

Abstract
Large conductance, Ca2+-activated K+ (maxi-K) channel activity was recorded in excised, inside-out patches from HEK 293 cells stably co-expressing the α- and β-subunits of human brain maxi-K channels. At +50 mV, and in the presence of 300 nm , single channel activity was acutely and reversibly suppressed upon reducing PO2 from 150 to > 40 mmHg by over 30 %. The hypoxia-evoked reduction in current was due predominantly to suppression in NPo, although a minor component was attributable to reduced unitary conductance of 8–12 %. Hypoxia caused an approximate doubling of the time constant for activation but was without effect on deactivation. At lower levels of (30 and 100 nm), hypoxic inhibition did not reach significance. In contrast, 300 nm and 1 μm both sustained significant hypoxic suppression of activity over the entire activating voltage range. At these two levels, hypoxia evoked a positive shift in the activating voltage (by ∼10 mV at 300 nm and ∼25 mV at 1 μm). At saturating [Ca2+]i (100 μm), hypoxic inhibition was absent. Distinguishing between hypoxia-evoked changes in voltage- and/or -sensitivity was achieved by evoking maximal channel activity using high depolarising potentials (up to +200 mV) in the presence of 300 nm or 100 μm or in its virtual absence (> 1 nm). Under these experimental conditions, hypoxia caused significant channel inhibition only in the presence of 300 nm . Thus, since regulation was observed in excised patches, maxi-K channel inhibition by hypoxia does not require soluble intracellular components and, mechanistically, is voltage independent and sensitive.
Acute modulation of ion channel activity by oxygen is crucial to the mechanisms which underlie chemosensing in carotid body (Peers, 1990), neuroepithelial body (Cutz & Jackson, 1999; Peers & Kemp, 2001) and vascular smooth muscle (Coppock et al. 2001) and is believed to play a significant role in modulation of excitability in several cellular components of the mammalian nervous system (Vergara et al. 1998). Crucial to these adaptive physiological and cellular responses is the acute inhibition of K+ channels by hypoxia. Although O2-sensitive tissues express a wide variety of channel types, central to the cellular mechanism of O2 sensing in many tissues is hypoxic suppression of large conductance Ca2+-activated K+ channels (maxi-K, BKCa or Slo channels). Thus, hypoxic inhibition of maxi-K channels has been demonstrated in carotid body (Peers et al. 1990; Riesco-Fagundo et al. 2001), pulmonary smooth muscle (Cornfield et al. 1996), chromaffin cells (Thompson & Nurse, 1998) and central neurones (Liu et al. 1999). Although their contribution to carotid body, chromaffin cell and central neuronal function is well supported, some controversy still surrounds their involvement in pulmonary vasoconstriction (Coppock et al. 2001). Thus, there is good evidence for both delayed rectifier (Tristani-Firouzi et al. 1996) and tandem P domain K+ channels in the response (Gurney et al. 2002). The later observation is fully supported by our recent report of O2 sensitivity of the tandem P domain channel, hTASK1, in a recombinant mammalian system similar to that employed in the present study (Lewis et al. 2001).
Although it has been suggested that the mechanism of hypoxic regulation of K+ channels in general, and maxi-K channels in particular, may involve the O2-dependent modulation of cellular redox potential, the nature of the sensor is still unclear and the mechanism whereby decreased PO2 evokes channel inhibition in different tissues remains controversial. Indeed, there are varying reports which have suggested involvement of either cytosolic factors (e.g. Jiang & Haddad, 1994; Wyatt & Peers, 1995) or more direct channel modulation (e.g. Liu et al. 1999; Riesco-Fagundo et al. 2001). In favour of potential regulation by cellular redox state are recent data demonstrating activation by oxidising agents in a recombinant cellular system (Tang et al. 2001) and inhibition by reduced glutathione in rat hippocampal neurones (Soh et al. 2001) of these channels. However, to date, direct demonstration of hypoxia-induced inhibition of human recombinant maxi-K channels has been lacking.
In the present study, we have employed human embryonic kidney (HEK 293) cells stably expressing both the α- and β-subunits of human maxi-K and have examined the O2 sensitivity of these recombinant K+ channels at the single channel level. Our findings demonstrate directly that human brain αβ-maxi-K is an O2-sensitive K+ channel and support strongly the notion that hypoxic inhibition of this channel occurs primarily via a reduction in Ca2+-sensitivity with an additional, minor reduction in unitary conductance.
METHODS
Cell culture
HEK 293 cells which express human brain αβ-maxi-K channels (Ahring et al. 1997) were maintained in Earle's minimal essential medium (containing l-glutamine) supplemented with 10 % fetal calf serum, 1 % antibiotic antimycotic, 1 % non-essential amino acids and 0.2 % gentamicin (all purchased from Gibco BRL, Paisley, Strathclyde, UK) in a humidified incubator gassed with 5 % CO2-95 % air. Cells were passaged every 7 days in a ratio of 1:25 using Ca2+- and Mg2+-free phosphate-buffered saline (Gibco BRL, Paisley, Strathclyde, UK). The co-expressed α- and β-subunits were KCNMA1 (Genbank accession number U11717) and KCNMB1 (Genbank accession number U42600), respectively.
Patch-clamp recording
Cells were grown for 24 h on glass coverslips before being transferred to a continuously perfused (5 ml min−1) recording chamber (volume ca 200 μl) mounted on the stage of an inverted microscope. For inside-out, excised patch clamp recordings, the standard perfusate was composed of (mm): 10 NaCl, 117 KCl, 2 MgCl2, 11 Hepes, (pH 7.2) with Ca2+ buffered to 30 nm, 100 nm, 300 nm, 1 μm and 100 μm using EGTA and CaCl2 in appropriate ratios. The Na+-rich pipette solution was composed of (mm): 135 NaCl, 5 KCl, 1.2 MgCl2, 5 Hepes, 2.5 CaCl2, 10 d-glucose (pH 7.4). The K+-rich pipette solution was composed of (mm): 10 NaCl, 117 KCl, 2 MgCl2, 2.0 CaCl2 (pH 7.2). When filled with these solutions, pipettes were of resistance 7–14 MΩ. Unless otherwise stated, all experiments were conducted in asymmetrical solutions. Hypoxic solutions were bubbled with N2 (g) for at least 30 min prior to perfusion of cells, which produced no shift in pH. PO2 was measured (at the cell) using a polarised carbon fibre electrode (Mojet et al. 1997) and found to be in the range 30–40 mmHg. Normoxic solutions were equilibrated with room air. All K+ currents were recorded at a bath temperature of 22 ± 0.5 °C. Current recordings were made using an Axopatch 200A amplifier and Digidata 1320 A/D interface (Axon Instruments, Foster City, CA, USA). To evoke macro-patch K+ currents, one of four protocols was employed. (1) Standard ramp, −70 mV holding potential, voltage ramps from −100 to +60 mV (2000 ms, 0.1 Hz). (2) Extended ramp, −70 mV holding potential, voltage ramps from −100 to +200 mV (2000 ms, 0.1 Hz). (3) Activating step, −70 mV holding potential, voltage steps from holding to between 0 and +50 mV in 10 mV increments (100 ms, 0.1 Hz). (4) Deactivating step, −70 mV holding potential, voltage step from holding to 130 mV (50 ms) followed by steps in the range −70 mV to −130 mV in 10 mV increments (1000 ms, 0.1 Hz)
All patches from stably expressing cells contained at least six maxi-K channels. Wild-type HEK 293 cells do not express maxi-K channels, see Fig. 4E, inset. Mean wild-type currents, recorded from inside-out patches with [Ca2+]i buffered to 100 μm, are shown in Fig. 4E.
Figure 4. and voltage dependence of hypoxic channel inhibition.
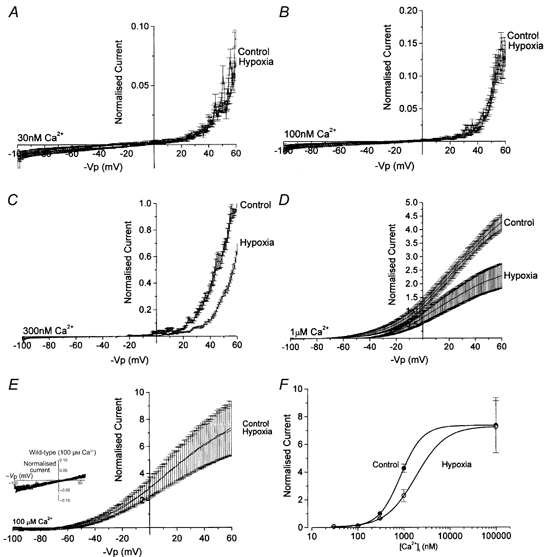
Mean (± s.e.m) normalised, standard ramp-evoked currents in normoxia (control) and hypoxia at 30 nm(A), 100 nm (B), 300 nm (C), 1 μm (D) and 100 μm (E) [Ca2+]i. F, the [Ca2+]i of hypoxia-evoked channel inhibition by plotting mean normalised currents in normoxia and hypoxia against [Ca2+]i. Also shown in E, inset, are mean ramp-evoked currents in wild-type HEK 293 cells in normoxia and at 100 μm [Ca2+]i; n = 7. Note the scale difference in the inset. F, dependence of currents (measured at +50 mV) on [Ca2+]i in normoxia (•) and hypoxia (○), as indicated.
Data handling
Data analyses were performed using the pCLAMP 8.0 suite of software. The product of channel number and open state probability (NPo) at any voltage was calculated from 30 s sections of current recording using the following equation, where I is the mean current and i is the single channel amplitude at any voltage (determined from all-points histograms):
NPo values were normalised to the values during the normoxic perfusion period (control) and NPo data are reported as means ± s.e.m. values. Statistical comparisons were made using Student's paired or unpaired t tests, as appropriate. All-points histograms were generated using Fetchan 6.03 and single channel amplitudes were calculated using PStat 6.03 by iterative Gaussian fitting protocols. Means (±s.e.m.) and subtracted current-voltage relationships were generated using Microsoft Excel and are normalised to the current amplitude obtained at +50 mV in the presence of 300 nm [Ca2+]i. Activation/deactivation kinetics were determined from iterative fitting using a standard exponential routine (ClampFit 8.0.2). Concentration-response curves for Ca2+ were fitted to the Hill equation.
RESULTS
HEK 293 cells stably expressed recombinant human maxi-K channels. Figure 1 shows exemplar currents recorded at +50 mV in the presence of 300 nm [Ca2+]i from an inside-out, excised patch containing at least six maxi-K channels (although transitions to the fifth and sixth levels represented, in this example, merely 0.44 % of the total observable events and so do not appear in either the expanded traces or the all-points histograms). The continuous current recording shown in Fig. 1A clearly demonstrates that hypoxia (30–40 mmHg) evoked a dramatic and reversible reduction in patch activity. All patches demonstrated similar hypoxic channel inhibition (Fig. 1B); mean normalised NPo was reduced significantly (P < 0.01) by 32.0 ± 6.5 % in hypoxia (n = 9). Those patches which remained intact for sufficient time demonstrated recovery when returned to normoxia. Traces shown in Fig. 1C, D and E (200 ms, arrows on Fig. 1A) and the all-points histograms (derived from 30 s sections of trace, horizontal bars on Fig. 1A) exemplify the reduction in channel activity evoked by hypoxia. These data demonstrate clearly that hypoxic inhibition of maxi-K currents does not have an absolute requirement for soluble cytosolic factors.
Figure 1. Hypoxic suppression of single channel activity.
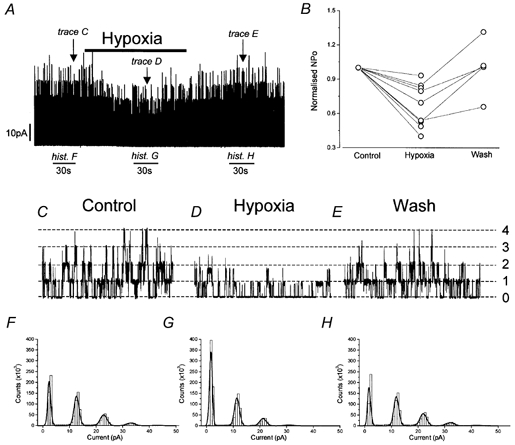
A, exemplar continuous current recording from an inside-out, excised membrane patch bathed in physiological solutions with [Ca2+]i buffered to 300 nm and held at +50 mV. During the time indicated by the horizontal bar above the trace, perfusate PO2 was reduced from 150 to 30–40 mmHg. B, normalised NPo values from nine separate cells under the conditions described in A during normoxia (control), hypoxia and following re-establishment of normoxia (wash). C, D and E, single channel currents on an extended time-base (200 ms per trace) from the recording shown in A at the arrows in normoxia (C), hypoxia (D) and following re-introduction of normoxic perfusate (E). F, G and H, all-points histograms generated from 30 s segments (shown by horizontal bars below the recording of the trace shown in A) in normoxia (F), hypoxia (G) and following re-introduction of normoxic perfusate (H).
In addition to the hypoxia-evoked reduction in patch current, inspection of Fig. 1 suggested that reduction in PO2 also decreased single channel unitary current (see Fig. 1CversusFig. 1D). This possibility was investigated and the data are presented in Fig. 2. In the presence of symmetrical K+-rich solutions ( 300 nm), hypoxia significantly (P < 0.05, n = 4) reduced single channel conductance by ∼12 % from 215.4 ± 2.0 to 188.5 ± 10 pS (Fig. 2A). Similarly, in asymmetrical solutions, single channel currents at all activating test potentials (from −40 to +60 mV in 300 nm [Ca2+]i) were also reduced by hypoxia (Fig. 2A). These differences were significant (P < 0.05, n = 5) at test potentials positive to 0 mV. Single channel conductance (measured between 0 and +40 mV) was mildly decreased by hypoxia from 106.0 ± 3.9 to 97.2 ± 3.4 pS (n = 5); a reduction of ∼8 %. This observation of reduced unitary conductance is highlighted in Fig. 2B where the channel amplitudes, calculated from the all-points histograms of an exemplar patch in asymmetrical solutions and held at +50 mV, are seen to be reduced at all open levels.
Figure 2. Hypoxic modulation of single channel conductance.
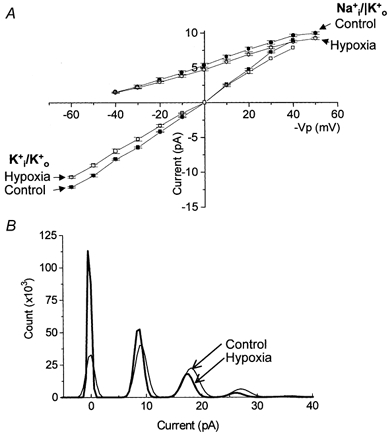
A, current-voltage relationships derived from single channels recorded in normoxia (control, filled symbols) and hypoxia (open symbols) with either Na+-rich (circles, n = 6) or K+-rich pipette solutions (squares, n = 4) with the intracellular membrane leaflet bathed in K+-rich solution containing 300 nm [Ca2+]i. B, exemplar all-points histogram taken from a patch which was bathed in the asymmetrical solution conditions described above and held at +50 mV in normoxia (control, fine line) and hypoxia (bold line). −Vp in this and subsequent figures refers to the potential with respect to inner membrane leaflet.
Figure 3 shows the effects of hypoxia on channel activation and deactivation kinetics. In order to study activation/ deactivation at test potentials which were on the rising phase of the current-voltage relationship (i.e. before the apparent inactivation seen at high potentials; see Fig. 5), we employed inside-out patches in 1 μm . All activating and deactivating currents were best described by a single exponential function. Under these conditions, hypoxia decreased the rate of activation at all test potentials (Fig. 3A and B) in addition to reducing the steady-state current amplitude (by between 3.3 and 35.9 % at +50 mV, consistent with the reduced NPo values shown in Fig. 1B). Proportionally, this decreased rate of activation was most pronounced at +50 mV. At +50 mV, the mean time constant was significantly (P < 0.02, n = 10) increased from 1.34 ± 0.15 ms (control) to 2.82 ± 0.64 ms (hypoxia). In contrast, deactivation was unaffected by hypoxia at all test potentials (Fig. 3C and D).
Figure 3. Activation and deactivation channel kinetics.
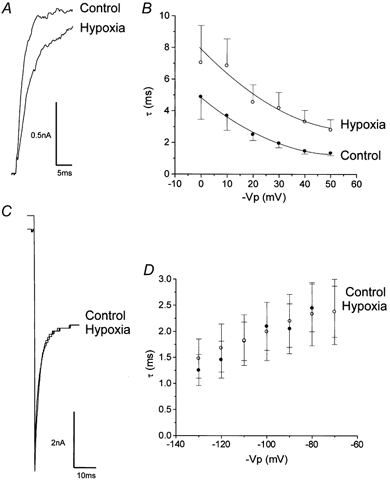
A, exemplar activation current (from holding potential to +50 mV) in the presence (hypoxia) and absence (control) of hypoxia. B, voltage dependence of mean activation time constants in the presence (hypoxia, ○) and absence (control, •) of hypoxia (n = 10). C, exemplar deactivating tail current (stepped from 130 mV to −130 mV) in the presence (hypoxia) and absence (control) of hypoxia. D, voltage dependence of mean deactivation time constants in the presence (hypoxia, ○) and absence (control, •) of hypoxia (n = 5). All recordings were performed in asymmetrical solutions with 1 μm .
Figure 5. Dependence of hypoxic channel inhibition on [Ca2+]i.
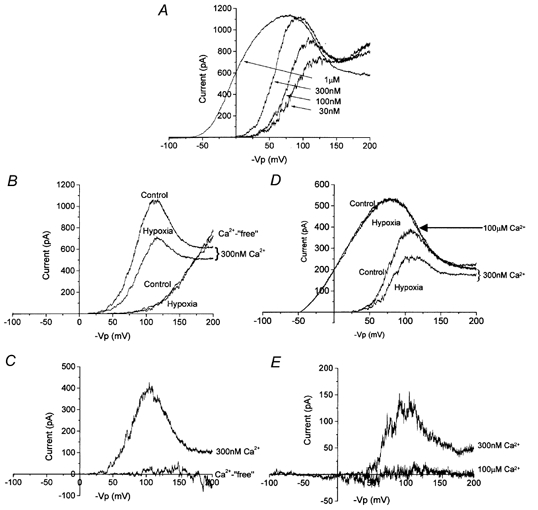
A, exemplar currents evoked using the extended-ramp protocol in the presence of varying [Ca2+]i as indicated. B, effect of hypoxia in the extended voltage range in the presence (300 nm) and absence (Ca2+-free) of [Ca2+]i. C, hypoxia-sensitive currents in the presence (300 nm) and absence (Ca2+-free) [Ca2+]i. Data for A–C are representative of four such recordings. D, effect of hypoxia in the extended voltage range in the presence of 300 nm and 100 μm [Ca2+]i as indicated. E, hypoxia-sensitive currents in the presence of 300 nm and 100 μm [Ca2+]i as indicated. Data for D and E are representative of four further recordings.
Figure 4 illustrates the dependence of hypoxic channel inhibition on [Ca2+]i. At 30 nm (Fig. 4A) and 100 nm [Ca2+]i (Fig. 4B), hypoxia had no significant effect on channel activity. Normalised currents (see Methods, above), measured at +50 mV in 30 nm [Ca2+]i, were 0.059 ± 0.008 (n = 4) in control and 0.031 ± 0.004 (n = 5) in hypoxia (P = 0.6) whilst in 100 nm [Ca2+]i they were 0.090 ± 0.011 (n = 4) in control and 0.084 ± 0.016 (n = 5) in hypoxia (P = 0.9). However, as [Ca2+]i was raised to 300 nm (Fig. 4C) and 1 μm (Fig. 4D), channel inhibition became apparent at all activating voltages. Normalised currents measured at +50 mV in 300 nm [Ca2+]i were 0.537 ± 0.024 (n = 4) in normoxia and 0.322 ± 0.019 (n = 5) in hypoxia (P < 0.0001) whilst in 1 μm [Ca2+]i they were 3.88 ± 0.25 (n = 4) in normoxia and 2.13 ± 0.43 (n = 5) in hypoxia (P < 0.0001). Thus, in normoxia, increasing [Ca2+]i caused the expected decrease in voltage-activation threshold but, importantly, this threshold was shifted to more positive potentials by reducing bath PO2 (by ∼10 mV at 300 nm and ∼25 mV at 1 μm). Furthermore, increasing [Ca2+]i to 100 μm resulted in a complete saturation of the hypoxic inhibition (Fig. 4E); mean normalised currents in normoxia and hypoxia were 6.91 ± 1.85 and 6.82 ± 1.73, respectively (n = 4). The Ca2+ dependence of hypoxic inhibition is quantified in Fig. 4F, which shows the normalised currents in normoxia and hypoxia plotted against [Ca2+]i; calculated EC50 values (employing the Hill equation) were 840 ± 15 nm in hypoxia versus 1805 ± 112 nm in hypoxia.
The data in Fig. 4 suggest that hypoxia evokes channel inhibition by either: (1) increasing the voltage threshold for channel activation; (2) decreasing Ca2+ sensitivity of the channels; or (3) a combination of the two. To distinguish between these possibilities, we employed a ramp protocol which was able to activate channels at different levels of [Ca2+]i. Interestingly, evoked currents displayed an apparent ‘inactivation’ at larger potentials of ca +70 mV to +120 mV. Since this effect was more striking with increasing Ca2+ levels, and was absent in Ca2+-‘free’ solutions (Fig. 5B), we attribute this effect to Ca2+ blockade of maxi-K channels, although more detailed studies are required to resolve this issue. This protocol was able to evoke similar degrees of channel activity, albeit at different voltages, regardless of [Ca2+]i (up to 100 μm). When [Ca2+]i was buffered to less than 1 nm (zero added Ca2+ plus 11 mm EGTA), the large depolarising ramp was able to evoke activity which was not significantly different from that in the presence of 300 nm or 1 μm [Ca2+]i (Fig. 5B, control). Most importantly, in the presence of 300 nm [Ca2+]i, hypoxia caused a significant decrease in current at all activating potentials (Fig. 5B, hypoxia) whereas hypoxic current suppression was completely absent when [Ca2+]i was buffered to below 1 nm; subtracted, hypoxia-sensitive currents in the ‘absence’ and ‘presence’ of [Ca2+]i are plotted in Fig. 5C. These data suggest strongly that hypoxia inhibits maxi-K channels by reducing their Ca2+ sensitivity, and to confirm this possibility we employed a saturating concentration of in the extended ramp protocol. At 100 μm [Ca2+]i, hypoxia was ineffective at all voltages of the extended ramp protocol (Fig. 5D). This is shown clearly in Fig. 5E which plots the hypoxia-sensitive currents in an exemplar patch when [Ca2+]i was buffered to either 300 nm or 100 μm.
DISCUSSION
Through the use of a recombinant expression system, our single channel analyses with inside-out membrane patches have demonstrated that hypoxia acutely inhibits maxi-K channels of known molecular identity. Furthermore, this work has revealed important new findings concerning the mechanism of maxi-K channel inhibition. Thus, hypoxia decreases channel sensitivity to , slows activation and reduces unitary conductance. Furthermore, hypoxic inhibition occurs independently of soluble cytosolic factors.
Maxi-K channels are widely distributed and, in excitable tissues are involved in a variety of physiological mechanisms including, controversially, control of vascular tone (Brenner et al. 2000; Pluger et al. 2000). Their involvement in neuronal excitability (Vergara et al. 1998) and chemosensing (Peers, 1997; Riesco-Fagundo et al. 2001) is less controversial and this channel type is clearly important to carotid body and central neuronal function (Lopez-Barneo et al. 2001). The α- and β-subunits examined in the present study were first cloned from human brain but are essentially ubiquitously expressed (Tseng-Crank et al. 1994, 1996). In chemosensory tissues, episodes of acute hypoxia result in modulation of ventilation and optimisation of ventilation-perfusion matching via the concerted actions of the carotid bodies, the neuroepithelial bodiese of the lung and the pulmonary vasculature (Cutz & Jackson, 1999; Peers & Kemp, 2001; Lopez-Barneo et al. 2001; Coppock et al. 2001). Evidence supports the idea that hypoxic inhibition of maxi-K channels is central to some of these effects. There is clear evidence for divergent mechanisms underlying the initial steps in O2 signal transduction in different chemosensory tissues (for recent review see Peers & Kemp, 2001). Thus, even though carotid body (Wyatt et al. 1995; Buckler et al. 2000), pulmonary vasculature (Weir & Archer, 1995) and airway O2 chemosensory pathways (Wang et al. 1996; Fu et al. 2000; O'Kelly et al. 2000, 2001) contain different O2 sensors, they each express specific K+ channels which are inhibited by acute hypoxia. In the neuroepithelial body of the lung, and its immortal cellular counterpart the H146 cell line, there is accumulating evidence to suggest that central to cellular O2 sensing is hypoxic inhibition of members of the K2P class of K+ channels (O'Kelly et al. 1999; Hartness et al. 2001), although the contribution of voltage-activated K+ channels to the responses is still a matter of some debate (Wang et al. 1996). Similarly, in pulmonary smooth muscle, there is clear evidence for the involvement of maxi-K and other voltage-activated K+ channels (Post et al. 1992; McCulloch et al. 1999) and emerging evidence to suggest a role for K2P channels (Gurney et al. 2002). In the carotid body, there is also some controversy as to the identity of the O2-sensitive channel, with both a K2P channel (Buckler et al. 2000) and a maxi-K channel (Peers, 1990; Wyatt & Peers, 1995) suggested as potential targets for O2 (Peers, 1990; Wyatt & Peers, 1995; Pardal et al. 2000; Lopez-Barneo et al. 2001). However, the notion that there is large contribution to the physiological response in carotid body from maxi-K channel inhibition has recently been reinforced by the observation that glomus cells cultured in clusters, and slices of intact carotid body, release catacholamines both in hypoxia and following application of iberiotoxin (Jackson & Nurse, 1998; Pardal et al. 2000).
Central neurones also respond rapidly to reduced PO2, and such responses include inhibition of a variety of different K+ channels. However, none of these have been identified at the molecular level, and different mechanisms have been proposed to account for hypoxic inhibition. For example iberiotoxin and charybdotoxin-insensitive maxi-K channels are inhibited by hypoxia via a mechanism which requires cytosolic factors (Liu et al. 1999). In contrast, other maxi-K-like channels, which are also regulated by ATP levels, are inhibited by hypoxia in a membrane-delimited manner (Jiang & Haddad, 1994).
A number of studies in both native tissues and recombinant systems have demonstrated that maxi-K channels are modulated by oxidising and reducing agents (Liu et al. 1999; Soh et al. 2001; Tang et al. 2001). This has led to the proposal that hypoxic inhibition is through reduction of the channel protein. Thus, where redox modulators evoke regulation of recombinant maxi-K channels in a manner which is consistent with this proposal (i.e. activation by oxidising agents and inhibition by reducing agents), there are clear differences in the mechanism of modulation, suggesting that these direct chemical interventions cannot explain the effect of environmental hypoxia which we report here. For example recombinant maxi-K channels are activated by specific oxidation of methionine residues with chloramine T (Tang et al. 2001). Such an increase in channel activity, which is observed in the absence of Ca2+, is abrogated by channel blockade with tetraethylammonium and accompanied by a shift in the voltage dependence and slowed deactivation without a change in unitary conductance. Reducing agents evoke the opposite effects and it seems likely, therefore, that the principal mechanism of regulation by such chemical interventions is covalent modification of a methionine in the channel pore (Tang et al. 2001). These effects are in marked contrast to the decreased unitary conductance, decreased activation (in the absence of modulation of deactivation) and Ca2+ dependence of hypoxic inhibition which we report.
That the use of such agents in attempting to define the mechanism of hypoxic inhibition is not entirely appropriate has been reinforced by observations of native maxi-K channels. Thus, neocortical neurones are activated by the reducing agents dithiothreitol and glutathione (Liu et al. 1999), whilst glutathione promotes maxi-K activity in rat hippocampal neurones (Soh et al. 2001). This issue is clouded by the lack of molecular identification of the channel under investigation (indeed numerous splice variants of the maxi-K α-subunit have been identified, which may co-express with various β-subunits (Toro et al. 1998)), together with clearly different requirements for cytosolic factors and probable cell-specific responses. Indeed, a recent study of native maxi-K channels expressed in rat carotid body has shown membrane-restricted hypoxic inhibition of channel activity. However, this response was only investigated at [Ca2+]i concentrations exceeding those seen physiologically and the molecular identity of the subunits constituting the functional channel was unknown (Riesco-Fagundo et al. 2001). Our data demonstrate that a brain-type maxi-K channel complex is sensitive to hypoxia at between 100 and 300 nm [Ca2+]i. In living cells, [Ca2+]i is close to 100 nm which suggests that hypoxic inhibition of this channel type may be of physiological significance; further study at levels bracketed by those employed here should shed further light on such issues. Absolute [Ca2+]i notwithstanding, our recombinant study significantly extends earlier studies in native tissues in that it employed clearly defined and identified channels in a membrane-delimited preparation. In so doing, it clarifies some of the issues described above and suggests that different isoforms of maxi-K channels may behave differently. Therefore, cell-specific expression of these isoforms will probably determine the response of an individual cell to hypoxia.
Since modulation by redox reagents appears to be inconsistent with the channel regulation which we report herein, it seems likely that changes in environmental PO2 proceeds via a different mechanism. In support of this idea are data from native neocortex which show that hypoxia inhibits activity by decreasing Ca2+ sensitivity (Liu et al. 1999). However, in contrast to our findings, hypoxic inhibition of neocortical maxi-K channels is dependent upon cytosolic components. One explanation for this difference (which supports the notion of cell-specific channel expression detailed above) appears to be that the native neuronal channels are a different class (since they are charybdotoxin/iberiotoxin insensitive) from the recombinant channels which were used in the present study. Thus, the iberiotoxin-sensitive maxi-K channel itself, or a tightly associated membrane-delimited protein(s), is directly sensitive to the inhibitory actions of hypoxia. Whether the presence of an intact intracellular milieu will alter the mechanism and/or degree of hypoxic inhibition will be important to investigate before the full cell-specific physiological consequences of decreased PO2 can be determined. However, this study shows that the presence of αβ-maxi-K channel protein within a biological membrane is sufficient for a hypoxic response. This notwithstanding, our data clearly demonstrate that hypoxic inhibition involves reduced Ca2+ sensitivity. Although we have no direct evidence for the mechanism of this hypoxic modulation, we speculate that there are a number of possibilities worthy of consideration. Based on the observations that mitochondria-derived reactive oxygen species (ROS) have been implicated in hypoxic modulation in a number of tissues (Chandel & Schumacker, 2000; Leach et al. 2001), together with the possibility that mitochondria can be contained within excised patches (Levitan & Kramer, 1990) it is possible that local production of ROS may be responsible for this effect. Another possibility, which currently remains speculative yet attractive (since the probability of mitochondria being retained in excised-membrane patches is not consistent with the highly reproducible effects of hypoxia as shown here), is that hypoxia alters channel activity by modulating the direct interaction of an, as yet, unidentified sensor with the channel protein. Additionally, it may simply be that the amino acid composition of the subunit per se is the site of O2 interaction (Lopez-Barneo, 1996) and this could be addressed by examining the actions of hypoxia on pure α-subunit protein in an artificial lipid bilayer system. It is also plausible that the hypoxic stimulus results in functional dissociation of the β-subunit from the α-subunit, resulting in slowing of activation and reduced Ca2+ sensitivity (Toro et al. 1998). Further alternatives include activation of protein kinases embedded in the isolated patch (Schubert & Nelson, 2001) or hypoxia-driven modulation of cytoskeletal structure (Friedman et al. 1998) which has been demonstrated previously to modify channel function (Mironov & Richter, 1999).
The consequences of inhibiting maxi-K channels via reduced Ca2+ sensitivity are probably both physiologically and pathologically important. In central neurones, the known pathological effect of prolonged hypoxia, which can lead to focal cell death during cerebral ischaemia, may be augmented by prevention of channel reactivation due to rises of [Ca2+]i. Physiologically, prevention of reactivation may contribute to the persistent excitation of the carotid body by hypoxia; initially, hypoxia inhibits maxi-K channels, thereby causing membrane depolarisation and voltage-gated Ca2+ entry. Without a decrease in the Ca2+ sensitivity of maxi-K channels this would lead to reactivation of these channels and consequently a transient hypoxic response in the tissue, but this has not been observed (e.g. Landauer et al. 1995).
Acknowledgments
This work was supported by the Wellcome Trust and the British Heart Foundation.
REFERENCES
- Ahring PK, Strobaek D, Christophersen P, Oleson SP, Johansen TE. Stable expression of the human large-conductance Ca2+-activated K+ channel alpha- and beta-subunits in HEK293 cells. FEBS Letters. 1997;415:67–70. doi: 10.1016/s0014-5793(97)01096-x. [DOI] [PubMed] [Google Scholar]
- Brenner R, Perez GJ, Bonev AD, Eckman DM, Kosek JC, Wiler SW, Patterson AJ, Nelson MT, Aldrich RW. Vasoregulation by the β1 subunit of the calcium-activated potassium channel. Nature. 2000;407:870–876. doi: 10.1038/35038011. [DOI] [PubMed] [Google Scholar]
- Buckler KJ, Williams BA, Honore E. An oxygen-, acid- and anaesthetic-sensitive TASK-like background potassium channel in rat arterial chemoreceptor cells. Journal of Physiology. 2000;525:135–142. doi: 10.1111/j.1469-7793.2000.00135.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandel NS, Schumacker PT. Cellular oxygen sensing by mitochondria: old questions, new insight. Journal of Applied Physiology. 2000;88:1880–1889. doi: 10.1152/jappl.2000.88.5.1880. [DOI] [PubMed] [Google Scholar]
- Coppock EA, Martens JR, Tamkun MM. Molecular basis of hypoxia-induced pulmonary vasoconstriction: role of voltage-gated K+ channels. American Journal of Physiology - Lung Cellular and Molecular Physiology. 2001;281:L1–8. doi: 10.1152/ajplung.2001.281.1.L1. [DOI] [PubMed] [Google Scholar]
- Cornfield DN, Reeve HL, Tolarova S, Weir EK, Archer S. Oxygen causes fetal pulmonary vasodilation through activation of a calcium-dependent potassium channel. Proceedings of the National Academy of Sciences of the USA. 1996;93:8089–8094. doi: 10.1073/pnas.93.15.8089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cutz E, Jackson A. Neuroepithelial bodies as airway oxygen sensors. Respiration Physiology. 1999;115:201–214. doi: 10.1016/s0034-5687(99)00018-3. [DOI] [PubMed] [Google Scholar]
- Friedman JE, Chow EJ, Haddad GG. State of actin filaments is changed by anoxia in cultured rat neocortical neurons. Neuroscience. 1998;82:421–427. doi: 10.1016/s0306-4522(97)00217-0. [DOI] [PubMed] [Google Scholar]
- Fu XW, Wang D, Nurse C, Dinauer MC, Cutz E. NADPH oxidase is an O2 sensor in airway chemoreceptors: evidence from K+ current modulation in wild type and oxidase-deficient mice. Proceedings of the National Academy of Sciences of the USA. 2000;97:4374–4379. doi: 10.1073/pnas.97.8.4374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurney AM, Osipenko ON, MacMillan D, Kempsill FEJ. Potassium channels underlying the resting membrane potential of pulmonary artery smooth muscle cells. Clinical and Experimental Pharmacology and Physiology. 2002;29:330–333. doi: 10.1046/j.1440-1681.2002.03653.x. [DOI] [PubMed] [Google Scholar]
- Hartness ME, Lewis A, Searle GJ, O'Kelly I, Peers C, Kemp PJ. Combined antisense and pharmacological approaches implicate hTASK as an airway O2 sensing K+ channel. Journal of Biological Chemistry. 2001;276:26499–26508. doi: 10.1074/jbc.M010357200. [DOI] [PubMed] [Google Scholar]
- Jackson A, Nurse CA. Role of acetylcholine receptors and dopamine transporter in regulation of extracellular dopamine in rat carotid body cultures grown in chronic hypoxia or nicotine. Journal of Neurochemistry. 1998;70:653–662. doi: 10.1046/j.1471-4159.1998.70020653.x. [DOI] [PubMed] [Google Scholar]
- Jiang C, Haddad GG. Oxygen deprivation inhibits a K+ channel independently of cytosolic factors in rat central neurons. Journal of Physiology. 1994;481:15–26. doi: 10.1113/jphysiol.1994.sp020415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landauer RC, Pepper DR, Kumar P. Effect of chronic hypoxaemia from birth upon chemosensitivity in the adult rat carotid body in vitro. Journal of Physiology. 1995;485:543–550. doi: 10.1113/jphysiol.1995.sp020750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leach RM, Hill HM, Snetkov VA, Robertson TP, Ward JP. Divergent roles of glycolysis and the mitochondrial electron transport chain in hypoxic pulmonary vasoconstriction of the rat: identity of the hypoxic sensor. Journal of Physiology. 2001;536:211–224. doi: 10.1111/j.1469-7793.2001.00211.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levitan ES, Kramer RH. Neuropeptide modulation of single calcium and potassium channels detected with a new patch clamp configuration. Nature. 1990;348:545–547. doi: 10.1038/348545a0. [DOI] [PubMed] [Google Scholar]
- Lewis A, Hartness ME, Chapman CG, Fearon IM, Meadows HJ, Peers C, Kemp PJ. Recombinant hTASK1 is an O2-sensitive K+ channel. Biochemical and Biophysical Research Communications. 2001;285:1290–1294. doi: 10.1006/bbrc.2001.5310. [DOI] [PubMed] [Google Scholar]
- Liu H, Moczydlowski E, Haddad GG. O2 deprivation inhibits Ca2+-activated K+ channels via cytosolic factors in mice neocortical neurons. Journal of Clinical Investigation. 1999;104:577–588. doi: 10.1172/JCI7291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Barneo J. Oxygen sensing by ion channels and the regulation of cellular function. Trends in Neurosciences. 1996;19:435–440. doi: 10.1016/0166-2236(96)10050-3. [DOI] [PubMed] [Google Scholar]
- Lopez-Barneo J, Pardal R, Ortega-Saenz P. Cellular mechanism of oxygen sensing. Annual Review of Physiology. 2001;63:259–287. doi: 10.1146/annurev.physiol.63.1.259. [DOI] [PubMed] [Google Scholar]
- McCulloch KM, Osipenko ON, Gurney AM. Oxygen-sensing potassium currents in pulmonary artery. General Pharmacology. 1999;32:403–411. doi: 10.1016/s0306-3623(98)00219-5. [DOI] [PubMed] [Google Scholar]
- Mironov SL, Richter DW. Cytoskeleton mediates inhibition of the fast Na+ current in respiratory brainstem neurons during hypoxia. Journal of Neuroscience. 1999;11:1831–1834. doi: 10.1046/j.1460-9568.1999.00584.x. [DOI] [PubMed] [Google Scholar]
- Mojet MH, Mills E, Duchen MR. Hypoxia-induced catecholamine secretion in isolated newborn rat adrenal chromaffin cells is mimicked by inhibition of mitochondrial respiration. Journal of Physiology. 1997;504:175–189. doi: 10.1111/j.1469-7793.1997.175bf.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Kelly I, Lewis A, Peers C, Kemp PJ. O2 sensing by airway chemoreceptor-derived cells: protein kinase C activation reveals functional evidence for involvement of NADPH oxidase. Journal of Biological Chemistry. 2000;275:7684–7692. doi: 10.1074/jbc.275.11.7684. [DOI] [PubMed] [Google Scholar]
- O'Kelly I, Peers C, Kemp PJ. NADPH oxidase does not account fully for O2 sensing in model airway chemoreceptor cells. Biochemical and Biophsyical Research Communications. 2001;283:1131–1134. doi: 10.1006/bbrc.2001.4919. [DOI] [PubMed] [Google Scholar]
- O'Kelly I, Stephens RH, Peers C, Kemp PJ. Potential identification of the O2-sensitive K+ current in a human neuroepithelial body-derived cell line. American Journal of Physiology. 1999;276:L96–104. doi: 10.1152/ajplung.1999.276.1.L96. [DOI] [PubMed] [Google Scholar]
- Pardal R, Ludewig U, Garcia-Hirschfeld J, Lopez-Barneo J. Secretory responses of intact glomus cells in thin slices of rat carotid body to hypoxia and tetraethylammonium. Proceedings of the National Academy of Sciences of the USA. 2000;97:2361–2366. doi: 10.1073/pnas.030522297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peers C. Hypoxic suppression of K+ currents in type-I carotid-body cells - selective effect on the Ca2+-activated K+ current. Neuroscience Letters. 1990;119:253–256. doi: 10.1016/0304-3940(90)90846-2. [DOI] [PubMed] [Google Scholar]
- Peers C. Oxygen-sensitive ion channels. Trends in Pharmacological Sciences. 1997;18:405–408. doi: 10.1016/s0165-6147(97)01120-6. [DOI] [PubMed] [Google Scholar]
- Peers C, Kemp PJ. Acute oxygen sensing: diverse but convergent mechanisms in airway and arterial chemoreceptors. Respiration Research. 2001;2:145–149. doi: 10.1186/rr51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pluger S, Faulhaber J, Furstenau M, Lohn M, Waldschutz R, Gollasch M, Haller H, Luft FC, Ehmke H, Pongs O. Mice with disrupted BK channel beta1 subunit gene feature abnormal Ca2+ spark/STOC coupling and elevated blood pressure. Circulation Research. 2000;87:E53–60. doi: 10.1161/01.res.87.11.e53. [DOI] [PubMed] [Google Scholar]
- Post JM, Hume JR, Archer SL, Weir EK. Direct role for potassium channel inhibition in hypoxic pulmonary vasoconstriction. American Journal of Physiology. 1992;262:C882–890. doi: 10.1152/ajpcell.1992.262.4.C882. [DOI] [PubMed] [Google Scholar]
- Riesco-Fagundo AM, Perez-Garcia MT, Gonzalez C, Lopez-Lopez JR. O2 modulates large-conductance Ca2+-dependent K+ channels of rat chemoreceptor cells by a membrane-restricted and CO-sensitive mechanism. Circulation Research. 2001;89:430–436. doi: 10.1161/hh1701.095632. [DOI] [PubMed] [Google Scholar]
- Schubert R, Nelson MT. Protein kinases: tuners of the BKCa channel in smooth muscle. Trends in Pharmacological Sciences. 2001;22:505–512. doi: 10.1016/s0165-6147(00)01775-2. [DOI] [PubMed] [Google Scholar]
- Soh H, Jung W, Uhm DY, Chung S. Modulation of large conductance calcium-activated potassium channels from rat hippocampal neurons by glutathione. Neuroscience Letters. 2001;298:115–118. doi: 10.1016/s0304-3940(00)01737-7. [DOI] [PubMed] [Google Scholar]
- Tang XD, Daggett H, Hanner M, Garcia ML, McManus OB, Brot N, Weissbach H, Heinemann SH, Hoshi T. Oxidative regulation of large conductance calcium-activated potassium channels. Journal of General Physiology. 2001;117:253–274. doi: 10.1085/jgp.117.3.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson RJ, Nurse CA. Anoxia differentially modulates multiple K+ currents and depolarizes neonatal rat adrenal chromaffin cells. Journal of Physiology. 1998;512:421–434. doi: 10.1111/j.1469-7793.1998.421be.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toro L, Wallner M, Meera P, Tanaka Y. Maxi-K(Ca), a unique member of the voltage-gated K channel superfamily. News in Physiological Sciences. 1998;13:112–117. doi: 10.1152/physiologyonline.1998.13.3.112. [DOI] [PubMed] [Google Scholar]
- Tristani-Firouzi M, Reeve HL, Tolarova S, Weir EK, Archer SL. Oxygen-induced constriction of rabbit ductus arteriosus occurs via inhibition of a 4-aminopyridine-, voltage-sensitive potassium channel. Journal of Clinical Investigation. 1996;98:1959–1965. doi: 10.1172/JCI118999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng-Crank J, Foster CD, Krause JD, Mertz R, Godinot N, Dichiara TJ, Reinhart PH. Cloning, expression, and distribution of functionally distinct Ca2+-activated K+ channel isoforms from human brain. Neuron. 1994;13:1315–1330. doi: 10.1016/0896-6273(94)90418-9. [DOI] [PubMed] [Google Scholar]
- Tseng-Crank J, Godinot N, Johansen TE, Ahring PK, Strobaek D, Mertz R, Foster CD, Olesen SP, Reinhart PH. Cloning, expression, and distribution of a Ca2+-activated K+ channel beta-subunit from human brain. Proceedings of the National Academy of Sciences of the USA. 1996;93:9200–9205. doi: 10.1073/pnas.93.17.9200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergara C, Latorre R, Marrion NV, Adelman JP. Calcium-activated potassium channels. Current Opinion in Neurobiology. 1998;8:321–329. doi: 10.1016/s0959-4388(98)80056-1. [DOI] [PubMed] [Google Scholar]
- Wang D, Youngson C, Wong V, Yeger H, Dinauer MC, Vega-Saenz de Miera E, Rudy B, Cutz E. NADPH-oxidase and hydrogen peroxide sensitive K+ channel may function as an oxygen sensor complex in airway chemoreceptors and small cell lung carcinoma cell lines. Proceedings of the National Academy of Sciences of the USA. 1996;93:13182–13187. doi: 10.1073/pnas.93.23.13182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weir EK, Archer SL. The mechanism of acute hypoxic pulmonary vasoconstriction: the tale of two channels. FASEB Journal. 1995;9:183–189. doi: 10.1096/fasebj.9.2.7781921. [DOI] [PubMed] [Google Scholar]
- Wyatt CN, Peers C. Ca2+-activated K+ channels in isolated type-I cells of the neonatal rat carotid-body. Journal of Physiology. 1995;483:559–565. doi: 10.1113/jphysiol.1995.sp020606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyatt CN, Wright C, Bee D, Peers C. O2-sensitive K+ currents in carotid-body chemoreceptor cells from normoxic and chronically hypoxic rats and their roles in hypoxic chemotransduction. Proceedings of the National Academy of Sciences of the USA. 1995;92:295–299. doi: 10.1073/pnas.92.1.295. [DOI] [PMC free article] [PubMed] [Google Scholar]