of compounds. C, representative traces of the effects of ACh alone (1 nm and 1 μm) and of ACh (1 nm) plus SNAP (1 μm) in HEK293 cells. A square symbol (▪) indicates the trace obtained in the presence of ACh 1 μm, and the other traces are labelled with a circle (•). The dotted lines indicate the zero-current level. D, mean effects of SNAP (1 μm) in the absence or in the presence of ACh (1 nm or 10 nm) on IK,ACh in HEK293 cells. The amplitude of IK,ACh (at −110 mV) in the presence of SNAP was normalised to the amplitude of the end pulse current in the corresponding experimental condition. In B and D, bars are the means and vertical lines are the s.e.m. of the number of experiments indicated near the bars. Significant differences from the basal level: *P≤ 0.05; ***P≤ 0.005.
The effect of SNAP (1 μm) was examined in HEK293 cells (Fig. 11C). As expected, SNAP did not modify the basal current in these cells. However, to our surprise, SNAP did not modify the ACh-stimulated (1 nm) current in these cells. On average (Fig. 11D), SNAP had no effect on IK,ACh in the absence or in the presence of ACh (1 or 10 nm). Altogether, our results indicate that a functional muscarinic receptor-G-protein-potassium-channel pathway is necessary, but not sufficient for the potentiating effect of NO donors on IK,ACh to take place.
DISCUSSION
The main finding of our present study is that low concentrations of NO donors can modulate the amplitude of ICa,L, a major trigger of cardiac contraction, and IK,ACh, a major modulator of cardiac beating frequency. The effects of the NO donors involved NO release, but did not require cGMP production by the NO-sensitive guanylyl cyclase. As discussed below, the effect of NO seemed to take place at the G-protein level, although the exact target of NO remains to be determined unambiguously.
NO donors were found to elicit stimulatory effects on ICa,L or to potentiate the effects of β-adrenergic agonists in cardiac myocytes. In several studies, the effects of the NO donors were attributed to cGMP-dependent inhibition of PDE3 (Kirstein et al. 1995; Vandecasteele et al. 1998; Wang et al. 1998, 2000). In another study in newborn rabbit ventricular myocytes, the stimulatory effect of NO donors on ICa,L was attributed to the cGMP-dependent activation of cG-PK (Kumar et al. 1997). Here, we found that in adult rat ventricular myocytes the effects of NO donors on the Iso-stimulated ICa,L, although involving NO release, occurred in a cGMP-independent manner since they were not modified by ODQ, a guanylyl cyclase inhibitor. Furthermore, the involvement of PDE3 was ruled out since (1) cilostamide, a PDE3 inhibitor, but not SNAP, potentiated the effect of the forskolin analogue L858051 on ICa,L and (2) SNAP did not enhance ICa,L in the presence of rolipram, a PDE4 inhibitor, while cilostamide did (Verde et al. 1999). Finally, internal dialysis of rat ventricular myocytes with cGMP or cG-PK was never found to enhance ICa,L (Méry et al. 1991; Sumi & Sperelakis, 1995). Thus, in this study, it was found that the cGMP pathway is not involved in the potentiating effect of low concentrations of NO donors on ICa,L. In contrast, a cGMP-dependent mechanism was shown to be involved in the inhibitory effect of higher concentrations of NO donors on the β-adrenergic stimulation of ICa,L in rat myocytes (Vila-Petroff et al. 1999; Abi-Gerges et al. 2001a). Likewise, we occasionally found an attenuation of the stimulatory effect of low concentrations of SNAP (≤ 1 μm) on the Iso-stimulated ICa,L when the concentration of the NO donor was raised to ≥ 1 μm (Fig. 1 and Fig. 3).
cA-PK inhibition blunted the potentiating effect of NO donors, demonstrating an absolute requirement for cAMP in this effect. Nevertheless, cAMP and cA-PK activation were not sufficient, since NO donors did not modify the effect of L858051, an adenylyl cyclase activator, on ICa,L. The lack of effect of NO donors on the L858051-stimulated ICa,L could be attributed to a superimposition of two opposite effects that match each other perfectly. However, when tested on the Iso-stimulated ICa,L, the potentiating effect of SNAP was always found to be strong enough to overcome the inhibitory effect. Since this potentiating effect was never observed at low SNAP concentrations in the presence of L858051, one can conclude that adenylyl cyclase per se is unlikely to be the target of NO in rat myocytes. Although NO donors were found to enhance adenylyl cyclase activity in a cellular context (Vila-Petroff et al. 1999), inhibitory rather than stimulatory effects on adenylyl cyclase were found in vitro (Hill et al. 2000). Thus, the stimulatory effect of NO donors on Iso-stimulated ICa,L (as well as the GTP-γS-stimulated ICa,L) must take place at an early step in the β-adrenergic signalling cascade (Vila-Petroff et al. 1999).
NO donors also potentiated the muscarinic activation of IK,ACh, which does not involve adenylyl cyclase, but rather the muscarinic receptor-dependent activation of a Gi/o protein, which ultimately opens the K+ channel (Wickman & Clapham, 1995). With the limit of our perfusion system, the effect of either SNAP or DEANO on IK,ACh appeared as fast as the effect of ACh (i.e. maximal within two pulses separated by 5 s). In this short pathway, the effector of the G protein (i.e. the K+ channel) was not the target of NO either, since: (1) NO donors did not modify the basal current (i.e. in the absence of ACh) and (2) NO donors did not enhance IK,ACh in HEK293 cells where the K+ channel had been expressed. All of our data suggest that the mechanisms involved in the potentiating effects of NO on ICa,L and IK,ACh are similar, since: (1) they are inhibited by cPTIO, a NO scavenger, (2) they are unaffected by ODQ, a guanylyl cyclase inhibitor, (3) they take place in the presence of non-saturating concentrations of receptor ligand or non-hydrolysable analogue of GTP, (4) they are blunted by saturating concentrations of receptor ligand and (5) they occur in the same range of concentrations of NO donors. Although similar, the effects of NO donors on ICa,L and IK,ACh are not identical. Indeed, while NO donors potentiated the effects of low ACh concentrations on IK,ACh, DEANO did not strengthen the effects of threshold ACh concentrations on the L858051-stimulated ICa,L (data not shown). In addition, treatment with pertussis toxin, which inactivates Gi/o proteins (Molyvdas & Sperelakis, 1989; Wickman & Clapham, 1995), did not antagonise the potentiating effect of DEANO on ICa,L (data not shown). Thus, the cGMP-independent effect of NO did not involve Gi proteins coupled to adenylyl cyclase. The effects of NO seemed to be restricted to activation of the effectors (adenylyl cyclase and K+ channel) by the G proteins (respectively, Gs and Gi/o proteins).
Taken together, these data support the view that NO modulates ICa,L and IK,ACh at a step located upstream from the G protein effectors. However, NO might not act at the receptor level. Indeed, whereas NO reduces β2-adrenergic receptor palmitoylation, this mechanism leads to inhibition rather than stimulation of the adenylyl cyclase pathway (Adam et al. 1999). In addition, the lack of effect of SNAP on IK,ACh in HEK293 cells also suggests that its potentiating effect (observed in cardiomyocytes) requires more than the muscarinic receptor or the G protein. Preliminary data exclude the involvement of regulators of G protein signalling (RGS) proteins as candidates since co-expression of RGS4 with the other components of the heterologous IK,ACh in HEK293 cells did not induce the potentiating effect of SNAP (data not shown). Furthermore, proteins involved in the agonist-dependent desensitisation of the receptors (i.e. receptor-activated kinases and arrestin) are unlikely candidates, since NO donors did not modify the rate of desensitisation of the ACh-activated IK,ACh. Thus, while the mechanism of action of NO remains to be established, its target protein must be involved intimately in the molecular mechanisms of G-protein activation.
Several findings support the view that the stimulatory effect of NO donors is of physiological significance. First, this effect occurred at very low concentrations of NO donors, since 1 pm to 1 μm SNAP is expected to release as low as 0.01 pm-10 nm NO (Ferrero et al. 1999; Muller-Strahl et al. 2000; Sarkar et al. 2000). According to Malinski and co-workers, maximal local NO levels can raise up to ∼0.5–1 μm in the heart, but decline very sharply away from the site of secretion (Kanai et al. 1997; Brovkovich et al. 1999). Altogether, these data suggest that the potentiating effect of NO plays a role in cardiac function at threshold NO levels, or in cells located far from the site of NO release. In contrast, the potentiating effect might reach saturation during peaks of NO secretion, and the inhibitory effect of NO might then be recruited. Second, NO donors potentiated the effects of low concentrations of Iso in a physiological range of activity of the β-adrenergic receptors. NO donors were also found to potentiate the effect of Iso on Ca2+ transients and cell shortening in rat myocytes (Vila-Petroff et al. 1999). In addition, it has been shown that a basal, constitutive release of NO by the endothelium exerts a tonic positive inotropic effect in the rat heart (Muller-Strahl et al. 2000). So far, these and other studies support the recently held view that NO exerts a biphasic effect on the β-adrenergic pathway, stimulatory at low or threshold levels of β-adrenergic agonists and inhibitory at higher concentrations (Méry et al. 1997; Shah & MacCarthy, 2000; Abi Gerges et al. 2001a; Cotton et al. 2001). However, the potentiation of the muscarinic activation of IK,ACh induced by low NO levels also needs to be taken into account at the whole-heart level. Therefore, the identification of the mechanisms underlying the cGMP-independent potentiation of ICa,L and IK,ACh in cardiac myocytes is of major importance in our understanding of the cardiac effects of NO.
Acknowledgments
We thank Patrick Lechêne for skilful technical assistance, Florence Lefebvre for preparation of the cells, Drs Michel Chesnais, Bernd K. Fleischmann and Renzo Levi for helpful discussions. We are grateful to Dr Xinqiang Han for sharing unpublished results. N. Abi-Gerges was supported by the Fondation pour la Recherche Medicale (F.R.M.) and the Association Française contre les Myopathies (A.F.M.).
REFERENCES
- Abi-Gerges N, Eschenhagen T, Hove-Madsen L, Fischmeister R, Méry PF. Methylene blue is a muscarinic antagonist in cardiac myocytes. Molecular Pharmacology. 1997;52:482–490. doi: 10.1124/mol.52.3.482. [DOI] [PubMed] [Google Scholar]
- Abi-Gerges N, Fischmeister R, Méry PF. G protein-mediated inhibitory effect of a nitric oxide donor on the L-type Ca2+ current in rat ventricular myocytes. Journal of Physiology. 2001a;531:117–130. doi: 10.1111/j.1469-7793.2001.0117j.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abi-Gerges N, Fischmeister R, Méry PF. Regulation of acetylcholine-activated potassium current by nitric oxide in isolated rat atrial myocytes. Biophysical Journal. 2001b;80:628a–629a. [Google Scholar]
- Adam L, Bouvier M, Jones TLZ. Nitric oxide modulates β2-adrenergic receptor palmitoylation and signaling. Journal of Biological Chemistry. 1999;274:26337–26343. doi: 10.1074/jbc.274.37.26337. [DOI] [PubMed] [Google Scholar]
- Akaike T, Yoshida M, Miyamoto Y, Sato K, Kohno M, Sasamoto K, Miyazaki K, Ueda S, Maeda H. Antagonistic action of imidazolineoxyl N-oxides against endothelium-derived relaxing factor/. NO through a radical reaction. Biochemistry. 1993;32:827–832. doi: 10.1021/bi00054a013. [DOI] [PubMed] [Google Scholar]
- Brovkovych V, Stolarczyk E, Oman J, Tomboulian P, Malinski T. Direct electrochemical measurement of nitric oxide in vascular endothelium. Journal Pharmaceutical Biomedical Analysis. 1999;19:135–143. doi: 10.1016/s0731-7085(98)00090-9. [DOI] [PubMed] [Google Scholar]
- Bunemann M, Hosey MM. Regulators of G protein signaling (RGS) proteins constitutively activate G βγ-gated potassium channels. Journal of Biological Chemistry. 1998;273:31186–31190. doi: 10.1074/jbc.273.47.31186. [DOI] [PubMed] [Google Scholar]
- Campbell DL, Stamler JS, Strauss HC. Redox modulation of L-type calcium channels in ferret ventricular myocytes-Dual mechanism regulation by nitric oxide and S-nitrosothiols. Journal of General Physiology. 1996;108:277–293. doi: 10.1085/jgp.108.4.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotton JM, Keamey MT, MacCarthy PA, Grocott-Mason RM, McClean DR, Heymes C, Richardson PJ, Shah AM. Effects of nitric oxide synthase inhibition on basal function and the force-frequency relationship in the normal and failing human heart in vivo. Circulation. 2001;104:2318–2323. doi: 10.1161/hc4401.098515. [DOI] [PubMed] [Google Scholar]
- Ferrero R, Rodriguez-Pascual F, Miras-Portugal MT, Torres M. Comparative effects of several nitric oxide donors on intracellular cyclic GMP levels in bovine chromaffin cells: correlation with nitric oxide production. British Journal of Pharmacology. 1999;127:779–787. doi: 10.1038/sj.bjp.0702607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross WL, Bak MI, Ingwall JS, Arstall MA, Smith TW, Balligand JL, Kelly RA. Nitric oxide inhibits creatine kinase and regulates rat heart contractile reserve. Proceedings of the National Academy of Sciences of the USA. 1996;93:5604–5609. doi: 10.1073/pnas.93.11.5604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han X, Kobzik L, Balligand JL, Kelly RA, Smith TW. Nitric oxide synthase (NOS3)-mediated cholinergic modulation of Ca2+ current in adult rabbit atrioventricular nodal cells. Circulation Research. 1996;78:998–1008. doi: 10.1161/01.res.78.6.998. [DOI] [PubMed] [Google Scholar]
- Han X, Shimoni Y, Giles WR. An obligatory role for nitric oxide in autonomic control of mammalian heart rate. Journal of Physiology. 1994;476:309–314. doi: 10.1113/jphysiol.1994.sp020132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartzell HC, Budnitz D. Differences in effects of forskolin and an analog on calcium currents in cardiac myocytes suggest intracellular and extracellular sites of action. Molecular Pharmacology. 1992;41:880–888. [PubMed] [Google Scholar]
- Hill J, Howlett A, Klein C. Nitric oxide selectively inhibits adenylyl cyclase isoforms 5 and 6. Cellular Signalling. 2000;12:233–237. doi: 10.1016/s0898-6568(99)00082-0. [DOI] [PubMed] [Google Scholar]
- Hu H, Chiamvimonvat N, Yamagishi T, Marban E. Direct inhibition of expressed cardiac L-type Ca2+ channels by S-nitrosothiol nitric oxide donors. Circulation Research. 1997;81:742–752. doi: 10.1161/01.res.81.5.742. [DOI] [PubMed] [Google Scholar]
- Kanai AJ, Mesaros S, Finkel MS, Oddis CV, Birder LA, Malinski T. Beta-adrenergic regulation of constitutive nitric oxide synthase in cardiac myocytes. American Journal of Physiology. 1997;273:C1371–C1377. doi: 10.1152/ajpcell.1997.273.4.C1371. [DOI] [PubMed] [Google Scholar]
- Kan K, Xie ZR, Finkel MS. Norepinephrine-stimulated MAP kinase activity enhances cytokine-induced NO production by rat cardiac myocytes. American Journal of Physiology. 1999;45:H47–H52. doi: 10.1152/ajpheart.1999.276.1.H47. [DOI] [PubMed] [Google Scholar]
- Kirstein M, Rivet-Bastide M, Hatem S, Bénardeau A, Mercadier JJ, Fischmeister R. Nitric oxide regulates the calcium current in isolated human atrial myocytes. Journal of Clinical Investigation. 1995;95:794–802. doi: 10.1172/JCI117729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojda G, Kottenberg K. Regulation of basal myocardial function by NO. Cardiovascular Research. 1999;41:514–523. doi: 10.1016/s0008-6363(98)00314-9. [DOI] [PubMed] [Google Scholar]
- Kojda G, Kottenberg K, Nix P, Schluter KD, Piper HM, Noack E. Low increase in cGMP induced by organic nitrates and nitrovasodilators improves contractile response of rat ventricular myocytes. Circulation Research. 1996;78:91–101. doi: 10.1161/01.res.78.1.91. [DOI] [PubMed] [Google Scholar]
- Kumar R, Namiki T, Joyner RW. Effects of cGMP on L-type calcium current of adult and newborn rabbit ventricular cells. Cardiovascular Research. 1997;33:573–582. doi: 10.1016/s0008-6363(96)00258-1. [DOI] [PubMed] [Google Scholar]
- Levi RC, Alloatti G, Penna C, Gallo MP. Guanylate-cyclase-mediated inhibition of cardiac I-Ca by carbachol and sodium nitroprusside. Pflügers Archiv. 1994;426:419–426. doi: 10.1007/BF00388305. [DOI] [PubMed] [Google Scholar]
- Martynyuk AE, Kane KA, Cobbe SM, Rankin AC. Role of nitric oxide, cyclic GMP and superoxide in inhibition by adenosine of calcium current in rabbit atrioventricular nodal cells. Cardiovascular Research. 1997;34:360–367. doi: 10.1016/s0008-6363(97)00043-6. [DOI] [PubMed] [Google Scholar]
- McVey M, Hill J, Howlett A, Klein C. Adenylyl cyclase, a coincidence detector for nitric oxide. Journal of Biological Chemistry. 1999;274:18887–18892. doi: 10.1074/jbc.274.27.18887. [DOI] [PubMed] [Google Scholar]
- Méry PF, Abi-Gerges N, Vandecasteele G, Jurevicius J, Eschenhagen T, Fischmeister R. Muscarinic regulation of the L-type calcium current in isolated cardiac myocytes. Life Sciences. 1997;60:1113–1120. doi: 10.1016/s0024-3205(97)00055-6. [DOI] [PubMed] [Google Scholar]
- Méry P-F, Lohmann SM, Walter U, Fischmeister R. Ca2+ current is regulated by cyclic GMP-dependent protein kinase in mammalian cardiac myocytes. Proceedings of the National Academy of Sciences of the USA. 1991;88:1197–1201. doi: 10.1073/pnas.88.4.1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Méry P-F, Pavoine C, Belhassen L, Pecker F, Fischmeister R. Nitric oxide regulates cardiac Ca2+ current - involvement of cGMP-inhibited and cGMP-stimulated phosphodiesterases through guanylyl cyclase activation. Journal of Biological Chemistry. 1993;268:26286–26295. [PubMed] [Google Scholar]
- Molyvdas PA, Sperelakis N. Acetylcholine inhibition in rabbit sinoatrial node is prevented by pertussis toxin. Canadian Journal of Physiology and Pharmacology. 1989;67:522–525. doi: 10.1139/y89-083. [DOI] [PubMed] [Google Scholar]
- Muller-Strahl G, Kottenberg K, Zimmer HG, Noack E, Kojda G. Inhibition of nitric oxide synthase augments the positive inotropic effect of nitric oxide donors in the rat heart. Journal of Physiology. 2000;522:311–320. doi: 10.1111/j.1469-7793.2000.00311.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peralta EG, Ashkenazi A, Winslow JW, Smith DH, Ramachandran J, Capon DJ. Distinct primary structures, ligand-binding properties and tissue-specific expression of four human muscarinic acetylcholine receptors. EMBO Journal. 1987;6:3923–3929. doi: 10.1002/j.1460-2075.1987.tb02733.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poteser M, Romanin C, Schreibmayer W, Mayer B, Groschner K. S-nitrosation controls gating and conductance of the alpha 1 subunit of class C L-type Ca2+ channels. Journal of Biological Chemistry. 2001;276:14797–14803. doi: 10.1074/jbc.M008244200. [DOI] [PubMed] [Google Scholar]
- Sarkar D, Vallance P, Amirmansour C, Harding SE. Positive inotropic effect of NO donors in isolated guinea-pig and human cardiomyocytes independent of NO species and cyclic nucleotides. Cardiovascular Research. 2000;48:430–439. doi: 10.1016/s0008-6363(00)00202-9. [DOI] [PubMed] [Google Scholar]
- Sandirasegarane L, Diamond J. The nitric oxide donors, SNAP and DEA/NO, exert a negative inotropic effect in rat cardiomyocytes which is independent of cyclic GMP elevation. Journal of Molecular and Cellular Cardiology. 1999;31:799–808. doi: 10.1006/jmcc.1998.0919. [DOI] [PubMed] [Google Scholar]
- Shah AM, MacCarthy PA. Paracrine and autocrine effects of nitric oxide on myocardial function. Pharmacology and Therapeutics. 2000;86:49–86. doi: 10.1016/s0163-7258(99)00072-8. [DOI] [PubMed] [Google Scholar]
- Stojanovic MO, Ziolo MT, Wahler GM, Wolska BM. Anti-adrenergic effects of nitric oxide donor SIN-1 in rat cardiac myocytes. American Journal of Physiology - Cell Physiology. 2001;281:C342–C349. doi: 10.1152/ajpcell.2001.281.1.C342. [DOI] [PubMed] [Google Scholar]
- Striessnig J. Pharmacology, structure and function of cardiac L-type Ca2+ channels. Cellular Physiology and Biochemistry. 1999;9:242–269. doi: 10.1159/000016320. [DOI] [PubMed] [Google Scholar]
- Sumii K, Sperelakis N. cGMP-dependent protein kinase regulation of the L-type Ca2+ current in rat ventricular myocytes. Circulation Research. 1995;77:803–812. doi: 10.1161/01.res.77.4.803. [DOI] [PubMed] [Google Scholar]
- Vandecasteele G, Eschenhagen T, Fischmeister R. Role of the NO-cGMP pathway in the muscarinic regulation of the L-type Ca2+ current in human atrial myocytes. Journal of Physiology. 1998;506:653–663. doi: 10.1111/j.1469-7793.1998.653bv.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verde I, Vandecasteele G, Lezoualc'h F, Fischmeister R. Characterization of the cyclic nucleotide phosphodiesterase subtypes involved in the regulation of the L-type Ca2+ current in rat ventricular myocytes. British Journal of Pharmacology. 1999;127:65–74. doi: 10.1038/sj.bjp.0702506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vila-Petroff MG, Younes A, Egan J, Lakatta EG, Sollott SJ. Activation of distinct cAMP- and cGMP-dependent pathways by nitric oxide in cardiac myocytes. Circulation Research. 1999;84:1020–1031. doi: 10.1161/01.res.84.9.1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahler GM, Dollinger SJ. Nitric oxide donor SIN-1 inhibits mammalian cardiac calcium current through cGMP-dependent protein kinase. American Journal of Physiology. 1995;37:C45–C54. doi: 10.1152/ajpcell.1995.268.1.C45. [DOI] [PubMed] [Google Scholar]
- Wang YG, Rechenmacher CE, Lipsius SL. Nitric oxide signaling mediates stimulation of L-type Ca2+ current elicited by withdrawal of acetylcholine in cat atrial myocytes. Journal of General Physiology. 1998;111:113–125. doi: 10.1085/jgp.111.1.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YG, Wagner MB, Joyner RW, Kumar R. cGMP-dependent protein kinase mediates stimulation of L-type calcium current by cGMP in rabbit atrial cells. Cardiovascular Research. 2000;48:310–322. doi: 10.1016/s0008-6363(00)00178-4. [DOI] [PubMed] [Google Scholar]
- Wickman K, Clapham DE. Ion channel regulation by G proteins. Physiological Reviews. 1995;75:865–885. doi: 10.1152/physrev.1995.75.4.865. [DOI] [PubMed] [Google Scholar]
- Wolin MS, Hintze TH, Shen W, Mohazzab-H KM, Xie YW. Involvement of reactive oxygen and nitrogen species in signalling mechanisms that control tissue respiration in muscle. Biochemical Society Transactions. 1997;25:934–939. doi: 10.1042/bst0250934. [DOI] [PubMed] [Google Scholar]
- Xu L, Eu JP, Meissner G, Stamler JS. Activation of the cardiac calcium release channel (ryanodine receptor) by poly-S-nitrosylation. Science. 1998;279:234–237. doi: 10.1126/science.279.5348.234. [DOI] [PubMed] [Google Scholar]
- Xu L, Murphy J, Otero AD. Participation of nucleoside-diphosphate kinase in muscarinic K+ channel activation does not involve GTP formation. Journal of Biological Chemistry. 1996;271:21120–21125. doi: 10.1074/jbc.271.35.21120. [DOI] [PubMed] [Google Scholar]