

Abstract
The dynamics of Ca2+ release and Ca2+-activated Cl− currents in two related, but functionally distinct exocrine cells, were studied to gain insight into how the molecular specialization of Ca2+ signalling machinery are utilized to produce different physiological endpoints: in this case, fluid or exocytotic secretion. Digital imaging and patch-clamp methods were used to monitor the temporal and spatial properties of changes in cytosolic Ca2+ concentration ([Ca2+]c) and Cl− currents following the controlled photolytic release of caged-InsP3 or caged-Ca2+. In parotid and pancreatic acinar cells, changes in [Ca2+]c and activation of a Ca2+-activated Cl− current occurred with close temporal coincidence. In parotid, a rapid global Ca2+ signal was invariably induced, even with low-level photolytic release of threshold amounts of InsP3. In pancreas, threshold stimulation generated an apically delimited [Ca2+]c signal, while a stronger stimulus induced a global [Ca2+]c signal which exhibited characteristics of a propagating wave. InsP3 was more effective in parotid, where [Ca2+]c signals initiated with shorter latency and exhibited a faster time-to-peak than in pancreas. The increased potency of InsP3 in parotid probably results from a four-fold higher number of InsP3 receptors as measured by radiolabelled InsP3 binding and western blot analysis. The Ca2+ sensitivity of the Cl− channels in parotid and pancreas was determined from the [Ca2+]-current relationship measured during a dynamic ‘Ca2+ ramp’ produced by the continuous, low-level photolysis of caged-Ca2+. In addition to a greater number of InsP3 receptors, the Cl− current density of parotid acinar cells was more than four-fold greater than that of pancreatic cells. Whereas activation of the current was tightly coupled to increases in Ca2+ in both cell types, local Ca2+ clearance was found to contribute substantially to the deactivation of the current in parotid. These data reveal specializations of common modules of Ca2+-release machinery and subsequent effector activation that are specifically suited to the distinct functional roles of these two related cell types.
The spatial and temporal properties of changes in cytosolic calcium concentration ([Ca2+]c) are known to mediate the activation of multiple, distinct, often compartmentalized Ca2+-dependent signalling cascades which control a wide variety of physiological and pathophysiological events (Clapham, 1995; Hardingham et al. 1997; Crabtree, 1999; Duchen, 2000; Bootman et al. 2001). For example, a rise in [Ca2+]c is known to activate distinct Ca2+-dependent ion channels, which are selective for either K+, Cl− or non-selective for cations. Specifically, Ca2+-activated Cl− currents (IClCa) have been observed in secretory cells of exocrine glands (Randriamampita et al. 1988; Hassoni & Gray, 1994; Arreola et al. 1996; Kidd & Thorn, 2000). In these cells, IClCa are functionally coupled to localized increases in [Ca2+]c generated by Ca2+ release from internal Ca2+ stores. This arrangement is particularly evident in the exocrine cells of salivary glands and pancreas where Ca2+-activated Cl− channels (ClCa) are thought to be localized to the luminal membrane and in close apposition to the subplasmalemmal Ca2+-release machinery (Kasai et al. 1993; Nathanson et al. 1994; Tojyo et al. 1997; Lee et al. 1997b; Takemura et al. 1999). As such, the IClCa have been extensively used as an index of localized Ca2+ dynamics in these cells (Petersen et al. 1991; Park et al. 1999; Giovannucci et al. 2000).
The primary function of mouse parotid acinar cells is to secrete fluid and electrolytes. The activation of IClCa and Ca2+-activated cation currents by Ca2+-mobilizing agonists has been postulated to be a critical component of the fluid secretion process. Cholinergic stimulation in response to autonomic nerve input induces a rise in [Ca2+]c and activates Ca2+-dependent Cl− and K+ conductances (Iwatsuki et al. 1985). Activation of these ionic fluxes, which are thought to be on the apical and basolateral membranes, respectively, results in primary electrolyte secretion into the lumen (Kasai & Augustine, 1990; Zdebik et al. 1997; Park et al. 2001). In contrast, the primary function of pancreatic acinar cells is the synthesis, packaging and exocytotic secretion of digestive enzymes in response to stimulation by the gastrointestinal hormone cholecystokinin or by cholinergic stimulation (Williams et al. 1997), although the pancreatic acinar cells of some species also secrete an isotonic, NaCl-rich fluid. The rate of pancreatic acini fluid secretion, however, is reported to be several orders of magnitude less than that of the salivary gland (Iwatsuki et al. 1985; Petersen, 1992).
Salivary gland and pancreatic acinar cells share a similar morphology and have been used extensively as models of Ca2+ signalling in non-excitable cells. They are thought to possess the same Ca2+-release channels (both inositol trisphosphate receptors (InsP3R) and ryanodine receptors (RyR) (Lee et al. 1997b; Zhang et al. 1999), Ca2+ influx pathways (Xu et al. 1997) and Ca2+ clearance mechanisms (Lee et al. 1997a)). Despite these structural similarities, we postulated that there may be differences in release kinetics and wave properties of Ca2+ signals or in ion channel properties that correlate with their respective primary roles in fluid/electrolyte secretion or more divergent roles in protein secretion and protein synthesis, as well as fluid secretion. Thus, comparing the properties of Ca2+ signals in these cell types should provide general insight into the cellular specializations that affect Ca2+ release and, ultimately, regulate distinct physiological events. To date, however, there has been no direct comparison of InsP3-evoked Ca2+-release events and IClCa dynamics between these related, but distinct, secretory cell models. Studies addressing aspects of Ca2+ signalling in these cell models have differed widely in their experimental conditions and in modes of InsP3R and ClCa activation. The focus of this study was therefore to investigate and directly compare the specific properties of the Ca2+ signals in cells that make use of similar architecture and common molecular modules to achieve rather different end results.
METHODS
Materials
Purified CLSPA-grade collagenase was purchased from Worthington Biochemicals (Lakewood, NJ, USA), and collagenase P was from Boehringer Mannheim (Indianapolis, IN, USA). Oregon Green 488 Bapta-2 (OGB-2), Oregon Green 488 Bapta-5N (OGB-5N), d-myo-inositol 1,4,5-trisphosphate, P4(5)-1-(2-nitrophenyl)-ethylester (caged-InsP3), and o-nitrophenyl EGTA (NP-EGTA) were purchased from Molecular Probes (Eugene, OR, USA). Basal Medium Eagle (BME) and bovine serum albumen (BSA) were purchased from Gibco (Rockville, MD, USA). All other materials were obtained from Sigma Chemical Co. (St Louis, MO, USA).
Preparation of mouse pancreatic and parotid salivary gland acinar cells
All animals were fed ad libitum, and handled in accordance with NIH policy and established protocol with the Division of Laboratory Medicine, University of Rochester, USA.
Mouse pancreatic acini were prepared as described previously (Williams et al. 1978). Briefly, following CO2 asphyxiation and cervical dislocation, pancreata were removed from male NIH-Swiss mice (25 g). The tissue was enzymatically digested with collagenase in BME with 0.1 % BSA and 1 mg ml−1 soybean trypsin inhibitor for 30 min and subsequently dispersed by gentle trituration. Acini were then filtered through 100 μm nylon mesh, centrifuged at 100 g for 2 min, and re-suspended in 1 % BSA in BME.
Single and small clusters of parotid acinar cells were isolated from freshly dissected parotid glands from wild type Swiss Black mice (25 g) by sequential digestion with trypsin and collagenase P, as described previously (Evans et al. 1999). Single isolated acinar cells were re-suspended in BSA-free BME supplemented with 2 mm glutamine and penicillin/streptomycin, and then attached to poly-l-lysine-coated coverslips. Qualitatively similar data regarding the temporal and spatial characteristics of Ca2+ signalling events reported below were obtained from parotid acinar cells prepared from NIH Swiss mice.
Electrophysiology
Ca2+-activated Cl− currents were recorded at 1 kHz using an Axopatch 200A patch-clamp amplifier (Axon Instruments, Foster City, CA, USA), ITC-16 digital interface (Instrutech, Port Washington, NY, USA), and IGOR Pro (Wavemetrics, Lake Oswego, OR, USA) and Pulse Control XOP software (kindly provided by Dr Richard Bookman, University of Miami Medical School, Coral Gables, FL, USA). The standard intracellular recording solution contained (mm): 140 CsCl, 10 Hepes-Tris, 1.5 MgCl2, 3 Mg-ATP, 1 n-hydroxyethylethylenediaminetriacetic acid (HEDTA), 0.075 OGB-2 or OGB-5N, and 0.001–0.1 caged-InsP3, pH 7.3. The intracellular recording solution for the photolytic release of caged-Ca2+ contained (mm): 130 CsCl, 10 Hepes-Tris, 10 NP-EGTA, 5 CaCl2, 2 Mg-ATP, 1.2 MgCl2, pH 7.2. Under whole cell conditions, resting [Ca2+]c and free [Mg2+] were estimated at 175 nm and 1 mm, respectively, using Patcher's Power Tools XOP software (Dr Francisco Mendez, Max Planck Institute for Biophysical Chemistry, Gottingen, Germany). Intervals of 4 min were maintained following patch rupture prior to and between stimuli to allow for sufficient equilibration with the patch pipette solution. The IClCa were isolated from Ca2+-dependent K+ and non-selective currents using an extracellular solution that contained (mm): 140 tetraethylammonium chloride (TEA-Cl), 16 Hepes-Tris, 5.5 d-glucose, 0.56 MgCl2, 1.28 CaCl2, pH 7.3. Cells were voltage clamped at a holding potential of −20 mV (except where noted otherwise) and the Cl− reversal potential was ∼0 mV. All recordings were performed at room temperature. The mean capacitance of single parotid and pancreatic acinar cells was 8.9 ± 0.5 pF (n = 30) and 13.3 ± 0.8 pF (n = 25), respectively.
Flash photolysis and digital imaging
The controlled photolysis of caged compounds was performed using a pulsed Xenon arc lamp (T.I.L.L. Photonics, Eugene, OR, USA) equipped with a fibre optic guide fed to a dual port epifluorescence condenser attached to a Nikon TE-200 fluorescence microscope. A high intensity 0.5 ms flash of UV light (360 ± 7.5 nm) was reflected onto the plane of focus with a DM400 dichroic mirror and Nikon ×40 oil immersion objective with a numerical aperture of 1.3. At the high intensity setting 80 J were discharged, which was estimated to photolyse ∼10 % of caged-InsP3. In other experiments, a low intensity (0.1 J) continuous strobe discharge was used to induce a ramped increase in cytosolic Ca2+ or InsP3. For simultaneous current recording and direct measurement of Δ[Ca2+]c, 75 μm of either OGB-2 or OGB-5N was added to the patch pipette solution and fluorescence changes were monitored using a monochrometer-based illumination system and high-speed CCD camera (T.I.L.L. Photonics). Cells were illuminated at 488 ± 15 nm and fluorescence collected through a 525 ± 25 nm band-pass filter (Chroma Technologies, Brattleboro, VT, USA). Images were acquired at 27 to 31 ms intervals, depending on image size, and displayed as ΔF = 100[(F - Frest)/Frest], where F was the recorded fluorescence and Frest was the average fluorescence of the first 15 sequential frames of the image series. Images (12-bit) were scaled to 4096 grey levels and pseudocoloured. A modified in situ calibration method was used to estimate [Ca2+]c levels reported by non-ratiometric dyes (Neher & Augustine, 1992). Fluorescence changes were converted using the equation:
| (1) |
where ΔFmin and ΔFmax were the relative fluorescence changes obtained during equilibration with ‘zero’ and saturating concentrations of Ca2+, respectively. For estimates of the change in [Ca2+]c, the values for ΔF, ΔFmin and ΔFmax were normalized to the time-dependent component of the relative fluorescence change, which is ΔFmin. Thus, ΔFmin is 1 and the value for ΔFmax, representing the relative fluorescence due to saturating [Ca2+] at 4 min, was about 110 % for OGB-2 and 119 % for OGB-5N of ΔFmin. These values represented a dynamic range of 409 and 738 grey levels (for OGB-2 and OGB-5N respectively) for the 12-bit images captured. Average values for ΔFmin and ΔFmax determined 4 min following attainment of whole cell configuration were 1 and 1.10 ± 0.02 (n = 5) for OGB-2-loaded cells, and 1 and 1.19 ± 0.02 (n = 6) for OGB-5N-loaded cells respectively. Only evoked changes that fell within this range were included in subsequent analysis. The resting [Ca2+]c value for cells loaded with either caged-InsP3 or caged-Ca2+ was estimated to be 100 and 175 nm, respectively. Minimum and maximum ΔF was determined using patch pipette solutions with a set [Ca2+] and a dye concentration of 75 μm. Typically, following membrane patch rupture, ΔF rose exponentially and reached a new steady state. The time-dependent ΔF in the presence of ‘zero’ or saturating Ca2+ was fitted with an exponential line. The exponential line obtained for the ‘zero’ Ca2+ solution was subtracted from the fit for Ca2+-containing solution, and the residual was interpreted to reflect the ΔF due to saturating concentration of Ca2+. The intracellular ΔFmax solution for high- and low-affinity dye calibrations contained (mm): 133 CsCl, 10 Hepes-NaOH, 3 HEDTA, 5 CaCl2, 1 Na-ATP, 1.13 MgCl2, pH 7.3. Free [Ca2+] and [Mg2+] were estimated at 1.34 mm and 734 μm, respectively. For determination of the ΔFmin value, the intracellular solution contained (mm): 128 CsCl, 10 Hepes-NaOH, 3 HEDTA, 5 Bapta, 1 Na-ATP, 4.7 MgCl2, pH 7.3. (Free [Ca2+] and [Mg2+] were estimated at 2 pm and 740 μm, respectively.) The intermediate value pipette solution for calculation of Keff contained (mm): 140 CsCl, 10 Hepes-NaOH, 1 EGTA, 0.9 CaCl2, 2 Na-ATP, 3.2 MgCl2, pH 7.3. (Free [Ca2+] and [Mg2+] were estimated at 642 μm and 1.24 mm, respectively.) The Keff of the OGB-2 dye for pancreatic and parotid acinar cells was estimated by substitution of the intermediate [Ca2+] value into eqn (1) and solving for Keff. The estimated Keff for pancreas and parotid were 229 nm and 549 nm, respectively. Based on the relative deviation of the OGB-2 dye from solution-based calibration values, we estimated that the Keff of the OGB-5N dye for pancreatic and parotid acinar cells was about 8 μm and 19 μm, respectively.
Biochemical analysis of InsP3R content
[3H]InsP3 binding to parotid and pancreatic membranes was assayed as described previously for rat cerebellar membranes (Bredt et al. 1989). Parotid and pancreatic glands were dissected from six Swiss Black mice and homogenized in 20 ml of ice-cold wash buffer for 30 s using a Polytron tissue grinder. The wash buffer contained 50 mm Tris-HCl, 1 mm EDTA, 1 mm 2-mercaptoethanol, 10 μg ml−1 phenylmethylsulphonyl fluoride, 1 μg ml−1 leupeptin, 0.7 μg ml−1 pepstatin A, 1 μg ml−1 antipain, 1 mm benzamidine and 2 μg ml−1 trypsin inhibitor, pH 7.7. The homogenate was then pelleted by centrifugation (15 min at 20 000 g and 4 °C) and re-suspended in fresh ice-cold wash buffer. This wash step was repeated an additional three times. Following the final centrifugation, pellets were re-suspended in 5 ml ice-cold incubation buffer (50 mm Tris-HCl, 1 mm EDTA, 1 mm 2-mercaptoethanol, 10 μg ml−1 PMSF and 2 μg ml−1 trypsin inhibitor, pH 8.8) and a protein assay of this membrane suspension was performed (Bio-Rad, Hercules, CA, USA). Prior to assay, various displacing concentrations of unlabelled InsP3 (0.3 nm-100 nm) were added to the membrane suspensions. To assay binding, 200 μl of the membrane suspension (0.5 mg − 1.2 mg protein) was added to 300 μl incubation buffer containing 1 nm[3H]InsP3 with a specific activity of 22 Ci mmol−1 (NEN-DuPont, Boston, MA, USA) giving 11 nCi per assay. Each assay was incubated for 10 min at 4 °C, vortexed every 2 min and terminated by centrifugation at 10 000 g for 15 min at 4 °C. The supernatant was aspirated and each pellet was re-suspended in 100 μl H2O and mixed with scintillant. Radioactivity was counted using a 6000 LSC liquid scintillation counter (Beckman, Fullerton, CA, USA). Non-specific binding was defined as the residual binding after displacing 1 nm[3H]InsP3 with 10 μm unlabelled InsP3. Scatchard analysis of the data was used to yield estimates of Bmax and KD.
For western blot analysis of InsP3 receptor types, acinar cell samples were prepared as above and then sonicated in 0.5 ml of ice cold lysis buffer (50 mm NaF; 50 mm Tris; 250 mm NaCl; 5 mm EDTA; 1 mm benzamidine; 1 % Triton X-100; 10 mg ml−1 leupeptin and 10 mg ml−1 pepstatin, at pH 7.4). After 30 min on ice the samples were centrifuged for 30 min at 10 000 g and the supernatant was assayed for protein concentration. The proteins were separated by SDS-PAGE and transferred to nitrocellulose (Schleicher & Schuell, Keene, NH, USA) prior to immunoblotting with appropriate InsP3R-specific antibodies (type-I InsP3R: CT1; type-II InsP3R: CT2 (gifts from Dr Richard Wojcikiewicz) and type-III InsP3R monoclonal antibody (PharMingen/ Transduction Labs, San Diego, CA, USA)). Immunoreactivity was visualized using peroxidase-conjugated secondary antibodies followed by detection using the Super Signal detection system (Pierce) exposed on X AR film (Kodak).
Curve-fitting and statistical analysis
Time-matched data segments encompassing the activation and saturation of the rises in [Ca2+]c and IClCa evoked by Ca2+ ramps (see Results) were related and subsequently fitted to the Hill equation:
| (2) |
where KD is the apparent dissociation constant and nH is the Hill coefficient.
Scatchard plots were fitted using linear regression, and statistical analysis of the data was performed using Student's unpaired t test (Graph Pad Software, San Diego, CA, USA). Averaged data are expressed as mean ±s.e.m.
RESULTS
Divergence of InsP3-evoked calcium dynamics in parotid and pancreatic acinar cells
Flash photolysis methods in combination with real-time digital fluorescence imaging (∼33 Hz image acquisition) were used to evoke Ca2+ release and to assess the spatial and temporal properties of Ca2+ signalling in isolated, OGB-2-loaded mouse pancreatic (PAC) or parotid (PAR) acinar cells. Nearly instantaneous and spatially uniform flash photolysis of varying amounts of caged-InsP3 in PAC and PAR cells maintained under identical recording conditions induced a transient [Ca2+]c rise in both cell types. Representative data corresponding to the photolysis of 3 μm caged-InsP3 are shown in Fig. 1. Characteristically, an increase in [Ca2+]c was initiated following a brief latency, as a spatially localized signal in a discrete portion of the apical region of the cell. Focal initiation of the Ca2+ signal has been previously reported to arise from a ‘trigger zone’ (Kasai et al. 1993). The trigger zone may reflect a subpopulation of the most sensitive InsP3 receptors (InsP3R) and is indicative of the functional and morphological (subluminal) localization of InsP3R in these cells (Lee et al. 1997b; Nathanson et al. 1994; Yule et al. 1997).
Figure 1. Effect of flash photolysis of caged-InsP3 in PAC and PAR.
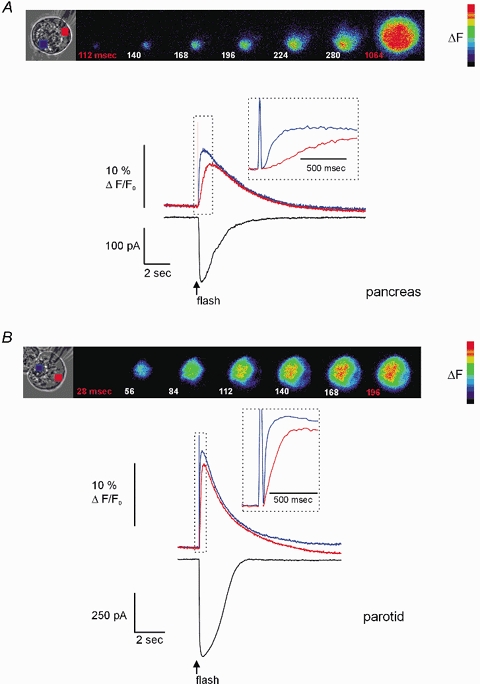
A, PAC, shown in brightfield, was loaded via the patch pipette with 3 μm caged-InsP3 and 75 μm OGB-2. Pseudocoloured digital fluorescence images at the indicated times are shown in series and reflect the Δ[Ca2+]c, evoked by flash photolysis delivered throughout the entire image plane. The Δ[Ca2+]c was initiated ∼112 ms after the flash in a region within the extreme apical pole of the cell. The [Ca2+]c becomes global in ∼1 s after photolysis. Upper traces depict the Δ[Ca2+]c initiated by photolysis. The blue trace was taken from a region of interest (ROI) within the luminal region and the red trace was from a ROI in the basal pole of the cell. Inset shows the rising phase of the response indicated by the hashed box on an expanded time scale to illustrate the delay between the initiation of the apical and basal signal. The lower black trace shows the IClCa activated by Δ[Ca2+]c. Note the high temporal coincidence with the apical [Ca2+]c signal. B, PAR shown in brightfield was subjected to an identical experimental paradigm. The pseudocoloured images show that a [Ca2+]c signal is again initiated in the extreme apical portion of the cell after a short latency (∼1 image frame) and in contrast to PAC within 200 ms becomes a global signal. The traces illustrate the apical [Ca2+]c (blue trace), basal [Ca2+]c (red trace) and corresponding IClCa (black trace) evoked by flash photolysis. Cells were held at −20 mV and external NaCl and KCl were replaced with TEA-Cl.
A comparison of the temporal characteristics of the Ca2+ signal between PAC and PAR, however, revealed significant divergence in both the latency of activation and in the progression of the Ca2+ rise from a localized to global signal. For example, as shown in the representative fluorescence images in Fig. 1A, the increase in [Ca2+]c in PAC began ∼112 ms following application of the UV flash and required nearly 1 s to result in a global signal. The kinetics of the ΔF evoked in both apical and basal regions of PAC are shown in the corresponding inset (Fig. 1A). In contrast, the Δ[Ca2+]c in PAR initiated with a substantially shorter latency (28–58 ms) and rapidly produced a global signal in < 200 ms. Accordingly, the corresponding ΔF selected from apical and basal regions peaked with relatively close temporal coincidence (inset Fig. 1B). In addition, the amplitude of ΔF was generally of greater magnitude in PAR compared with PAC (see below). The average latency in response to the photolysis of 3 μm caged-InsP3 for PAR and PAC was 40 ± 4 ms and 150 ± 30 ms, respectively (n = 17 and 8, P≤ 0.001). Furthermore, activation of Ca2+ release by flash photolysis of 3 μm caged-InsP3 in PAR consistently evoked a global rise in [Ca2+]c. In contrast, an identical photolytic stimulus produced either a global or apically localized response in PAC. For example, in 20 % of PAC, flash photolysis produced a Ca2+ signal that remained largely limited to the apical region of the cell. These data indicate a clear difference between PAR and PAC in both the activation and propagation of the InsP3-evoked Ca2+ signal. Photolysis of InsP3 in PAC and PAR also evoked a robust IClCa spike that correlated with the apical Ca2+ signal (Fig. 1, black traces). These IClCa were similar in amplitude to those evoked during hormonal- or neurotransmitter-induced Ca2+ oscillations (Hassoni & Gray, 1994; Ito et al. 1997).
The relative difference between PAR and PAC in the amplitude and speed of propagation of the Ca2+ signal evoked by a relatively low concentration of caged-InsP3 prompted us to investigate whether this indicated a divergence in the functional properties of the Ca2+-release machinery or in InsP3 sensitivity. We therefore determined the concentration dependence of the (i) amplitude, (ii) latency, and (iii) propagation of the Ca2+ signal evoked by the photolysis of varying concentrations of caged-InsP3.
Amplitude of the Δ[Ca2+]c
The ability of higher concentrations of caged-InsP3 to substantially decrease the latency of initiation (see below) suggested that PAR and PAC differ in their sensitivity to InsP3. To test this hypothesis, peak apical and basal Δ[Ca2+]c were determined using both high-affinity and low-affinity dyes. The low-affinity dye OGB-5N was used to track the robust Δ[Ca2+]c induced by the photolysis of 10–100 μm caged-InsP3.
Following attainment of whole-cell configuration the patch pipette solution containing 1, 3, 10, 30 or 100 μm caged-InsP3 was allowed to equilibrate for 4 min prior to photorelease. Photolysis of 1–10 μm caged-InsP3 induced a transient Δ[Ca2+]c that was steeply dependent on the concentration of caged-InsP3. Figure 2 shows the relationship for peak apical Δ[Ca2+]cvs. caged [InsP3] in PAC and PAR. The potency with which InsP3 evoked a [Ca2+]c response was significantly greater in PAR than PAC with an estimated KD for caged-InsP3 of 3 μm and 7.8 μm, respectively. For example, photolysis of 3 μm caged-InsP3 evoked an apical Δ[Ca2+]c nearly five-fold that of PAC (1523 nmvs. 357 nm; P≤0.0001).
Figure 2. Concentration vs. response relationships for the Δ[Ca2+]c evoked by flash photolysis of caged-InsP3 in PAC and PAR.
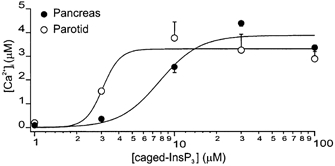
Peak Δ[Ca2+]c (in μm) evoked by photolysis of different concentrations of caged-InsP3 plotted from ROIs at the apical zone of PAC (•) and PAR (○). Flash photolysis induced changes in apical [Ca2+]c with peak amplitudes of 198 ± 24, 1523 ± 28, 3765 ± 674, 3251 ± 677 and 2887 ± 576 nm (PAR, 4 ≤n≥ 17) and 96 ± 2, 357 ± 29, 2538 ± 233, 4383 ± 126 and 3360 ± 171 nm (PAC, 4 ≤n≥ 8). Data for 1 and 3 μm caged-InsP3 were obtained using the high-affinity dye OGB-2; and for 10–100 μm caged-InsP3, the low-affinity dye OGB-5N was used. Lines represent Hill plot fits to the data.
Latency of the Δ[Ca2+]c
As shown in Fig. 3A, the latency of ΔF in PAR exhibited a dramatic decrease between 1 and 3 μm caged-InsP3. A further increase in [caged-InsP3] induced only a minor reduction in the latency suggesting that in PAR initiation of the [Ca2+]c signal was maximally activated by > 3 μm caged-InsP3. (The absolute latency values at the highest concentrations tested should be considered estimates given the limitation of the imaging sampling rate.) In contrast to PAR, the mean latencies of the evoked ΔF in PAC were significantly longer at all but the highest concentration of caged-InsP3, and decreased by nearly ten-fold over the concentration range.
Figure 3. Characteristics of [Ca2+]c signals evoked by flash photolysis of caged-InsP3 in PAC and PAR.
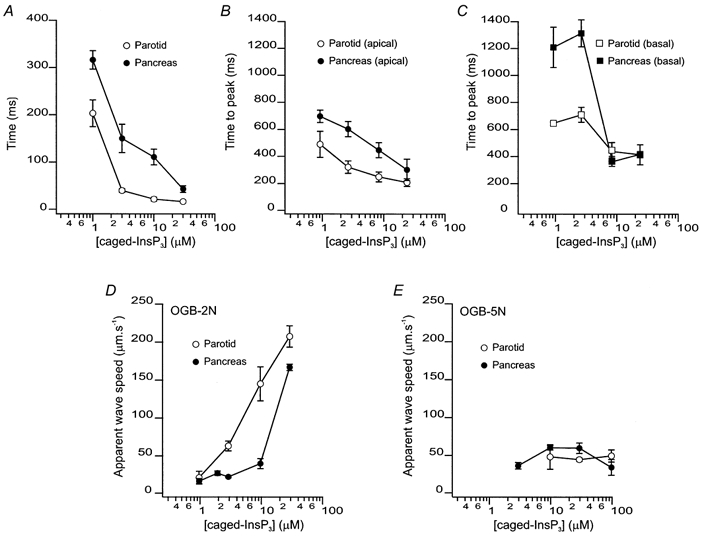
A, latency before a detectable Δ[Ca2+]c following photolysis of caged-InsP3 in PAC (•) and PAR (○). The mean latencies for 1, 3, 10 and 30 μm caged-InsP3 in PAC were 316 ± 20, 150 ± 30, 111 ± 17 and 43 ± 7 ms (4 ≤n≥ 10). The mean latencies for 1, 3, 10 and 30 μm caged-InsP3 in PAR were 203 ± 28, 40 ± 4, 22 ± 4 and 17 ± 3 ms (5 ≤n≥ 17). The latency was significantly shorter in PAR at all concentrations of caged-InsP3. B, time-to-peak Δ[Ca2+]c following photolysis determined from ROIs in the apical zone of PAC (•) and PAR (○). The time-to-peak was significantly shorter in PAR when compared with PAC. C, corresponding data illustrating time-to-peak determined from ROIs from the basal region of PAC (•) and PAR (□). Data illustrate that at all concentrations of caged-InsP3 utilized a rapid global wave is initiated in PAR, whilst only higher concentrations exhibit this property in PAC. D, apparent wave speeds measured with OGB-2 upon photolysis of caged-InsP3 on PAC (•) and PAR (○). Apparent wave speed increased in a concentration-dependent fashion in PAR, but remains relatively constant in PAC until high concentrations of caged-InsP3 are photolysed. E, apparent wave speeds measured with OGB-5N upon photolysis of caged-InsP3 on PAC (•) and PAR (○). Wave speed remains constant in both PAR and PAC at all [caged-InsP3] tested.
Apparent wave propagation
Although the global spread of a [Ca2+]c signal is often thought to represent the propagation of a [Ca2+]c wave through sequential activation of neighbouring release sites by CICR, [Ca2+]c signals may also simply reflect the simultaneous activation of individual or clusters of InsP3R, acting as autonomous release sites throughout the cytoplasm. In this scenario, it is likely that, in an area of a cell where release sites are at low density, Ca2+ release induced by InsP3 would be slower to rise to detectable levels because release is not efficiently entrained by CICR. To investigate this possibility we quantified the time-to-peak of the evoked fluorescence rise in the apical and basal regions as a function of concentration. The regional time-to-peak value was determined by designating the first post-flash frame where there was ΔF≥ 0.5 % above the regional resting value as time-zero for activation of the signal and measuring the time interval of the [Ca2+]c signal to reach peak amplitude. As shown in Fig. 3B and C, the time-to-peak in both the apical and basal regions of PAC were significantly slower than in PAR at [caged-InsP3] below 10 μm. However, at higher concentrations, the respective apical and basal rise times converged and were not significantly different between cell types. Additionally, the apical and basal time-to-peak values within a particular cell type also converged at higher concentrations. These observations are consistent with the idea that the simultaneous activation of individual or clusters of InsP3R acting as autonomous release sites contributes to the ‘apparent wave’ observed in PAR over a broad range of [caged-InsP3] and in PAC at concentrations above 10 μm.
To investigate this hypothesis further, we quantified the spread of the ΔF signal, using OGB-2, as a function of [caged-InsP3] for PAR and PAC by selecting a threshold value and converting the changes in fluorescence above and below this threshold to a binary signal. Using this method, we could follow the advancing edge of the apparent [Ca2+]c wave as it propagated outward from the trigger zone. As shown in Fig. 3D, the apparent wave speed in PAR increased from 21 ± 9 μm s−1 to 203 ± 14 μm s−1 when the [caged-InsP3] was raised from 1 to 30 μm. In contrast, PAC wave speed remained relatively constant at ∼26 μm s−1 between 1 and 10 μm caged-InsP3. However, at the highest concentration of caged-InsP3, apparent wave speed was markedly enhanced and approached that of PAR. Interestingly, using the low-affinity dye OGB-5N and performing similar analysis wave speed remained constant at ∼50 μm s−1 in both PAR and PAC through the caged-InsP3 concentration range tested (Fig. 3E).
The idea that autonomous activation of Ca2+ release channels contributes significantly to signalling in PAR is further supported by the following set of experiments using continuous low-level photolytic stimulation to produce ‘threshold’ receptor activation (Giovannucci et al. 2000). In PAC, continuous low-level photolysis of 10 μm caged-InsP3 induced multiple, localized Ca2+-release events. As shown in the blue trace in Fig. 4A, these events were largely confined to the apical region of the cell, arose from discrete sites, and generally correlated with IClCa spikes (presumably increases in IClCA activity in the absence of any apparent changes in [Ca2+]c results from [Ca2+]c changes occurring above/below the focal plane). These discrete Ca2+-release events appeared as local, apically limited [Ca2+]c‘wavelets’ similar to those previously described (Kidd et al. 1999). These events were interpreted to reflect the activation of a few or small clusters of InsP3R and ranged in amplitude from 10 to 55 nm above resting levels. However, these estimates of [Ca2+]c signal amplitude may be significantly biased by the focal plane at which the release event occurred. Current spikes (black trace) ranged from −7 to −50 pA and, based on previous estimates of a single ClCa conductance of ∼2 pS (Zdebik et al. 1997), corresponded to the activation of ∼175–1250 ClCa. In contrast, continuous, low-level photolysis of 10 μm caged-InsP3 in PAR following apical initiation invariably produced a global rise in [Ca2+]c (Fig. 4B). The amplitude of this increase was 350 nm above resting value and activated a steady-state current with a plateau amplitude of −20 pA. In some PAR, additional current spikes corresponding to apical release events could be resolved on top of the plateau current.
Figure 4. Threshold release of InsP3 in PAC and PAR.
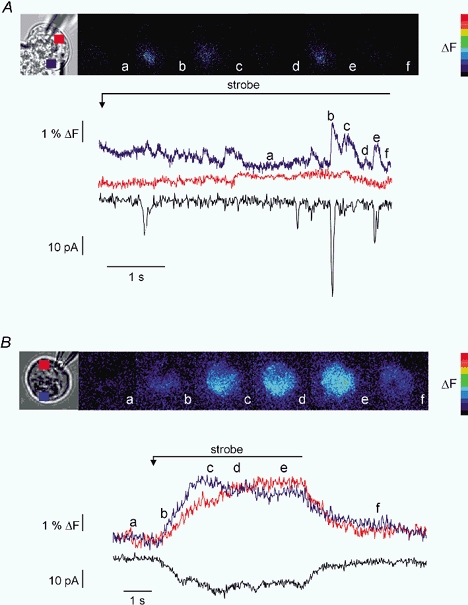
Continuous, low-level photolysis was used to uncage threshold-activating levels of InsP3 in PAC and PAR. A, PAC was loaded with 10 μm caged-InsP3 and 75 μm OGB-2. The pseudocolour panel shows images taken at the times indicated by the corresponding letters on the trace and images. Low-level photolysis evokes highly localized, transient increases in [Ca2+]c reminiscent of ‘puffs’ or ‘blips’. These increases in [Ca2+]c only occur in the apical region of the cell and the larger increases are generally coincident with activation of IClCA (increases in IClCA activity in the absence of [Ca2+]c may be the result of [Ca2+]c changes above/below the focal plane). Traces show the temporal data for a ROI in the apical region (blue trace) the basal region (red trace), which shows no increase, and corresponding IClCa (black trace). B, PAR shown in brightfield was subjected to an identical experimental paradigm. The images demonstrate that although the signal is initiated in the apical zone it becomes global rapidly. The traces illustrate the apical region (blue), basal region (red) and IClCa (black) resulting from the photolysis.
Biochemical analysis of InsP3R content in PAR and PAC
Having demonstrated clear differences in the kinetics of Ca2+ signalling between PAR and PAC, we extended our investigation to determine whether differences in InsP3R density may in part account for these differences. To quantify the relative density of Ca2+ release channels between PAR and PAC we performed experiments using radioligand binding in Swiss Black mice. This was performed primarily to quantify total binding capacity (Bmax), as an indirect measure of InsP3R content. However, estimates of apparent binding affinity for InsP3 to its receptors (KD) were also obtained, which may relate to differences in Ca2+ signalling dynamics between PAR and PAC. Within the concentration range of 0.3–30 nm InsP3, Scatchard plots yielded estimates of Bmax and apparent KD which represented the densities and functional average affinities of the three detected InsP3R types. Representative Scatchard plots of [3H]InsP3 binding to PAR and PAC membranes shown in Fig. 5A revealed that the PAR expressed nearly four-fold more InsP3R (Bmax = 1.08 ± 0.13 pmol mg−1; n = 4) than PAC (Bmax = 0.29 ± 0.06 pmol mg−1; n = 4). However the apparent affinity for InsP3 binding between PAR and PAC was statistically indistinguishable (KD = 6.3 ± 0.6 nm, n = 6; and KD = 6.1 ± 1 nm; n = 4, respectively).
Figure 5. Biochemical comparison of InsP3R protein in PAR and PAC.
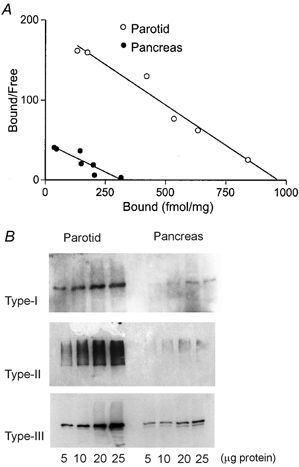
A, Scatchard plot of [3H]InsP3 equilibrium binding to PAR (○) and PAC (•) membranes. Data shown are representative of four experiments performed in duplicate on both tissues in parallel from up to six mice per experiment. These data yield estimates of Bmax (1.08 pmol mg−1 for PAR; 0.29 pmol mg−1 for PAC) and KD (6.3 nm for PAR; 6.1 nm for PAC). B, western blots of InsP3R protein in PAR and PAC. Increasing amounts of protein from corresponding tissue homogenates were loaded on the same gel and probed with polyclonal antisera raised against the C-terminal domain of the type-I (CT1) or the type-II (CT2) InsP3R, or type-III-specific monoclonal antibody. These data demonstrate that PAR expresses relatively more of each InsP3R type compared with PAC.
To determine if one or all InsP3R types were enriched in PAR, we performed western blot analysis using type-specific antisera. Representative western blots of InsP3R are shown in Fig. 5B. These data reveal that PAR express significantly higher levels of all three subtypes of InsP3R compared with PAC. Functional data showing decreased latency together with the global increase in [Ca2+]c upon low-level photolytic stimuli are consistent with increased numbers of InsP3R localized in both the trigger zone and on sites throughout the cytoplasm in PAR.
Kinetics of IClCa activation
The latency and kinetics of IClCa matched that of the rise in the [Ca2+]c signal, suggesting that this was the rate-limiting process for activation. Using the photolysis of caged-Ca2+, we tested this by directly and uniformly raising [Ca2+]c to saturating levels (> 1 μm) nearly instantaneously to activate the IClCa. Figure 6A compares representative examples of the activation of PAC IClCa evoked by the flash photolysis of caged-Ca2+ (traces 1 and 2) or caged-InsP3 (traces 3 in A and B). Currents were normalized and the InsP3-evoked signal trace is offset −100 ms for comparison. The IClCa evoked by the flash photolysis of caged-Ca2+ activated within 1 ms and had a much faster rate of rise than the InsP3-evoked signal. The time course of activation of the IClCa evoked by the photolysis of caged-Ca2+ was fitted by a single exponential line with a time constant (τ) of ∼15 ms and probably reflected the kinetics of Ca2+-dependent channel gating by saturating Ca2+ levels. For example, direct elevation of [Ca2+]c in cells held at the Cl− reversal potential (∼0 mV) produced little or no detectable current (trace 1). However, when cells were step-hyperpolarized to −30 mV 100 ms post-flash, a nearly instantaneous (< 1 ms) current was apparent (trace 1), indicating that channels were already maximally activated (kinetically) at 0 mV. The kinetics of IClCa evoked by elevating [Ca2+]c by flash photolysis of caged-Ca2+ were not significantly different between PAC and PAR (τPAC = 15.3 ± 3.8 ms vs.τPAR = 10.53 ± 2.4 ms; n = 4, P = 0.33). Thus, in both cell types, IClCa activation was primarily dependent on InsP3 receptor activation and the rate of rise of the [Ca2+]c signal (Fig. 6A and B, traces 3). Thus, rather than a fundamental difference in the molecular attributes of the ClCa, divergence in the mechanism of Ca2+ signalling is likely to play a major role in according functional specialization.
Figure 6. Kinetics of Ca2+-activated Cl- current activation evoked by flash photolytic release of saturating levels of Ca2+ or by the flash photolysis of 10 μm caged-InsP3.
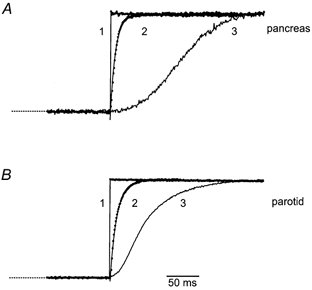
For comparison, the representative currents from patch-clamped PAC (A) or PAR (B) were normalized and aligned. Trace 1 shows that there was no substantial current evoked by the flash photolysis of caged-Ca2+ when cells were held at 0 mV (the reversal potential for Cl−), but current was nearly instantaneously and maximally activated when Vm was stepped to −30 mV. Trace 2 shows examples of IClCa activation (symbols) evoked by flash photolysis of caged-Ca2+ in cells held at −30 mV. The continuous lines represent single exponential fits to the data. Time constants determined from the fits were 15.3 ms for PAC and 10.5 ms for PAR. Currents produced by the flash photolysis of 3 μm caged-InsP3 in PAC (trace 3 in A) and PAR (trace 3 in B) activated following a delay and rose with complex kinetics. InsP3-induced currents were offset −100 ms and −25 ms for PAC and PAR, respectively, to align the rises in current with those evoked by the photolytic release of caged-Ca2+.
Probing the Ca2+-dependence of the activation and inactivation of IClCa with caged-Ca2+
Even though ClCa activation is commonly used as an index of apical, submembraneous Ca2+ release, there are few quantitiative estimates of the Ca2+ required for activation of the IClCa under physiologically relevant conditions. For example, most estimates of the Ca2+-dependence of the IClCa have relied on steady-state Ca2+ activation. Thus, to assess the Ca2+-dependence of ClCa under non-steady-state conditions of [Ca2+]c, we developed a [Ca2+]c-ramp method.
PAR and PAC were loaded via the patch pipette with caged-Ca2+ (10 mm NP-EGTA, 50 % bound with Ca2+) and 75 μm OGB-2. Caged-Ca2+ and a continuous low-level photolytic stimulus was used to ramp [Ca2+]c to micromolar levels over tens of seconds. The IClCa were monitored simultaneously and a relationship describing the Ca2+-dependence of the IClCa activation was constructed (Fig. 7). Representative time-matched rises in [Ca2+]c and whole-cell IClCa from PAC (A) and PAR (B) are shown. Continuous photolysis induced changes in [Ca2+]c with similar kinetics and amplitude between PAR and PAC. A comparison of the activation of the IClCa with the changes in [Ca2+]c revealed that the IClCa lagged behind the rise in [Ca2+]c, reflecting a threshold value for activation. On average, this threshold value was 41 ± 28 nm above resting level for PAR (n = 6) and 48 ± 13 nm for PAC (n = 6), where threshold for activation was defined as the Δ[Ca2+]c where there was a deviation from the baseline equal to 0.01 % peak amplitude of the IClCa. In addition, the IClCa were found to reach a maximum value prior to the peak [Ca2+]c response, indicating saturation of the IClCa. This is consistent with the data obtained using high intensity flash photolysis, which increases [Ca2+]c to levels > 1 μm but did not result in larger currents. Concentration-response (Δ[Ca2+]c-I) relationships for individual cells (Fig. 7C) were obtained by relating the instantaneous, time-matched Δ[Ca2+]c and the rise in IClCa during the strobe stimulus (Fig. 7A and B, shaded regions). For the Δ[Ca2+]c-I relationships, currents were normalized to whole cell capacitance and inverted. The Δ[Ca2+]c represents the change from resting level and not the absolute [Ca2+]c value. These relationships were fitted with a single concentration-response function (continuous lines). On average, the Imax, KD and Hill coefficients were estimated to be 145 ± 35 pA pF−1, 352 ± 106 nm and 4.3 ± 0.3 for PAR (n = 6); and 33 ± 5 pA pF−1, 506 ± 57 nm and 3.7 ± 0.3 for PAC (n = 6), respectively. This indicated that the IClCa of both PAR and PAC are activated by Ca2+ in a higher-order fashion and would be maximally activated by InsP3-evoked Ca2+ release. Similarities between KD and Hill coefficient suggest both functional and perhaps molecular conservation of the ClCa of PAR and PAC. Peak current amplitudes were about four-fold greater in PAR compared with PAC (145 ± 35 pA pF−1vs. 33 ± 5 pA pF−1, P < 0.05). The difference in current densities between these two cell types may reflect a larger number of channels in PAR.
Figure 7. Determination of the Ca2+ sensitivity of ClCa during dynamic changes in [Ca2+]c using the [Ca2+]c-ramp method.
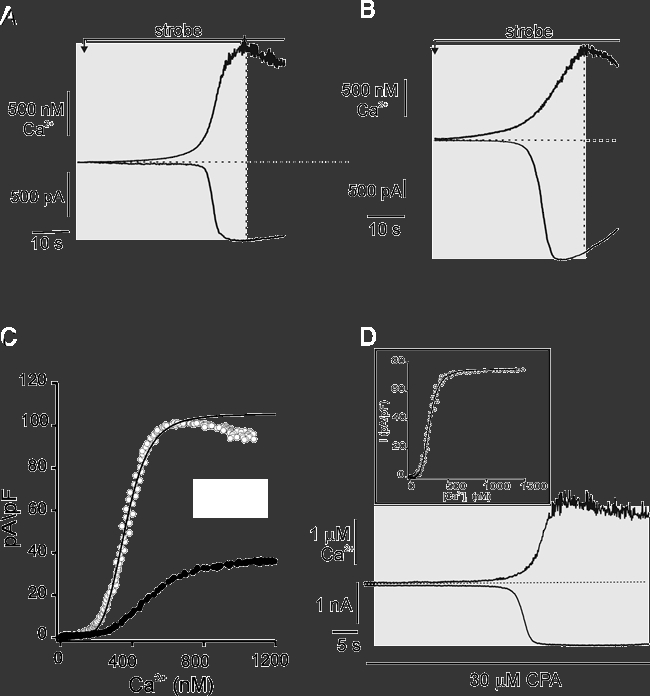
Representative changes in [Ca2+]c (upper traces) and corresponding IClCa (lower traces) in PAC (A) and PAR (B) induced by the continuous, low-level strobe photolysis of caged-Ca2+. Cells were held at −20 mV. C, plots of the instantaneous, single-cell Δ[Ca2+]c-I responses for PAC (filled symbols) and PAR (open symbols) from A and B.[Ca2+]c and IClCa traces within the shaded regions of cells in A and B were time-matched, and values at 100 ms intervals were related. Currents were inverted and normalized to whole-cell capacitance. Continuous lines are fits to the Hill equation. The KD values for PAC and PAR were estimated to be 500 nm and 360 nm, respectively. D, changes in [Ca2+]c and current in PAR was induced by Ca2+ ramp as in A and B. Treatment with 30 μm cyclopiazonic acid (CPA) largely abolished the apparent inactivation of the IClCa. Cells were held at −20 mV. Inset shows the corresponding time-matched, instantaneous concentration-response relationship for the CPA-treated cell in A. The KD value was estimated under these conditions to be 204 nm.
PAR and PAC exhibited subtle differences in the time courses of current relaxation. For example, in nearly all PAR tested there was found to be substantial divergence in the Δ[Ca2+]c-I relationship following peak activation of the IClCa. Despite a continuing rise in [Ca2+]c, the IClCa began to return towards pre-stimulus values. This uncoupling of the Ca2+-dependence of the IClCa is reminiscent of the ‘hysteresis’ in the I-Ca2+ relationship that has been noted previously for agonist-induced current dynamics (Kidd & Thorn, 2000; Kotlikoff & Wang, 1998; see also Fig. 1 of the current study). The mechanism underlying the apparent inactivation of IClCa is not known, although it has been suggested that this results from intrinsic Ca2+- or time-dependent inactivation of ClCa (Kidd & Thorn, 2000). An alternative explanation for this phenomenon is that the decrease in the current reflects local clearance of cytosolic Ca2+ in the vicinity of the ClCa that is not detectable with the spatial resolution of the imaging method used here. Work from our laboratory and others has indicated that SERCA pumps play an important role in Ca2+ clearance following activation of global changes in [Ca2+]c in PAC (Mogami et al. 1998; Straub et al. 2000).
To determine if local Ca2+ clearance by SERCA pumps contributed to the apparent inactivation of the IClCa in PAR, we performed [Ca2+]c ramps on PAR that were treated with the SERCA pump inhibitor cyclopiazonic acid (CPA). As shown in Fig. 7B, the IClCa induced by [Ca2+]c ramp under control conditions exhibited a typical Ca2+-dependent activation and the apparent inactivation during a continuing rise in [Ca2+]c. To deplete internal stores, 30 μm CPA was applied in standard recording saline with no added Ca2+ for approximately 10 min prior to [Ca2+]c ramp. Previous work has demonstrated that agonist- and second messenger-evoked increases in [Ca2+]c are largely unaffected by this Ca2+-free treatment (Straub et al. 2000). Initial experiments using Fura-2-loaded cells demonstrated that CPA treatment induced a transient rise in both IClCa and [Ca2+]c indicating SERCA pump inhibition and depletion of intracellular Ca2+ stores (data not shown). Following return of the [Ca2+]c and current to resting levels, and in the continuous presence of CPA, a [Ca2+]c ramp was applied. As shown by the representative traces in Fig. 7D, the induced current was maximally activated during the rising phase of the [Ca2+]c and, in contrast to control, showed no apparent inactivation. On average, there was a 1.2 ± 0.9 % decrease in current after reaching peak in CPA-treated PAR compared with 21.6 ± 6.2 % decrease in control cells during a 30 s interval following attainment of peak current (n = 4 and 5, respectively; P = 0.008).
These data indicate that local clearance by SERCA pumps is the dominant mechanism underlying the apparent inactivation of IClCa under these conditions. This suggested that local clearance may have occurred concurrently with [Ca2+]c ramp estimates of IClCa activation. Consistent with this, the average KD and Hill coefficient for CPA-treated cells was 202 ± 6 nm and 5.9 ± 2.5, respectively (n = 3), representing an apparent leftward shift compared with KD values obtained under control conditions (inset in Fig. 7D). These values, however, failed to achieve statistical significance, most likely reflecting the limitations in determining absolute [Ca2+]c values using the current methods.
This apparent inactivation of the IClCa was less prevalent in PAC (an average reduction in current of 9.8 ± 2.5 %, 30 s after reaching peak, n = 6). Similar to PAR, CPA treatment was found to abolish this apparent inactivation (1.5 ± 1.5 % decrease in peak current value, n = 3). The average KD and Hill coefficient for CPA-treated PAC was 447 ± 170 nm and 3.5 ± 0.4, respectively (n = 5). These data are consistent with the idea that IClCa dynamics are directly mediated by changes in [Ca2+]c and that, rather than inactivation, this decrease in current reflects deactivation of the channel by local removal of Ca2+.
DISCUSSION
InsP3R density and the properties of InsP3-evoked Ca2+ signals
In both PAR and PAC, oscillations in [Ca2+]c, induced by hormonal or neural input, activate to various extents fluid and/or protein secretion. Previous work has demonstrated that, in terms of Ca2+ release, PAR and PAC share many molecular components. For example, biochemical and functional studies from a variety of laboratories have shown that both PAR and PAC contain all three types of InsP3R, with type-II the most abundant receptor in PAR (Zhang et al. 1999) and type-II/III predominating in PAC (Wojcikiewicz & He, 1995). Similarly, in both cell types, immunocytochemical methods have demonstrated that the vast majority of all the InsP3R types are localized with a subluminal actin web at the extreme apical portion of the cell (Lee et al. 1997b; Nathanson et al. 1994; Yule et al. 1997). The functional consequence of this arrangement is that in both cell types, Ca2+ release has been shown to initiate at a discrete subset of these receptors - or ‘trigger zone’ - and propagate, probably via CICR, throughout the cell as a global rise in [Ca2+]c (Kasai et al. 1993). The propagation of this Ca2+ signal may be aided by the concurrent or subsequent activation of ryanodine receptors. Indeed, recent studies have provided molecular, biochemical and functional evidence that PAR and PAC contain RyR receptors (Zhang et al. 1997; Fitzsimmons et al. 2000; Straub et al. 2000).
In the present study we monitored various parameters of Ca2+ signalling in an effort to address how differences in the regulation of Ca2+ release in two ostensibly similar secretory cell types might impact the control of secretion. The use of flash photolysis and time-resolved fluorescence imaging methods to produce a controlled pulse of InsP3 and monitor the evoked Ca2+ signal allowed us to directly compare the time courses of [Ca2+]c change in PAR and PAC. Given the similarities in the Ca2+-release machinery of PAR and PAC, there was surprising divergence in both the kinetics and the spatial aspects of their Ca2+ signals.
For example, the latency between the photolysis of 3 μm caged-InsP3 and the ΔF was about three-fold shorter in duration in PAR compared with PAC, and once initiated rose at a rate that was two-fold faster. Moreover, in PAR, low concentrations of InsP3 produced larger amplitude [Ca2+]c signals than PAC and, unfailingly, induced a global rise in [Ca2+]c. Although photorelease of InsP3 had similar efficacy, potency was enhanced in PAR more than two-fold, as evidenced by a leftward shift in the concentration response.
There was also a distinct difference in the propagation velocity of the global apparent waves generated in PAC and PAR. This difference, observed when the Ca2+ signal was evoked by the photolysis of caged-InsP3 at concentrations less than 10 μm, was manifested by only a minor dependence of apparent wave speed on concentration in PAC and a steep dependence of apparent wave speed on concentration in PAR. Moreover, PAC exhibited a biphasic dependence of apparent wave speed on the concentration of caged-InsP3, such that at higher concentrations, the characteristics of Ca2+ signal propagation in PAC resembled those of PAR. The steep dependence of apparent wave speed on concentration in PAR, and in PAC at high [caged-InsP3], was greater than that expected given experimental and theoretical evidence that increases in [InsP3] can promote either little or modest increases in wave speed by decreasing InsP3R excitation threshold (Rooney et al. 1990; Bugrim et al. 1997; Marchant et al. 1999). A previous study has also addressed the InsP3 concentration-dependence of these parameters using the low-affinity dye BTC (Ito et al. 1999). These workers reported little dependence of wave speed on [InsP3]. In a similar fashion, using the low-affinity dye OGB-5N we too have made a similar observation: a minimal effect of InsP3 concentration on wave speed (Fig. 3E). We have interpreted this finding as the low-affinity dye acting as a ‘chemical filter’ to essentially detect a ‘wave-crest’, possibly reflecting a true propagating component of the [Ca2+]c signal. Experimental differences (high-affinity versus low-affinity dye, the use of threshold detection to track apparent wave spread, and the improved time resolution of the current study: about an order of magnitude greater) also probably contribute to some of the differences observed by Ito et al. and the present study.
Because of the limited range over which Ca2+ can diffuse within the cytoplasm, it is generally believed that there is a regenerative release component that underlies the generation of a global [Ca2+]c signal. The velocity at which a Ca2+ wave propagates is commonly determined from the distance over which the peak of the signal travels in a specific time interval, analogous to the regenerative propagation of a neuronal action potential. However, it is unclear whether such measurements reflect the propagation of a true wave (regenerative propagation at a fixed velocity) or a heterogeneous cellular distribution of Ca2+-release channels with differing functional sensitivities to InsP3.
Indeed, previous studies have provided evidence for such a distribution, and suggest that release from multiple subcellular compartments may also contribute to the spatial and temporal aspects of the Ca2+ signals of exocrine cells (Kasai et al. 1993; Tanimura et al. 1998; Fogarty et al. 2000; Giovannucci et al. 2000). Therefore, while Ca2+ release sites throughout a cell may be activated simultaneously, a ΔF may be detected initially in a region where release sites are at higher density, and then subsequently in an area where release sites are not as abundant. Presumably, in an area of a cell where there is a paucity of release sites, the initial Ca2+ release is slower to rise to detectable levels, as release is not as efficiently entrained by CICR as in high-density release sites. Thus, an observed Ca2+ signal may not necessarily propagate as a wave per se, but may also reflect the simultaneous activation of individual or clusters of InsP3R acting as autonomous release sites. Our data are consistent with this notion, and suggest that the simultaneous activation of autonomous cytoplasmic, non-apical release sites contribute substantially to the global Ca2+ signal in PAR.
The concentration dependence of the time course of InsP3-evoked [Ca2+]c signals suggested that a lower density of release channels in PAC compared with PAR accounts for the divergence in Ca2+-release kinetics between PAR and PAC. A longer latency, slower rise time, and slower relaxation of the Ca2+ transient in PAC are all consistent with this notion. Conversely, in PAR, the higher receptor density would be expected to produce short-lived transients with rapid activation and rate of rise, as both long- and short-range effects of Ca2+ are enhanced. Indeed, our data demonstrating that PAR express nearly three-fold more InsP3R than PAC is consistent with this assertion.
In addition to receptor density, other factors may contribute to these differences in the spatiotemporal aspects of Ca2+ signals in acinar cells. For example, differences in mobile and fixed buffers between PAR and PAC would impact the short- and long-range effects of Ca2+ on receptor activation, recruitment of additional release sites, and receptor inactivation. Moreover, differences in InsP3R type sensitivity, distribution or phosphoregulation may also play a role in determining release kinetics (LeBeau et al. 1999; Giovannucci et al. 2000).
Ca2+-activated Cl- current properties of PAR and PAC
Despite the ubiquitous distribution and recent cloning of several putative ClCa, their functional significance and molecular identity have not been conclusively established (Nehrke et al. 2000). Moreover, mechanistic differences in the Ca2+-dependent activation or inactivation of IClCa have been reported (Arreola et al. 1998). Previous studies have suggested a direct and higher-order dependence of the IClCa on Ca2+ (Arreola et al. 1998; Kidd & Thorn, 2000, 2001). However, in rat PAR and mouse PAC, the ‘steady-state’ Ca2+-dependence of IClCa has been investigated by introducing exogenous buffers with set Ca2+ concentrations via the patch pipette and applying voltage steps to activate IClCa. These studies have yielded estimates of the voltage and Ca2+ sensitivities of ClCa (Arreola et al. 1996; Kidd & Thorn, 2001). Yet, it is not certain that the values estimated under these conditions accurately reflect the Ca2+ sensitivity of the ClCa under the dynamic conditions encountered during a Ca2+ oscillation induced by physiological stimuli (Kidd & Thorn, 2000). To bridge this gap in our knowledge, we used the controlled photolysis of caged-Ca2+ and caged-InsP3 to induce changes in [Ca2+]c and monitored the Ca2+-dependence of the IClCa under non-steady-state conditions.
Using a novel [Ca2+]c ramp method, we assessed the Ca2+ sensitivity of ClCa activation. Accounting for a resting [Ca2+]c level of 170 nm (in the patch pipette solution), and using the Δ[Ca2+]c-I relationship determined in the presence of CPA, we obtained KD values for PAR and PAC of 370 nm and 620 nm, respectively. Whereas the PAR KD values were nearly identical to previous estimates (∼360 nm) obtained by steady-state [Ca2+] (Arreola et al. 1996), our estimates for PAC indicated ∼two-fold less potency of [Ca2+] value for activation than that recently reported (Kidd & Thorn, 2001). The reason for this discrepancy is not clear, although it is unlikely that it reflects a fundamental difference in steady-state [Ca2+]vs.[Ca2+]c-ramp methods for ClCa activation. In fact, our results with PAR validate the use of steady-state methods for determining the Ca2+-dependence of ClCa. We cannot, however, rule out confounding effects of an endogenous, immobile buffering component, recruitment of another Ca2+-activated Cl− channel induced at the high [Ca2+]c produced by photolytic release of Ca2+, or the difficulty in determining an absolute value for our dye calibration.
Divergence in the mechanisms of inactivation of IClCa has also been reported. For example, airway smooth muscle cells have been reported to require CaMKII for IClCa inactivation (Kotlikoff & Wang, 1998). In these cells, phosphoregulation of ClCa is proposed to uncouple the Ca2+ sensitivity of the channel. Agonist-induced oscillations of the IClCa in PAC have also been shown to decay more rapidly than the corresponding apical [Ca2+]c oscillations, although the underlying mechanism has not been established (Kidd & Thorn, 2000). In our experiments, the IClCa of both PAR and PAC often recovered to baseline more rapidly than the [Ca2+]c signal, as reported by high-affinity dye, suggesting an uncoupling of sensitivity to [Ca2+]c. Faster recovery may be indicative of enhanced Ca2+ clearance and/or termination of InsP3-evoked Ca2+ release. The former hypothesis is supported by experiments monitoring apparent inactivation of the IClCa in the presence of CPA to inhibit SERCA pump activity (see Fig. 7D). These experiments indicated that in PAR, and to a lesser degree in PAC, local clearance mechanisms play an important role in shaping the Ca2+ signal ‘seen’ by the ClCa, and thus the apparent inactivation. In addition, high flux of Ca2+ into the cytoplasm, such as that reported to occur in InsP3R-rich Purkinje neurons, has been shown to result in rapid termination of Ca2+ release, presumably via Ca2+-dependent inhibition of the InsP3R, effectively producing fast, brief Ca2+-release events (Ogden & Capiod, 1997). This would contribute to a local decline in [Ca2+]c following the termination of Ca2+ flux, particularly for PAR, which exhibit a greater density of release channels. We propose that the decline in the IClCa during persistent global increases in [Ca2+]c is largely due to decreases in [Ca2+]c in the vicinity of the ClCa, and thus reflects deactivation of the channel rather than inactivation.
Because neither the molecular identity nor single-channel properties of ClCa in PAR have been experimentally determined, we can only speculate as to the luminal density of these channels. However, given the similarities in channel activation and the functional correlation with apical release kinetics, suggesting a preponderance of channels in the luminal membrane, channel density would be ∼four-fold greater in PAR than in PAC, assuming a similar open probability and single-channel conductance.
Functional implications of divergent Ca2+-release kinetics for secretion in acinar cells
A common feature of models describing fluid secretion from exocrine cells is the unidirectional flux of Cl− across the luminal plasma membrane into the extracellular space (Greger, 1996). Accumulation of Cl− in the lumen is followed by diffusion of Na+ via a paracellular pathway, and the subsequent osmotic movement of water to create an isotonic, NaCl-rich fluid (Begenisich & Melvin, 1998). On agonist stimulation, an increase in [Ca2+]c is initiated in the luminal pole of the cell and is thus ideally positioned to rapidly activate luminal ClCa. The flux of Cl− across the luminal membrane leads to membrane depolarization and a collapse of the driving force for further secretion as the membrane potential (Vm) approaches the equilibrium potential for Cl− (ECl). To maintain Cl− secretion, the membrane potential must be re-established below ECl (Greger, 1996). In many exocrine cells, this is thought to be accomplished by the activation of basolaterally localized Ca2+-activated K+ channels. In PAR, and in PAC from some species, the activation of voltage-sensitive, Ca2+-dependent large conductance (maxi-K+; Maruyama et al. 1983; Suzuki & Petersen, 1988; Hassoni & Gray, 1994) and/or intermediate conductance K+ channels (Kim & Greger, 1999) may satisfy this requirement. In rodent PAC, which lack maxi-K+ channels, an intermediate Ca2+-sensitive, slowly activating (IKs; Kim & Greger, 1999) and inwardly rectifying K+ current (IKir; Kim et al. 2000) have been demonstrated, which could function to maintain PAC Vm at hyperpolarized potentials. Replenishment of intracellular Cl− occurs via basolateral Na+-K+-2Cl− co-transporters (Zhao & Muallem, 1995; Begenisich & Melvin, 1998; Evans et al. 2000), Cl−-HCO3− exchangers (Turner & George, 1988; Gillespie et al. 1989; Muallem & Loessberg, 1990; Melvin & Turner, 1992; Zhao et al. 1995), or alternatively by activation of flux through basolateral Cl− and non-specific cation channels (Kasai & Augustine, 1990).
Data presented in this study reveal functional specializations of PAR designed to maximize the efficiency of Cl− transport, and thus fluid secretion. Specifically, PAR express either more ClCa, or ClCa of larger unitary conductance, than PAC. In addition to the more than four-fold increase in IClCa density, the [Ca2+]c signal evoked in PAR is ‘tuned’ to provide nearly synchronous activation of luminal ClCa (for Cl− secretion) and basolateral K+ channels (to maintain the driving force for Cl− flux). This is accomplished by a high density of release channels, which predisposes PAR rapidly to initiate global [Ca2+]c signals and thus co-ordinate the activation of spatially separated effectors at the apical and basolateral membranes, resulting in efficient fluid secretion. The global signal appears in large part to result from recruitment of an autonomous component of release, such that after initiation in the ‘trigger zone’ a nearly synchronous increase in [Ca2+]c occurs. As a secondary consequence of this large global signal, [Ca2+]c is elevated to levels that would support exocytosis at the apical pole of the cell. In contrast, the lower density of release channels in PAC allows a continuum of Ca2+ signalling events to be generated: from localized apical signals at threshold stimulation, to rapid global signals (similar to PAR) under conditions of maximal stimulation. The spatial and temporal characteristics of [Ca2+]c signals in PAC at low stimulus strength are consistent with the primary exocytotic function of the cells. On the other hand, global signals evoked upon greater stimulus strength presumably reflect the fact that [Ca2+]c also plays import roles in PAC in such divergent processes as fluid secretion, protein synthesis and gene transcription. These observations underscore the similarity in the release modules and indicate that the differences in the signal are largely reflective of the number of release channels. Moreover, release channel density may provide a general mechanism whereby cells can tune the spatial and temporal characteristics of [Ca2+]c signals to efficiently activate the appropriate effectors.
Acknowledgments
We thank Jodi Pilato and Pauline Leakey for excellent technical assistance with some of the experiments, and Dr Ted Begenisich for helpful comments during these studies. InsP3R-specific antisera were generously provided by Dr Richard Wojcikiewicz. This work was supported by National Institutes of Health Grants DE-13539 (T.J.S. and D.I.Y.), DK-54568 (D.I.Y.) and GM 40457 (T.J.S.).
REFERENCES
- Arreola J, Melvin JE, Begenisich T. Activation of calcium-dependent chloride channels in rat parotid acinar cells. Journal of General Physiology. 1996;108:35–47. doi: 10.1085/jgp.108.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arreola J, Melvin JE, Begenisich T. Differences in regulation of Ca2+-activated Cl− channels in colonic and parotid secretory cells. American Journal of Physiology. 1998;274:C161–166. doi: 10.1152/ajpcell.1998.274.1.C161. [DOI] [PubMed] [Google Scholar]
- Begenisich T, Melvin JE. Regulation of chloride channels in secretory epithelia. Journal of Membrane Biology. 1998;163:77–85. doi: 10.1007/s002329900372. [DOI] [PubMed] [Google Scholar]
- Bootman MD, Collins TJ, Peppiatt CM, Prothero LS, MacKenzie L, De Smet P, Travers M, Tovey SC, Seo JT, Berridge MJ, Ciccolini F, Lipp P. Calcium signalling - an overview. Seminars in Cell and Developmental Biology. 2001;12:3–10. doi: 10.1006/scdb.2000.0211. [DOI] [PubMed] [Google Scholar]
- Bredt DS, Mourey RJ, Snyder SH. A simple, sensitive, and specific radioreceptor assay for inositol 1,4,5-trisphosphate in biological tissues. Biochemical and Biophysical Research Communications. 1989;159:976–982. doi: 10.1016/0006-291x(89)92204-3. [DOI] [PubMed] [Google Scholar]
- Bugrim AE, Zhabotinsky AM, Epstein IR. Calcium waves in a model with a random spatially discrete distribution of Ca2+ release sites. Biophysical Journal. 1997;73:2897–2906. doi: 10.1016/S0006-3495(97)78318-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clapham DE. Calcium signaling. Cell. 1995;80:259–268. doi: 10.1016/0092-8674(95)90408-5. [DOI] [PubMed] [Google Scholar]
- Crabtree GR. Generic signals and specific outcomes: signaling through Ca2+, calcineurin, and NF-AT. Cell. 1999;96:611–614. doi: 10.1016/s0092-8674(00)80571-1. [DOI] [PubMed] [Google Scholar]
- Duchen MR. Mitochondria and calcium: from cell signalling to cell death. Journal of Physiology. 2000;529:57–68. doi: 10.1111/j.1469-7793.2000.00057.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans RL, Bell SM, Schultheis PJ, Shull GE, Melvin JE. Targeted disruption of the Nhe1 gene prevents muscarinic agonist-induced up-regulation of Na+/H+ exchange in mouse parotid acinar cells. Journal of Biological Chemistry. 1999;274:29025–29030. doi: 10.1074/jbc.274.41.29025. [DOI] [PubMed] [Google Scholar]
- Evans RL, Park K, Turner RJ, Watson GE, Nguyen HV, Dennett MR, Hand AR, Flagella M, Shull GE, Melvin JE. Severe impairment of salivation in Na+/K+/2Cl− cotransporter (NKCC1)-deficient mice. Journal of Biological Chemistry. 2000;275:26720–26726. doi: 10.1074/jbc.M003753200. [DOI] [PubMed] [Google Scholar]
- Fitzsimmons TJ, Gukovsky I, McRoberts JA, Rodriguez E, Lai FA, Pandol SJ. Multiple isoforms of the ryanodine receptor are expressed in rat pancreatic acinar cells. Biochemical Journal. 2000;351:265–271. doi: 10.1042/0264-6021:3510265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fogarty KE, Kidd JF, Tuft DA, Thorn P. Mechanisms underlying InsP3-evoked global Ca2+ signals in mouse pancreatic acinar cells. Journal of Physiology. 2000;526:515–526. doi: 10.1111/j.1469-7793.2000.t01-1-00515.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillespie JI, Hedley C, Greenwell JR, Argent BE. Chloride-bicarbonate exchange in isolated rat pancreatic acini. Quarterly Journal of Experimental Physiology. 1989;74:883–895. doi: 10.1113/expphysiol.1989.sp003359. [DOI] [PubMed] [Google Scholar]
- Giovannucci DR, Groblewski GE, Sneyd J, Yule DI. Targeted phosphorylation of inositol 1,4,5-trisphosphate receptors selectively inhibits localized Ca2+ release and shapes oscillatory Ca2+ signals. Journal of Biological Chemistry. 2000;275:33704–33711. doi: 10.1074/jbc.M004278200. [DOI] [PubMed] [Google Scholar]
- Greger R. The membrane transporters regulating epithelial NaCl secretion. Pflügers Archiv. 1996;432:579–588. doi: 10.1007/s004240050173. [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Chawla S, Johnson CM, Bading H. Distinct functions of nuclear and cytoplasmic calcium in the control of gene expression. Nature. 1997;385:260–265. doi: 10.1038/385260a0. [DOI] [PubMed] [Google Scholar]
- Hassoni AA, Gray PT. Flash photolysis studies of the localization of calcium release sites in rat parotid isolated acinar cells. Journal of Physiology. 1994;478:461–467. doi: 10.1113/jphysiol.1994.sp020265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K, Miyashita Y, Kasai H. Micromolar and submicromolar Ca2+ spikes regulating distinct cellular functions in pancreatic acinar cells. EMBO Journal. 1997;16:242–251. doi: 10.1093/emboj/16.2.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K, Miyashita Y, Kasai H. Kinetic control of multiple forms of Ca2+ spikes by inositol trisphosphate in pancreatic acinar cells. Journal of Cell Biology. 1999;146:405–413. doi: 10.1083/jcb.146.2.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwatsuki N, Maruyama Y, Matsumoto O, Nishiyama A. Activation of Ca2+-dependent Cl− and K+ conductances in rat and mouse parotid acinar cells. Japanese Journal of Physiology. 1985;35:933–944. doi: 10.2170/jjphysiol.35.933. [DOI] [PubMed] [Google Scholar]
- Kasai H, Augustine GJ. Cytosolic Ca2+ gradients triggering unidirectional fluid secretion from exocrine pancreas. Nature. 1990;348:735–738. doi: 10.1038/348735a0. [DOI] [PubMed] [Google Scholar]
- Kasai H, Li YX, Miyashita Y. Subcellular distribution of Ca2+ release channels underlying Ca2+ waves and oscillations in exocrine pancreas. Cell. 1993;74:669–677. doi: 10.1016/0092-8674(93)90514-q. [DOI] [PubMed] [Google Scholar]
- Kidd JF, Fogarty KE, Tuft RA, Thorn P. The role of Ca2+ feedback in shaping InsP3-evoked Ca2+ signals in mouse pancreatic acinar cells. Journal of Physiology. 1999;520:187–201. doi: 10.1111/j.1469-7793.1999.00187.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidd JF, Thorn P. Intracellular Ca2+ and Cl− channel activation in secretory cells. Annual Reviews in Physiology. 2000;62:493–513. doi: 10.1146/annurev.physiol.62.1.493. [DOI] [PubMed] [Google Scholar]
- Kidd JF, Thorn P. The properties of the secretagogue-evoked chloride current in mouse pancreatic acinar cells. Pflügers Archiv. 2001;441:489–497. doi: 10.1007/s004240000451. [DOI] [PubMed] [Google Scholar]
- Kim SJ, Greger R. Voltage-dependent, slowly activating K+ currents (IKs) and its augmentation by carbachol in rat pancreatic acini. Pflügers Archiv. 1999;438:604–611. doi: 10.1007/s004249900071. [DOI] [PubMed] [Google Scholar]
- Kim SJ, Kerst G, Schreiber R, Pavenstadt H, Greger R, Hug MJ, Bleich M. Inwardly rectifying K+ channels in the basolateral membrane of rat pancreatic acini. Pflügers Archis. 2000;441:331–340. doi: 10.1007/s004240000427. [DOI] [PubMed] [Google Scholar]
- Kotlikoff MI, Wang YX. Calcium release and calcium-activated chloride channels in airway smooth muscle cells. American Journal of Respiratory Critical Care Medicine. 1998;158:S109–114. doi: 10.1164/ajrccm.158.supplement_2.13tac600. [DOI] [PubMed] [Google Scholar]
- Lebeau AP, Yule DI, Groblewski GE, Sneyd J. Agonist-dependent phosphorylation of the inositol 1,4,5-trisphosphate receptor: A possible mechanism for agonist-specific calcium oscillations in pancreatic acinar cells. Journal of General Physiology. 1999;113:851–872. doi: 10.1085/jgp.113.6.851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MG, Xu X, Zeng W, Diaz J, Kuo TH, Wuytack F, Racymaekers L, Muallem S. Polarized expression of Ca2+ pumps in pancreatic and salivary gland cells. Role in initiation and propagation of [Ca2+]i waves. Journal of Biological Chemistry. 1997a;272:15771–15776. doi: 10.1074/jbc.272.25.15771. [DOI] [PubMed] [Google Scholar]
- Lee MG, Xu X, Zeng W, Diaz J, Wojcikiewicz RJ, Kuo TH, Wuytack F, Racymaekers L, Muallem S. Polarized expression of Ca2+ channels in pancreatic and salivary gland cells. Correlation with initiation and propagation of [Ca2+]i waves. Journal of Biological Chemistry. 1997b;272:15765–15770. doi: 10.1074/jbc.272.25.15765. [DOI] [PubMed] [Google Scholar]
- Marchant J, Callamaras N, Parker I. Initiation of IP3-mediated Ca2+ waves in Xenopus oocytes. EMBO Journal. 1999;18:5285–5299. doi: 10.1093/emboj/18.19.5285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruyama Y, Petersen OH, Flanagan P, Pearson GT. Quantification of Ca2+-activated K+ channels under hormonal control in pig pancreas acinar cells. Nature. 1983;305:228–232. doi: 10.1038/305228a0. [DOI] [PubMed] [Google Scholar]
- Melvin JE, Turner RJ. Cl− fluxes related to fluid secretion by the rat parotid: involvement of Cl−-HCO3− exchange. American Journal of Physiology. 1992;262:G393–398. doi: 10.1152/ajpgi.1992.262.3.G393. [DOI] [PubMed] [Google Scholar]
- Mogami H, Tepikin AV, Petersen OH. Termination of cytosolic Ca2+ signals: Ca2+ reuptake into intracellular stores is regulated by the free Ca2+ concentration in the store lumen. EMBO Journal. 1998;17:435–442. doi: 10.1093/emboj/17.2.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muallem S, Loessberg PA. Intracellular pH-regulatory mechanisms in pancreatic acinar cells. I. Characterization of H+ and HCO3− transporters. Journal of Biological Chemistry. 1990;265:12806–12812. [PubMed] [Google Scholar]
- Nathanson MH, Fallon MB, Padfield PJ, Maranto AR. Localization of the type 3 inositol 1,4,5-trisphosphate receptor in the Ca2+ wave trigger zone of pancreatic acinar cells. Journal of Biological Chemistry. 1994;269:4693–4696. [PubMed] [Google Scholar]
- Neher E, Augustine GJ. Calcium gradients and buffers in bovine chromaffin cells. Journal of Physiology. 1992;450:273–301. doi: 10.1113/jphysiol.1992.sp019127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nehrke K, Begenisich T, Pilato J, Melvin JE. Into ion channel and transporter function. Caenorhabditis elegans ClC-type chloride channels: novel variants and functional expression. American Journal of Physiology. 2000;279:C2052–2066. doi: 10.1152/ajpcell.2000.279.6.C2052. [DOI] [PubMed] [Google Scholar]
- Ogden D, Capiod T. Regulation of Ca2+ release by InsP3 in single guinea pig hepatocytes and rat Purkinje neurons. Journal of General Physiology. 1997;109:741–756. doi: 10.1085/jgp.109.6.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park MK, Lomax RB, Tepikin AV, Petersen OH. Local uncaging of caged Ca2+ reveals distribution of Ca2+-activated Cl− channels in pancreatic acinar cells. Proceedings of the National Academy of Sciences of the USA. 2001;98:10948–10953. doi: 10.1073/pnas.181353798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park MK, Tepikin AV, Petersen OH. The relationship between acetylcholine-evoked Ca2+-dependent current and the Ca2+ concentrations in the cytosol and the lumen of the endoplasmic reticulum in pancreatic acinar cells. Pflügers Archiv. 1999;438:760–765. doi: 10.1007/s004249900128. [DOI] [PubMed] [Google Scholar]
- Petersen OH. Stimulus-secretion coupling: cytoplasmic calcium signals and the control of ion channels in exocrine acinar cells. Journal of Physiology. 1992;448:1–51. doi: 10.1113/jphysiol.1992.sp019028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen OH, Gallacher DV, Wakui M, Yule DI, Petersen CC, Toescu EC. Receptor-activated cytoplasmic Ca2+ oscillations in pancreatic acinar cells: generation and spreading of Ca2+ signals. Cell Calcium. 1991;12:135–144. doi: 10.1016/0143-4160(91)90015-7. [DOI] [PubMed] [Google Scholar]
- Randriamampita C, Chanson M, Trautmann A. Calcium and secretagogues-induced conductances in rat exocrine pancreas. Pflügers Archiv. 1988;411:53–57. doi: 10.1007/BF00581646. [DOI] [PubMed] [Google Scholar]
- Rooney TA, Sass EJ, Thomas AP. Agonist-induced cytosolic calcium oscillations originate from a specific locus in single hepatocytes. Journal of Biological Chemistry. 1990;265:10792–10796. [PubMed] [Google Scholar]
- Straub SV, Giovannucci DR, Yule DI. Calcium wave propagation in pancreatic acinar cells: functional interaction of inositol 1,4,5-trisphosphate receptors, ryanodine receptors, and mitochondria. Journal of General Physiology. 2000;116:547–560. doi: 10.1085/jgp.116.4.547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki K, Petersen OH. Patch-clamp study of single-channel and whole-cell K+ currents in guinea pig pancreatic acinar cells. American Journal of Physiology. 1988;255:G275–285. doi: 10.1152/ajpgi.1988.255.3.G275. [DOI] [PubMed] [Google Scholar]
- Takemura H, Yamashina S, Segawa A. Millisecond analyses of Ca2+ initiation sites evoked by muscarinic receptor stimulation in exocrine acinar cells. Biochemical and Biophysical Research Communications. 1999;259:656–660. doi: 10.1006/bbrc.1999.0818. [DOI] [PubMed] [Google Scholar]
- Tanimura A, Matsumoto Y, Tojyo Y. Polarized Ca2+ release in saponin-permeabilized parotid acinar cells evoked by flash photolysis of ‘caged’ inositol 1,4,5-trisphosphate. Biochemical Journal. 1998;332:769–772. doi: 10.1042/bj3320769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tojyo Y, Tanimura A, Matsumoto Y. Imaging of intracellular Ca2+ waves induced by muscarinic receptor stimulation in rat parotid acinar cells. Cell Calcium. 1997;22:455–462. doi: 10.1016/s0143-4160(97)90073-7. [DOI] [PubMed] [Google Scholar]
- Turner RJ, George JN. Cl−-HCO3− exchange is present with Na+-K+-Cl− cotransport in rabbit parotid acinar basolateral membranes. American Journal of Physiology. 1988;254:C391–396. doi: 10.1152/ajpcell.1988.254.3.C391. [DOI] [PubMed] [Google Scholar]
- Williams JA, Groblewski GE, Ohnishi H, Yule DI. Stimulus-secretion coupling of pancreatic digestive enzyme secretion. Digestion. 1997;58:42–45. doi: 10.1159/000201524. [DOI] [PubMed] [Google Scholar]
- Williams JA, Korc M, Dormer RL. Action of secretagogues on a new preparation of functionally intact, isolated pancreatic acini. American Journal of Physiology. 1978;235:517–524. doi: 10.1152/ajpendo.1978.235.5.E517. [DOI] [PubMed] [Google Scholar]
- Wojcikiewicz RJ, He Y. Type I, II and III inositol 1,4,5-trisphosphate receptor co-immunoprecipitation as evidence for the existence of heterotetrameric receptor complexes. Biochemical and Biophysical Research Communications. 1995;213:334–341. doi: 10.1006/bbrc.1995.2134. [DOI] [PubMed] [Google Scholar]
- Xu X, Zeng W, Diaz J, Lau KS, Gukovskaya AC, Brown RJ, Pandol SJ, Muallem S. nNOS and Ca2+ influx in rat pancreatic acinar and submandibular salivary gland cells. Cell Calcium. 1997;22:217–228. doi: 10.1016/s0143-4160(97)90015-4. [DOI] [PubMed] [Google Scholar]
- Yule DI, Ernst SA, Ohnishi H, Wojcikiewicz RJ. Evidence that zymogen granules are not a physiologically relevant calcium pool. Defining the distribution of inositol 1, 4,5-trisphosphate receptors in pancreatic acinar cells. Journal of Biological Chemistry. 1997;272:9093–9098. doi: 10.1074/jbc.272.14.9093. [DOI] [PubMed] [Google Scholar]
- Zdebik A, Hug MJ, Greger R. Chloride channels in the luminal membrane of rat pancreatic acini. Pflügers Archiv. 1997;434:188–194. doi: 10.1007/s004240050382. [DOI] [PubMed] [Google Scholar]
- Zhang X, Wen J, Bidasee KR, Besch H R, Jr, Rubin RP. Ryanodine receptor expression is associated with intracellular Ca2+ release in rat parotid acinar cells. American Journal of Physiology. 1997;273:C1306–1314. doi: 10.1152/ajpcell.1997.273.4.C1306. [DOI] [PubMed] [Google Scholar]
- Zhang X, Wen J, Bidasee KR, Besch HR, Jr, Wojcikiewicz RJ, Lee B, Rubin RP. Ryanodine and inositol trisphosphate receptors are differentially distributed and expressed in rat parotid gland. Biochemical Journal. 1999;340:519–527. [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Muallem S. Na+, K+, and Cl− transport in resting pancreatic acinar cells. Journal of General Physiology. 1995;106:1225–1242. doi: 10.1085/jgp.106.6.1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Xu X, Diaz J, Muallem S. Na+, K+, and H+/HCO3− transport in submandibular salivary ducts. Membrane localization of transporters. Journal of Biological Chemistry. 1995;270:19599–19605. [PubMed] [Google Scholar]