

Abstract
Chemical modification can inhibit ion channels either by reacting with pore-lining residues and directly occluding the channel or by closing the channel allosterically. A general method to distinguish between these two mechanisms does not exist. Previously, sulfhydryl (SH) modification has been shown to inhibit ATP-sensitive K+ (KATP) channels. The crucial modification has been localized to C42 near the N-terminus of Kir6.2, a pore-forming subunit of KATP channels, but little is known about how SH modification of C42 causes channel inhibition. To investigate this mechanism, we used the membrane-impermeable methanethiosulfonates, MTSET and MTS-TEAH, to modify Kir6.2 channels. While intracellular application of MTSET irreversibly inhibited channels, MTS-TEAH failed to do so. Instead, MTS-TEAH treatment prolonged channel openings and prevented the effect of subsequent MTSET treatment. Similar observations were made in mutants in which cysteines other than C42 had been mutated. Neither MTSET nor MTS-TEAH, however, affected mutant channels in which valines were substituted for C42 residues in all subunits. The reagents were effective when two of four C42 residues in the tetramer were replaced by valines. These results can be interpreted as indicating that both reagents modify C42. We then employed spermine, a known inner pore blocker, as a probe to examine whether MTS-TEAH modification alters pore accessibility. We found that spermine block was not changed by MTS-TEAH modification. Based on these data, we postulate that C42 faces either the cytoplasm or a vestibule section wide enough to allow spermine to pass freely after modification by MTS-TEAH. Our study suggests that channel inhibition caused by SH modification of Kir6.2 is an allosteric effect, and is not caused by direct pore blockage.
The substituted-cysteine-accessibility method (SCAM) (Akabas et al. 1992) can be used to identify pore-lining residues in ion channels (Akabas et al. 1994). In practice, an exposed pore-lining residue can be identified if sulfhydryl (SH) modification of its cysteine substitute causes irreversible reduction or inhibition of channel current. Channel inhibition is often assumed to be a consequence of direct occlusion of the pore by bound SH-reactive reagents. However, this approach to characterization of pore structure is often confounded by the possibility that modification may affect channels through allosteric mechanisms. In such cases, channel current could be affected even if the modified residue is not located inside the pore. Distinguishing direct pore occlusion from allosteric effects caused by SH modification is often difficult. In this study, we present an analysis that helps to distinguish between these two mechanisms.
Studies done on native ATP-sensitive K+ (KATP) channels have indicated that these channels are prone to react with SH-reactive chemicals such as N-ethylmaleimide (NEM) and 5,5′-dithiobis(2-nitrobenzoic acid) (DTNB) (Weik & Neumcke, 1989; Lee et al. 1994; Coetzee et al. 1995). p-Chloromercuriphenylsulfonate (pCMPS), another SH-reactive reagent, inhibited cloned KATP channels composed of SUR1 and Kir6.2 subunits (Trapp et al. 1998b). Because the inhibitory effect also occurs in channels composed entirely of Kir6.2 (Kir6.2ΔC26) subunits (which also confer ATP sensitivity), it is clear that inhibition of KATP channels takes place at the channel-forming subunit. The primary reactive site responsible for the inhibition is a single cysteine residue, C42, near the N-terminus. Mutant channels whose C42 was substituted by a valine or an alanine were no longer sensitive to the inhibitory effect of SH-reactive reagents. However, it is not known how modification of C42 leads to channel inhibition. Intriguingly, C42 is conserved in many, but not all, members of the inwardly rectifying K+ channel superfamily. SH modification at the equivalent position reportedly inhibits Kir1.1 (C49), Kir2.1 (C54) and Kir2.3 (C50) (Schulte et al. 1998; Lu et al. 1999b; Radeeke et al. 1999). These data may imply that this position is involved in a structural homology and/or a function shared by these channels. Understanding the mechanism of the inhibition by SH modification may help to uncover such functions.
Information on the location of C42 is key to determining whether SH modification directly blocks the channel, or closes the channel allosterically. According to hydropathy analysis, C42 extends into the cytoplasm by a distance of 30 residues from the transmembrane segment M1 (Inagaki et al. 1995), and therefore it seems unlikely that it could form part of the transmembrane region of the pore. Unlike the residues of transmembrane segments, whose roles in pore formation have been extensively studied, few clues are available as to the structure of the cytoplasmic vestibule that extends from the transmembrane pore. Interpretation of experiments in which charged SH-specific reagents react with residues of the cytoplasmic region is complicated because these residues can face either the pore lumen or the cytoplasm. In SCAM experiments carried out on Kir2.1 channels, cysteine substitution of the residues next to C54 exhibited periodic inhibition by SH modification, which was interpreted to be a consequence of the pore-lining structure of this region (Lu et al. 1999b). In contrast, C49 of the Kir1.1 channel was shown to be a key residue involved in a state-dependent modification process that results in channel inhibition, and it was proposed that SH modification of this residue may lock channels in a closed state (Schulte et al. 1998). In Kir6.2 channels, a study of SH modification hints that C42 is probably involved in channel gating (Trapp et al. 1998b). A clear explanation for this discrepancy has not been made (Lu et al. 1999b).
In order to determine whether channel inhibition caused by SH modification is due to occlusion or to allosteric effects, we designed a series of experiments to investigate whether SH modification of C42 is able to block the Kir6.2 pore. The study was spurred by findings that a membrane-impermeable methanethiosulfonate (MTS) reagent, (triethylammonium)hexyl methanethiosulfonate bromide (MTS-TEAH), modifies the Kir6.2 channel without inhibiting it. Charged MTS reagents react with exposed thiol groups at the aqueous interface of proteins more specifically and rapidly than other traditional SH-reactive chemicals. Because channels covalently linked to MTS-TEAH-derived molecules still conduct current, we used a pore-plugging reagent, spermine, to explore the possible structural change in the pore. Changes in channel block by spermine in response to SH modification would be expected if a pore-lining residue were modified. Our analysis does not support the hypothesis that C42 modified by SH reagents blocks the pore, but instead indicates that channel activity is affected allosterically.
METHODS
Cell culture and transfection
A monkey kidney COS-1 cell line was purchased from the American Type Culture Collection (Rockville, MD, USA) and cultured under the suggested conditions. The method for transient transfection was as previously published (Fan & Makielski, 1999). Kir6.2Δ a truncated form of Kir6.2 lacking the last 35 residues, and its mutants were subcloned in mammalian expression vectors and transfected with a NovaFECTOR kit (VennNova, LLC, Pompano Beach, FL, USA). Kir6.2Δ was used to express this subunit on the plasma membrane (Tucker et al. 1997). The expression of the channels usually peaked 48–72 h after transfection. The cells were then immediately used for electrophysiological experiments.
Site-directed mutagenesis
A double-stranded mutagenesis kit, Chameleon (Stratagene Inc., La Jolla, CA, USA), was used to generate the desired point mutations. A PCR-based site-directed mutagenesis kit, ExSite (Stratagene Inc.), was used to create the Kir6.2Δ construct (Fan & Makielski, 1999). To make a fused subunit, the 3′-end of Kir6.2Δ(C42V) cDNA was linked to the 5′-end of Kir6.2Δ cDNA by a nucleotide bridge encoding a linker region of six glycines. The final construct was subcloned into a pCR3.1-Uni mammalian expression vector (Invitrogen, Carlsbad, CA, USA). Maxi- or mini-scale production of plasmids containing the resulting mutants was used to amplify the cDNA for transfection.
Modification of expressed channels by SH-specific reagents
The membrane-impermeable SH-specific reagents [(trimethylammonium) ethyl] methanethiosulfonate bromide (MTSET) and MTS-TEAH (Toronto Research Chemicals Inc., North York, Ontario, Canada) were used in our experiments (Fig. 1A). Both reagents contain the same SH-specific moiety that can react with the side chain of a cysteine residue. Compared to MTSET, MTS-TEAH has a bigger, bulkier space-filling group which is connected to the reactive moiety by a longer connecter. Because these reagents dissociate rapidly in aqueous solution, they were used immediately (typically < 2 min) after dissolution. The MTS reagents were dissolved in DMSO (< 0.1 % final concentration) and then added into the bath solution (intracellular side of excised membrane patches) via a gravity-driven perfusion system. It took less than 20 s (an average of ∼5 s) to switch the bath solution. Other solution exchanges were performed through a pressurized perfusion system (DAD-12, ALA Scientific Instruments, Westbury, NY, USA). For recovery experiments, channel activity was measured 1 min after washout. All other chemicals were from Sigma (St Louis, MO, USA). Experiments were performed at room temperature.
Figure 1. Chemical structure of MTSET and MTS-TEAH (A) and their effects on current activity of Kir6.2Δ channels (B,D,E), and dimer channels (C).
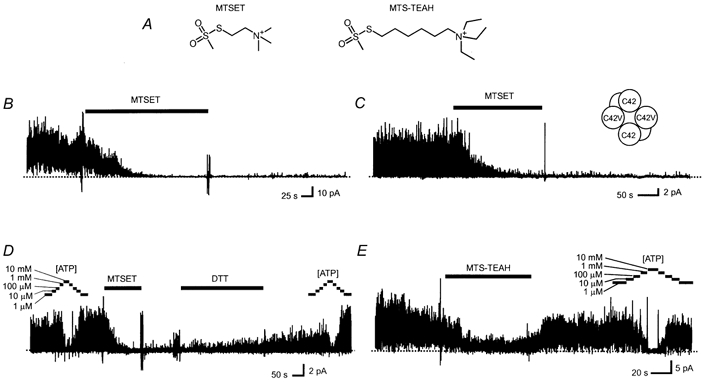
Multiple-channel currents were acquired from membranes in an inside-out patch configuration. The patch membranes were excised from COS-1 cells expressing transfected channels, bathed in an intracellular solution of 140 mm K+ and a pipette (extracellular) solution of 10 mm K+, and held at 0 mV throughout the recording periods. MTSET (0.6 mm), MTS-TEAH (0.6 mm), DTT (5 mm) and ATP (at the indicated concentrations) were applied during the periods marked by the corresponding bars above the current traces. The putative subunit composition of the Kir6.2Δ(C42V)-Kir6.2Δ dimer channel is illustrated in the inset of C. A dotted line through each current trace indicates the level where all channels were closed. The current recordings were digitally filtered at 200 Hz using a low-pass filtering algorithm.
Multiple and single channel recordings
Inside-out single or multiple channel currents were recorded in an intracellular solution containing (mm): 140 KCl, 2 EGTA, 0.2 MgCl2, 5 Hepes (pH 7.4), and ATP (1 or 10 μm, indicated in the text) and an extracellular solution containing (mm): 10 KCl, 120 NaCl, 1.8 CaCl2, 0.48 MgCl2, 5.5 glucose, and 5 Hepes (pH 7.2), using the inside-out patch-clamp recording technique described in detail elsewhere (Fan & Makielski, 1999). MgCl2 was omitted from the intracellular solution in experiments using spermine. In some experiments, NaCl in the extracellular solution was replaced with 130 mm KCl. Currents were usually recorded at a membrane potential of 0 mV, filtered through a 1 kHz digital low-pass filter, and digitized at 5 kHz. For better display, the current traces shown in the figures were refiltered through a 200 Hz filter. Open channel activity was assessed and expressed as the average current in a 5–30 s window in which the current was also refiltered though a 200 Hz filter, under the assumption that the single channel current amplitude was constant under the same recording conditions. Single channel current amplitude was measured by a conventional unitary amplitude histogram method (Fan et al. 1993). For analysis of open channel kinetics, currents were filtered through a 0.6 or 1 kHz filter, and digitized at 10 or 20 kHz. The open time distribution was analysed by a conventional histogram analysis method. In order to limit possible bias on open time distributions, recordings initially showing three or fewer active channels were selected for analysis. A 50 % threshold criterion was used to detect open channel events using visual confirmation. After the events were detected, distributions of open times were plotted against a logarithmic time scale with event duration log-binned at a resolution of 20 bins per log unit. In patches containing more than one active channel, the mean open times of non-overlapping events and the mean open times of all events at first level were measured separately and the results compared to ensure that the bias did not invalidate the conclusion (Colquhoun & Sigworth, 1995). The reported values include overlapping events; however, such events were rare. The mean open times are reported without processing or correction. In our experiments we found that treatment with MTS reagents tended to destabilize the patch seal at depolarized and hyperpolarized voltages; this effect limited the use of larger pipettes. Under our experimental conditions, after the formation of the inside-out patches, 5–20 active channels were present in a patch sealed with a pipette of > 3 MΩ. Because the fluctuating currents from these channels could interfere with accurate measurement of macroscopic current levels, 12–30 successive individual current traces were recorded and averaged. Data averaging significantly reduced the effects of current fluctuation, except in the presence of spermine, which profoundly reduced current levels. The destabilizing action of MTS reagents also precluded averaging a larger number of traces.
Statistical analysis of data
Experimental data are presented as mean ± s.e.m. A one-way ANOVA test followed by a post hoc Student-Newman-Keuls test was used to assess the statistical differences between data groups. Student's t test was used wherever two groups of data were compared.
RESULTS
Differential effects of MTSET and MTS-TEAH on Kir6.2 currents
Multiple or single channel currents were recorded in the absence of ATP from membrane patches detached from COS-1 cells expressing Kir6.2Δ channels. Outward currents were observed under asymmetric K+ concentrations at 0 mV as described in the Methods section. ATP inhibited channel activity, with a half-concentration of inhibition (Ki) as summarized in Table 1. MTSET and MTS-TEAH were individually applied to the intracellular side of the membranes at concentrations between 0.6 and 2 mm for at least 3 min. MTSET irreversibly inhibited the activity of Kir6.2Δ channels in all experiments. In most patches, channel currents were completely inhibited within 2 min of inhibitor application. The channels remained inactive after withdrawal of MTSET from the perfusing solution (Fig. 1B). We noticed that channel activity, however low, remained in some patches after removal of MTSET, especially at the concentration of 0.6 mm. These residual currents had single channel unitary current amplitude indistinguishable from that measured before MTSET application (2.0 ± 0.1 pA, n = 7vs. 2.0 ± 0.1 pA, n = 5, P > 0.05). The inhibited channels, however, could be partially reactivated by application of 5 to 50 mm dithiothreitol (DTT). The current trace in Fig. 1D is a representative recording of these experiments. In contrast to MTSET, MTS-TEAH only partially reduced channel activity during its application. Channel activity was almost fully restored after withdrawal of MTS-TEAH, and could still be inhibited by ATP with a Ki similar to that measured before MTS-TEAH application (Table 1). Reversible current reduction during MTSET treatment of Kir2.1 channels has been reported previously. The effect is attributed to a reversible pore block by the positively charged reagents (Lu et al. 1999a). This mechanism appears applicable to Kir6.2 as well, because we observed a reduced reversible effect at −80 mV in symmetric K+. It should also be noted that run-down usually occurred, and its magnitude varied from experiment to experiment. Run-down, though not negligible, does not affect our major conclusion, because the magnitude of the effects in which we are interested is much greater. An example of a current recorded from a patch treated by MTS-TEAH is given in Fig. 1E. The statistical significance of these results, together with the data described below, is presented in Fig. 2C.
Table 1.
Effect of MTSET and MTS-TEAH treatment on ATP inhibition of Kir6.2δC, Kir6.2δ(C42V), and Kir6.2δ(C81V/C166V/C197V) channels
| Bg | Bg MTS-TEAH | Bg MTS-TEAH/MTSET | C42V | C42V MTS-TEAH | C42V MTSET | C81V/C166V/C197V | C81V/C166V/C197V MTS-TEAH | |
|---|---|---|---|---|---|---|---|---|
| Ki (μm) | 341 ± 25 | 321 ± 22 | 324 ± 50 | 384 ± 53 | 433 ± 55 | 263 ± 68 | 396 ± 28 | 442 ± 40 |
| n | 14 | 4 | 4 | 9 | 5 | 3 | 4 | 3 |
Bg, Kir6.2δ channels; C42V, Kir6.2δ(C42V) channels; C81V/C166V/C197V, Kir6.2δ(C81V/C166V/C197V) channels. Ki, concentration for half-inhibition obtained by fitting a Hill function to the data. n, number of experiments. An ANOVA test revealed no significant difference in mean values between the listed groups.
Figure 2. The effect of MTSET and MTS-TEAH on the current activity of Kir6.2Δ(C42V) mutant channels.

Examples showing the effect of MTSET (A) or MTS-TEAH (B) on Kir6.2Δ(C42V) channel currents. The currents were acquired from membranes containing multiple Kir6.2Δ(C42V) channels in an inside-out patch configuration, under the same conditions as those described in Fig. 1. MTSET and MTS-TEAH (both at 0.6 mm), and ATP were applied as indicated. C, comparison of the effects of MTSET and MTS-TEAH treatment on Kir6.2Δ (denoted by Bg, background) and mutant channels; C42V denotes Kir6.2Δ(C42V) mutant channels; C42V-C42 denotes Kir6.2Δ(C42V)-Kir6.2Δ dimer channels. The effect is expressed as a relative current, i.e. the percentage of the average current after the specified treatment to the current prior to the treatment. The data denoted by Bg MTS-TEAH/MTSET are relative currents measured before and after MTSET treatment, in patches pre-treated with MTS-TEAH. All data are presented as mean ± s.e.m. A one-way ANOVA test followed by a post hoc Student-Newman-Keuls test was used to establish statistical differences between all data groups. The comparisons were statistically significant (P < 0.05) except those between bracketed data groups. D, schematic illustration of the approximate location of ‘internal’ cysteine residues in the Kir6.2 channel topology.
The Kir6.2 channel has five ‘internal’ cysteines that are potentially accessible to hydrophilic reagents in the cytoplasm (Fig. 2D). As noted earlier, it was previously reported that inhibition of channel activity by the SH-reactive reagent pCMPS was abolished when C42 was mutated (Trapp et al. 1998b). Therefore, we examined whether C42 is also critical to the inhibitory effect of MTSET. As expected, MTSET failed to cause significant irreversible inhibition of Kir6.2Δ(C42V) current (Fig. 2A). Not surprisingly, MTS-TEAH treatment did not affect C42V channel activity (Fig. 2B). Given that MTSET cannot inhibit channels in which all four subunits contain the C42V mutation, we tested channels formed by fusion subunits, each containing a Kir6.2Δ subunit linked to a Kir6.2Δ(C42V) mutant subunit. According to previous studies (e.g. Loussouarn et al. 2001), the functional channels formed when these two fusion subunits dimerize contain two Kir6.2Δ subunits and two Kir6.2Δ(C42V) subunits. MTSET (0.6 mm) inhibited the dimer channels and Kir6.2Δ channels with similar potency (Fig. 1C). Comparisons of the effects of MTSET and MTS-TEAH on the background Kir6.2Δ currents, the dimer channel currents, and the Kir6.2Δ(C42V) currents are given in Fig. 2C.
Although MTS-TEAH did not cause irreversible inhibition, we noticed that MTS-TEAH treatment induced a persistent increase in mean open time of single channel current of Kir6.2Δ channels. The mean open time of untreated Kir6.2Δ current recorded at 0 mV was 2.7 ± 0.3 ms which was prolonged to 5.2 ± 0.4 ms by MTS-TEAH treatment (0.6 mm for 3–5 min). Interestingly, mutation of C42V resulted in a significant shortening of the mean open time compared to that of Kir6.2Δ channels. Neither MTS-TEAH nor MTSET produced any detectable change in the mean open time of Kir6.2Δ(C42V) single channel current. Examples of the single channel currents and histogram analysis of open times are plotted in Figs 3A and B. Statistical results are summarized in Fig. 3C. Prolongation of mean open times was also observed in Kir6.2Δ(C166S) mutant channels (25.0 ms ± 2.0, n = 7vs. 40.7 ± 9.9 ms, n = 3, P < 0.05, before and after treatment, respectively), in Kir6.2Δ(C344S) mutant channels (3.7 ± 0.4 ms, n = 5, vs. 5.1 ± 0.4 ms, n = 3, P < 0.05), and in Kir6.2Δ(C81V/C166V/C197V) mutant channels (2.1 ± 0.2 ms vs. 3.8 ± 0.4 ms, n = 4, P < 0.05). Interestingly, mean open time (2.5 ± 0.4 ms, n = 3) of Kir6.2Δ(C42V)-Kir6.2Δ dimer channels was similar to that of Kir6.2Δ channels (2.7 ± 0.3 ms, n = 10), but differed from that of Kir6.2Δ(C42V) channels (1.4 ± 0.1 ms, n = 10, P < 0.05). MTS-TEAH treatment prolonged mean open time (4.0 ± 0.2 ms, n = 3, P < 0.05).
Figure 3. The effect of intracellular MTS-TEAH treatment on open channel kinetics of Kir6.2Δ and Kir6.2Δ(C42V) channels.
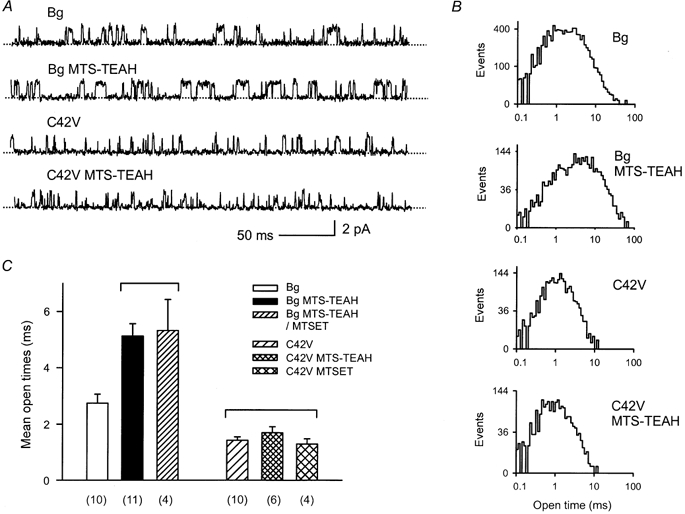
A, single channel currents recorded from inside-out patch-clamped membranes before and after MTS-TEAH treatment. Bg, background Kir6.2Δ channels; C42V, Kir6.2Δ(C42V) mutant channels. The current traces marked by MTS-TEAH were recorded after the intracellular side of the membrane was treated with 0.6 mm MTS-TEAH for > 3 min. B, histograms of open time distribution measured from currents, parts of which are shown in A. The mean open times for these histograms are 2.3, 5.5, 1.4 and 1.3 ms for Bg, Bg after MTS-TEAH, C42V and C42V after MTS-TEAH, respectively. The histograms were analysed from data recorded in a single patch. C, comparison of the effect of MTSET and MTS-TEAH treatment on mean open times. The mean open times were measured in the absence of the indicated reagents. In the case of Bg MTS-TEAH/MTSET, currents taken for analysis were recorded after MTS-TEAH (0.6 mm) pretreatment followed by MTSET (0.6 mm) treatment. Current events were filtered though a low-pass filter with a cut-off frequency of 600 Hz and subjected to no additional adjustment. A one-way ANOVA test followed by a post hoc Student-Newman-Keuls test was used to examine the statistical differences between all data groups. The number of analysed patches for each data set is indicated on the bottom of the plot. The comparisons were statistically significant (P < 0.05) except those between the bracketed data groups.
MTS-TEAH pretreatment prevents the inhibitory effect of MTSET on Kir6.2 channels
Results from the above experiments strongly suggest that Kir6.2Δ is subject to MTS-TEAH modification, and that C42 is the likely site of this modification. If this were true, we predict that pretreatment by MTS-TEAH would prevent further modification of the channel by MTSET. Indeed, in experiments in which MTS-TEAH and MTSET were applied sequentially, MTSET no longer produced any irreversible inhibition of Kir6.2Δ channel current after MTS-TEAH treatment. Figure 4A illustrates a current recording from these experiments. Neither treatment produced any significant change in the current inhibition by ATP (Fig. 4A and Table 1). The effects of double treatment on the overall activity of Kir6.2Δ current are summarized in Fig. 2C together with data from the experiments described earlier. Similarly, MTSET treatment following MTS-TEAH pretreatment failed to produce any noticeable change in open times that were already prolonged (Fig. 3C). When experiments were repeated in the Kir6.2Δ(C81V/C166V/C197V) mutant channels, MTSET inhibited current, but failed to do so after MTS-TEAH pretreatment (Figs 4B–D). Qualitatively similar observations were also made in Kir6.2Δ(C166S) mutant channels, Kir6.2Δ(C344S) mutant channels, and Kir6.2Δ(C42V)-Kir6.2Δ dimer channels. The actual current recordings of these experiments resemble those shown in Fig. 4.
Figure 4. MTS-TEAH pretreatment protects against irreversible channel inhibition by MTSET treatment.

A, record of multiple Kir6.2Δ channel currents. MTSET (0.6 mm), MTS-TEAH (0.6 mm), and ATP were applied as indicated. The lower traces are expanded displays. B, pretreatment with MTS-TEAH prevents the inhibitory effect of MTSET on Kir6.2Δ(C81V/C166V/C197V) channels. The current traces were recorded from the same patch membrane but isolated by ∼1 min for each gap. C, inhibition of Kir6.2Δ(C81V/C166V/C197V) currents by MTSET (0.6 mm). D, summary of the effect of MTSET, and the protective effect of MTS-TEAH pretreatment against MTSET inhibition of Kir6.2Δ(C81V/C166V/C197V) currents. The comparisons were statistically significant (P < 0.05) except that between the two bracketed data groups.
MTS-TEAH does not interfere with voltage-dependent block of Kir6.2Δ channels by spermine
The fact that modification of C42 by MTS-TEAH does not inhibit the channel provides a unique opportunity to probe whether the C42 residue faces the pore. Polyamines are known to insert into the pore of Kir channels and produce a voltage-dependent block. Modification of a residue in the pore may hinder the access of polyamines and alter blocking. To test this possibility, we examined whether MTS-TEAH treatment affects channel block induced by spermine. Spermine at either 20 or 100 μm was applied to the intracellular side of membrane patches that were voltage-clamped by a ramping protocol. Intracellular pH was set at 8.0 to enhance the blocking effect of spermine (Baukrowitz et al. 1999). We confirmed the effect of pH on Kir6.2Δ channels by varying pH between 7.3 and 8.0. Both potency and the voltage dependence of the block were much stronger at pH 8.0 (data not shown). In these experiments, macroscopic currents averaged from more than 12 successive traces of multiple channel currents were plotted against the ramping voltages (Fig. 5B, also see Methods section for details). Spermine blocked the macroscopic currents in a predicted voltage-dependent pattern both before and after MTS-TEAH treatment. Figure 5B summarizes our results showing that there is no detectable change in spermine block caused by MTS-TEAH treatment.
Figure 5. Voltage-dependent spermine block of Kir6.2Δ channel currents before and after MTS-TEAH treatment.

A, multiple channel currents responding to a constant voltage ramp from −80 to +100 mV. The currents were recorded in the absence and presence of spermine (20 or 100 μm), respectively, from two inside-out patches containing Kir6.2Δ channels, in asymmetrical K+ concentrations (140 mm at the intracellular side and 10 mm at the extracellular side) at an intracellular pH of 8.0. Three representative current traces are plotted for each condition. MTS-TEAH, these current traces were recorded after 3 min of MTS-TEAH treatment, either in the absence or presence of spermine. B, current-voltage (I-V) relations plotted from the experiment for which representative current traces are displayed in A. The I-V curves were obtained after averaging 12–21 current traces. SPM, spermine; MTS-TEAH, after MTS-TEAH treatment. The I-V curves are normalized to the maximal values in the absence of spermine. The bar plots summarize spermine block of Kir6.2Δ channels before (□, n = 5) and after (▪, n = 5) MTS-TEAH treatment. Averaged currents at the indicated voltages are presented as mean ± s.e.m. No statistically significant difference was detected between untreated and treated groups.
Next, the effect of MTS-TEAH on the blocking kinetics of spermine was analysed at the single channel level. Neither 100 μm spermine nor MTS-TEAH treatment affected the single channel unitary current amplitude at +100 mV, at which 100 μm spermine at pH 8.0 produced ∼80 % block of the macroscopic current (Fig. 6A; note that some smaller current amplitudes were caused by low-pass filtering and were present both in the absence and presence of spermine). Under the same conditions, however, spermine significantly reduced the open times of Kir6.2Δ currents (Fig. 6B) and thus shortened the mean open time (Fig. 6C). As the open time distribution of Kir6.2Δ currents can be fitted to a single exponential component, the channel appears to have one open state (Drain et al. 1998; Fan & Makielski, 1999) (Fig. 6D). It should also be noted that we fitted the open time distributions with single-exponential functions and converted the time constants of the exponentials to apparent blocking rate constants. A blocking rate constant obtained in this way is generally larger, but its response to MTS-TEAH treatment was still statistically insignificant.
Figure 6. Spermine blocking rate constant in control and MTS-TEAH-treated Kir6.2Δ channels.

A, the blocking effect of 100 μm spermine on single channel currents at +100 mV, before and after MTS-TEAH treatment. Bg, background Kir6.2Δ channels; SPM, in the presence of 100 μm spermine; MTS-TEAH, after treatment with MTS-TEAH; MTS-TEAH/SPM, in the presence of 100 μm spermine after MTS-TEAH treatment. The current traces were recorded from a patch that showed a single active channel after the initial recording started. The membrane patch was held at +100 mV. The intracellular solutions were maintained at pH 8.0 during the recording. MTS-TEAH treatment was performed by applying a 1 mm solution to the intracellular side of the patch for 3 min at pH 7.4. B, corresponding histograms of open time distribution. The mean open times for these histograms are 2.9, 5.2, 1.3 and 1.5 ms for Bg, SPM, MTS-TEAH, and MTS-TEAH/SPM, respectively. C, the effects of MTS-TEAH treatment on mean open times of Kir6.2Δ channel currents in the absence and presence of SPM. Currents were filtered though a low-pass filter at a cut-off frequency of 1 kHz and subject to no additional adjustment. A total of four experiments were included in each data set. D, apparent blocking rate constant of spermine at membrane potential of +100 mV before and after MTS-TEAH treatment. The blocking rate constants were converted from the mean open times given in C according to the general scheme shown in the inset, which has the relation: kb = 1/τ0′ − 1/τ0, where kb is the blocking rate constant, τ0′ is the mean open time in the presence of spermine, and τ0 is the mean open time in the absence of spermine. In the scheme, O is the open state of the channel, C is an intrinsic closed state, and B represents a state blocked by spermine. C and B may connect to other closed or blocked states. No statistical difference was found between the mean values of the two data groups.
DISCUSSION
Modification of Kir6.2 by MTS reagents
Modification of proteins by SH-specific reagents is a proven, useful approach that can provide structural information and reveal functional roles of target cysteine residues. When applied intracellularly, a few SH-reactive reagents reportedly inhibit the activity of KATP channels in β-cells and cardiac myocytes (Weik & Neumcke, 1989; Lee et al. 1994; Coetzee et al. 1995) as well as cloned Kir6.2/SUR1 and Kir6.2 (Kir6.2ΔC26) channels (Trapp et al. 1998b). MTS reagents used in this study are highly SH-specific and reactive. MTSET has effects typical of the other SH-reactive reagents reported previously, in that it irreversibly inhibits Kir6.2Δ channels in a manner that can be reversed by reducing reagents such as DTT. A major finding of the present study is that MTS-TEAH modification does not inhibit Kir6.2Δ channels. We demonstrate that MTS-TEAH modifies cysteines by showing that (1) MTSET has no effect when applied after MTS-TEAH pretreatment, and (2) MTS-TEAH persistently prolongs channel open times.
Does MTS-TEAH react with C42?
Since the effect of MTS-TEAH differs significantly from other SH-specific reagents, special consideration must be paid to whether it modifies the channel and, if so, whether this effect is caused by modification of C42. The larger size of MTS-TEAH raises the possibility that this reagent may not be able to access the reactive site of C42 as MTSET does. Indeed, the possibility of limited access to C42 has been suggested by ATP protection against the inhibitory effect of SH-reactive reagents (Weik & Neumcke, 1989; Trapp et al. 1998b) although in our experiments we were unable to see this protective effect of ATP (up to 50 mm) against MTSET modification of Kir6.2Δ channels (Z. Fan & Y. Cui, unpublished observation). Protection of channels from MTSET inhibition by MTS-TEAH pretreatment can be interpreted as indicating that both reagents target the same thiol group and therefore modification of the target by one reagent prevents the other's reaction. This protection, however, might also be attributed to limited access to C42 caused by an allosteric effect of MTS-TEAH modification of a separate residue of Kir6.2, or even other adjacent proteins. The possibility that MTS-TEAH modifies a cysteine on Kir6.2 other than C42 can be rejected with reasonable certainty because the protection was also observed in the Kir6.2Δ(C81V/C166V/C197V) and Kir6.2Δ(C344S) mutants. Collateral effects brought about via modification of other proteins also seem unlikely because MTS-TEAH did not prolong the open times of the Kir6.2Δ(C42V) mutant. Thus, we conclude that MTS-TEAH and MTSET competitively react with the same thiol group of C42.
Is C42 a pore-lining residue?
The simplest explanation for irreversible blockage of Kir channels by MTSET is that the reagent reacts with residue C42 which projects into the pore lumen, thus physically occluding the channel. However, several types of evidence suggest that this simple possibility cannot be correct. If MTSET were physically blocking the channel, the larger MTS-TEAH would be expected to do the same, yet our results show that it does not affect channel activity. To test the possibility that MTS-TEAH reacts with a pore-lining residue but is somehow ‘leaky’, we used the inner pore blocker, spermine. Spermine blocks Kir channels by entering deeply into the pore from the cytoplasmic side (Fakler et al. 1994; Ficker et al. 1994; Lopatin et al. 1994; Lopatin et al. 1995). In Kir6.2, spermine can interact with residue 160 of M2 (Shyng et al. 1997), a residue that projects into the inner cavity when aligned in the KscA channel structure (Loussouarn et al. 2000). As a residue outside the transmembrane segments, C42 is unlikely to reside in the inner cavity or the tunnel within the membrane field. If it were in the pore, this residue would most likely face the cytoplasmic vestibule where spermine must pass. Although MTS-TEAH has a larger head group than MTSET, it also has an extended hexyl chain ∼1.2 Å narrower than the diameter of the MTSET head. Although this size difference might cause MTS-TEAH to form a leaky occlusion, this possibility seems unlikely, because if we assume that two subunits are modified, a spermine molecule (diameter: ∼3.5 Å) would still be too large to pass freely through the channel. At this steeply narrowed pore region, access of spermine should be hindered significantly enough to alter the blockage and/or the blocking kinetics. Even in a wider pore region where change of pore size might have little effect, spermine accessibility could also be affected by the altered electrostatic profile in the pore due to the presence of positively charged MTS-TEAH derivatives. The sensitivity of spermine block to changes in charge due the putative pore-forming residues has been demonstrated experimentally (Yang et al. 1995; Kubo & Murata, 2001). In Kir6.2 channels, mutational study suggests that positively charged H216 at acidic pH inhibits spermine entry into the pore (Baukrowitz et al. 1999). In addition, modification of putative pore-facing residues causes graded changes in the unitary amplitude (Loussouarn et al. 2001). Change of unitary amplitude has not been observed in the patches treated with MTSET; we also showed that residual currents after MTSET treatment had the same unitary amplitude as untreated channels. In summary, because our results show that spermine block is not altered by MTS-TEAH treatment, we propose that the modified C42 is not located in the pore lumen critical to the channel permeation, but instead faces the cytoplasm, or a vestibule section that is wide enough to allow spermine to pass freely even after modification by MTS-TEAH.
Mechanism of inhibition by SH-specific reagents
Either of the two possibilities described above permits us to postulate that MTS modification of C42 causes channel inhibition not by physical occlusion, but rather by an allosteric effect. Although allosteric effects are often complex and generally difficult to characterize, the results of this study provide some clues indicating that the modification may affect the channel gating process. We showed that Kir6.2Δ(C42V) channels open more briefly than Kir6.2Δ channels, and that modification of C42 by MTS-TEAH prolongs the open times, although C42V mutation seems unable to affect the gating when only two of four subunits are mutated. It is also interesting that prolongation of the open channel durations by MTS-TEAH treatment is observed in various mutants, including Kir6.2Δ(C166S) which already exhibits long open times (Trapp et al. 1998a). Furthermore, the finding that at most two MTSET molecules are needed to inhibit the channel is in agreement with a preliminary report showing that two subunits were sufficient to gate the channel normally (Li et al. 1999). These observations imply that C42 may be part of, or closely associated with, the intrinsic gating process of the channel. This is consistent with other studies that have suggested that the N-terminus is involved in channel gating (Babenko et al. 1999; Koster et al. 1999; Reimann et al. 1999). It remains unclear how residues of the N-terminus could affect the gating process. It is not yet understood how modification of C42 causes the channel to close, or why a relatively small variation in size and shape of a modifying reagent added to C42 results in dramatically opposed effects on channel gating (i.e. inhibition vs. longer openings). Elucidation of these mechanisms may provide perspective for future studies of the gating mechanisms of KATP channels.
Acknowledgments
This work was supported by the following grants: National Institutes of Health Grants HL58133 and GM61943, and a Grant-in-Aid from the American Heart Association Southeast Affiliation.
REFERENCES
- Akabas MH, Kaufmann C, Archdeacon P, Karlin A. Identification of acetylcholine receptor channel-lining residues in the entire M2 segment of the alpha subunit. Neuron. 1994;13:919–927. doi: 10.1016/0896-6273(94)90257-7. [DOI] [PubMed] [Google Scholar]
- Akabas MH, Stauffer DA, Xu M, Karlin A. Acetylcholine receptor channel structure probed in cysteine-substitution mutants. Science. 1992;258:307–310. doi: 10.1126/science.1384130. [DOI] [PubMed] [Google Scholar]
- Babenko AP, Gonzalez G, Bryan J. The N-terminus of KIR6. 2 limits spontaneous bursting and modulates the ATP-inhibition of KATP channels. Biochemical and Biophysical Research Communications. 1999;255:231–238. doi: 10.1006/bbrc.1999.0172. [DOI] [PubMed] [Google Scholar]
- Baukrowitz T, Tucker SJ, Schulte U, Benndorf K, Ruppersberg JP, Fakler B. Inward rectification in KATP channels: a pH switch in the pore. EMBO Journal. 1999;18:847–853. doi: 10.1093/emboj/18.4.847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coetzee WA, Nakamura TY, Faivre JF. Effects of thiol-modifying agents on KATP channels in guinea pig ventricular cells. American Journal of Physiology. 1995;269:H1625–1633. doi: 10.1152/ajpheart.1995.269.5.H1625. [DOI] [PubMed] [Google Scholar]
- Colquhoun D, Sigworth FJ. Fitting and statistical analysis of single channel records. In: Sakmann B, Neher E, editors. Single Channel Recording. New York, London: Plenum Press; 1995. pp. 483–587. [Google Scholar]
- Drain P, Li L, Wang J. KATP channel inhibition by ATP requires distinct functional domains of the cytoplasmic C terminus of the pore-forming subunit. Proceedings of the National Academy of Sciences of the USA. 1998;95:13953–13958. doi: 10.1073/pnas.95.23.13953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fakler B, Brandle U, Bond C, Glowatzki E, Konig C, Adelman JP, Zenner HP, Ruppersberg JP. A structural determinant of differential sensitivity of cloned inward rectifier K+ channels to intracellular spermine. FEBS Letters. 1994;356:199–203. doi: 10.1016/0014-5793(94)01258-x. [DOI] [PubMed] [Google Scholar]
- Fan Z, Furukawa T, Sawanobori T, Makielski JC, Hiraoka M. Cytoplasmic acidosis induces multiple conductance states in ATP-sensitive potassium channels of cardiac myocytes. Journal of Membrane Biology. 1993;136:169–179. doi: 10.1007/BF02505761. [DOI] [PubMed] [Google Scholar]
- Fan Z, Makielski JC. Phosphoinositides decrease ATP sensitivity of the cardiac ATP-sensitive K+ channel. A molecular probe for the mechanism of ATP-sensitive inhibition. Journal of General Physiology. 1999;114:251–269. doi: 10.1085/jgp.114.2.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ficker E, Taglialatela M, Wible BA, Henley CM, Brown AM. Spermine and spermidine as gating molecules for inward rectifier K+ channels. Science. 1994;266:1068–1072. doi: 10.1126/science.7973666. [DOI] [PubMed] [Google Scholar]
- Inagaki N, Gonoi T, Clement JP, Namba N, Inazawa J, Gonzalez G, Aguilar-Bryan L, Seino S, Bryan J. Reconstitution of IKATP : an inward rectifier subunit plus the sulfonylurea receptor. Science. 1995;270:1166–1170. doi: 10.1126/science.270.5239.1166. [DOI] [PubMed] [Google Scholar]
- Koster JC, Sha Q, Shyng S, Nichols CG. ATP inhibition of KATP channels: control of nucleotide sensitivity by the N-terminal domain of the Kir6. 2 subunit. Journal of Physiology. 1999;515:19–30. doi: 10.1111/j.1469-7793.1999.019ad.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubo Y, Murata Y. Control of rectification and permeation by two distinct sites after the second transmembrane region in Kir2. 1 K+ channel. Journal of Physiology. 2001;531:645–660. doi: 10.1111/j.1469-7793.2001.0645h.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K, Ozanne SE, Hales CN, Ashford ML. Effects of chemical modification of amino and sulfhydryl groups on KATP channel function and sulfonylurea binding in CRI-G1 insulin-secreting cells. Journal of Membrane Biology. 1994;139:167–181. doi: 10.1007/BF00232621. [DOI] [PubMed] [Google Scholar]
- Li L, Wang J, Drain P. Multiple independent components and subunit interactions in the ATP-dependent inhibition gating of the KATP channel. Biophysical Journal. 1999;76:A72. [Google Scholar]
- Lopatin AN, Makhina EN, Nichols CG. Potassium channel block by cytoplasmic polyamines as the mechanism of intrinsic rectification. Nature. 1994;372:366–369. doi: 10.1038/372366a0. [DOI] [PubMed] [Google Scholar]
- Lopatin AN, Makhina EN, Nichols CG. The mechanism of inward rectification of potassium channels: “long-pore plugging” by cytoplasmic polyamines. Journal of General Physiology. 1995;106:923–955. doi: 10.1085/jgp.106.5.923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loussouarn G, Makhina EN, Rose T, Nichols C G. Structure and dynamics of the pore of inwardly rectifying KATP channels. Journal of Biological Chemistry. 2000;275:1137–1144. doi: 10.1074/jbc.275.2.1137. [DOI] [PubMed] [Google Scholar]
- Loussouarn G, Phillips LR, Masia R, Rose T, Nichols CG. Flexibility of the Kir6. 2 inward rectifier K+ channel pore. Proceedings of the National Academy of Sciences of the USA. 2001;98:4227–4232. doi: 10.1073/pnas.061452698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu T, Nguyen B, Zhang X, Yang J. Architecture of a K+ channel inner pore revealed by stoichiometric covalent modification. Neuron. 1999a;22:571–580. doi: 10.1016/s0896-6273(00)80711-4. [DOI] [PubMed] [Google Scholar]
- Lu T, Zhu YG, Yang J. Cytoplasmic amino and carboxyl domains form a wide intracellular vestibule in an inwardly rectifying potassium channel. Proceedings of the National Academy of Sciences of the USA. 1999b;96:9926–9931. doi: 10.1073/pnas.96.17.9926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radeke CM, Conti LR, Vandenberg CA. Inward rectifier potassium channel Kir 2. 3 is inhibited by internal sulfhydryl modification. NeuroReport. 1999;10:3277–3282. doi: 10.1097/00001756-199911080-00006. [DOI] [PubMed] [Google Scholar]
- Reimann F, Tucker SJ, Proks P, Ashcroft FM. Involvement of the N-terminus of Kir6. 2 in coupling to the sulphonylurea receptor. Journal of Physiology. 1999;518:325–336. doi: 10.1111/j.1469-7793.1999.0325p.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulte U, Hahn H, Wiesinger H, Ruppersberg JP, Fakler B. pH-dependent gating of ROMK (Kir1. 1) channels involves conformational changes in both N and C termini. Journal of Biological Chemistry. 1998;273:34575–34579. doi: 10.1074/jbc.273.51.34575. [DOI] [PubMed] [Google Scholar]
- Shyng S, Ferrigni T, Nichols CG. Control of rectification and gating of cloned KATP channels by the Kir6. 2 subunit. Journal of General Physiology. 1997;110:141–153. doi: 10.1085/jgp.110.2.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapp S, Proks P, Tucker SJ, Ashcroft FM. Molecular analysis of ATP-sensitive K channel gating and implications for channel inhibition by ATP. Journal of General Physiology. 1998a;112:333–349. doi: 10.1085/jgp.112.3.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapp S, Tucker SJ, Ashcroft FM. Mechanism of ATP-sensitive K channel inhibition by sulfhydryl modification. Journal of General Physiology. 1998b;112:325–332. doi: 10.1085/jgp.112.3.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker SJ, Gribble FM, Zhao C, Trapp S, Ashcroft FM. Truncation of Kir6. 2 produces ATP-sensitive K+ channels in the absence of the sulphonylurea receptor. Nature. 1997;387:179–183. doi: 10.1038/387179a0. [DOI] [PubMed] [Google Scholar]
- Weik R, Neumcke B. ATP-sensitive potassium channels in adult mouse skeletal muscle: characterization of the ATP-binding site. Journal of Membrane Biology. 1989;110:217–226. doi: 10.1007/BF01869152. [DOI] [PubMed] [Google Scholar]
- Yang J, Jan YN, Jan LY. Control of rectification and permeation by residues in two distinct domains in an inward rectifier K+ channel. Neuron. 1995;14:1047–1054. doi: 10.1016/0896-6273(95)90343-7. [DOI] [PubMed] [Google Scholar]