

Abstract
Emerging evidence has implicated a potential role for 5-HT4 receptors in cognition and anxiolysis. One of the main target structures of 5-HT4 receptors on ‘cognitive and emotional’ pathways is the prefrontal cortex (PFC). As GABAergic signalling plays a key role in regulating PFC functions, we examined the effect of 5-HT4 receptors on GABAA receptor channels in PFC pyramidal neurons. Application of 5-HT4 receptor agonists produced either an enhancement or a reduction of GABA-evoked currents in PFC neurons, which are both mediated by anchored protein kinase A (PKA). Although PKA phosphorylation of GABAA receptor β3 or β1 subunits leads to current enhancement or reduction respectively in heterologous expression systems, we found that β3 and β1 subunits are co-expressed in PFC pyramidal neurons. Interestingly, altering PKA activation levels can change the direction of the dual effect, switching enhancement to reduction and vice versa. In addition, increased neuronal activity in PFC slices elevated the PKA activation level, changing the enhancing effect of 5-HT4 receptors on the amplitude of GABAergic inhibitory postsynaptic currents (IPSCs) to a reduction. These results suggest that 5-HT4 receptors can modulate GABAergic signalling bidirectionally, depending on the basal PKA activation levels that are determined by neuronal activity. This modulation provides a unique and flexible mechanism for 5-HT4 receptors to dynamically regulate synaptic transmission and neuronal excitability in the PFC network.
The serotonergic system plays a key role in regulating cognitive behaviours and emotional processes in the central nervous system (Buhot, 1997; Deakin, 1998; Davidson et al. 2000). Dysfunction of serotonergic neurotransmission has been implicated in the pathogenesis of neuropsychiatric disorders including schizophrenia, depression and anxiety (Breier 1995; Dubovsky & Thomas 1995; Abi-Dargham et al. 1997; Stockmeier 1997). One of the main target structures of the serotonergic system is the prefrontal cortex (PFC), a brain region critically involved in a variety of functions, such as attention, response selection, planning, and particularly, a form of short-term information storage described as ‘working memory’ (Goldman-Rakic, 1995).
The PFC is composed of two major neuronal populations: glutamatergic pyramidal projection neurons and GABAergic interneurons. The axon terminals of local GABAergic neurons form numerous synapses with pyramidal projection neurons (Somogyi et al. 1983), exerting powerful inhibitory control over the excitatory output of the PFC. The balance in the excitatory (glutamatergic) and inhibitory (GABAergic) transmission determines the neuronal activity of the PFC. Serotonergic projections target both types of PFC neurons in a synaptic and non-synaptic manner (Smiley & Goldman-Rakic, 1996). Recent evidence shows that serotonin (5-HT) neurotransmission is predominantly paracrine, raising the possibility that 5-HT can act on receptors that are distant from its release site (Bunin & Wightman, 1999). Serotonin receptors mediate a modulatory response, which can be either excitatory or inhibitory (for review, see Andrade, 1998), implicating a role in information processing by controlling the signal-to-noise ratio. Specific changes in serotonin signalling and neuronal activity have been found in PFC neurons of subjects with various neuropsychiatric disorders (Jaffe et al. 1993; Gurevich & Joyce, 1997), suggesting that serotonin plays a crucial role in neural computation associated with execution of complex tasks involved in cognition and emotion.
Among the multiple G protein-coupled serotonin receptor subtypes, 5-HT4 receptors are highly enriched in the frontal cortex (Domenech et al. 1994). Emerging evidence argues for a significant role of 5-HT4 receptors in cognition and anxiolysis (for review, see Eglen et al. 1995b). Activation of 5-HT4 receptors exerts ameliorative effects on spatial memory tests and reverses cognitive performance deficits induced by cholinergic hypofunction (Eglen et al. 1995b). In addition, application of 5-HT4 receptor antagonists inhibits the anxiolytic effect of diazapam (an enhancer of GABA response), particularly under conditions of high serotonergic tone (Costall & Naylor, 1993). Since GABAA receptor-mediated inhibitory synaptic transmission is highly involved in controlling neuronal excitability, and GABAA receptors have been implicated in the pathogenesis of anxiety disorders (Macdonald & Olsen, 1994), these lines of evidence prompt the speculation that 5-HT4 receptors may exert some of their functions by acting on GABAergic signalling in PFC neurons. Here we report that activation of 5-HT4 receptors has a dual effect (enhancement or reduction) on GABAA receptor-mediated currents in PFC pyramidal neurons. This bidirectional regulation of postsynaptic GABAA receptors is through a PKA-mediated and phosphorylation state-dependent mechanism. Furthermore, the direction of the 5-HT4 effect on GABAA currents is determined by neuronal activity. Based on these experimental data, we have proposed a model which illustrates the potential roles that 5-HT4 receptors may play in neural computation in PFC pyramidal neurons. Our results suggest that serotonin, by acting on 5-HT4 receptors, can dynamically regulate neuronal excitability and synaptic transmission in PFC networks using a flexible and activity-guided mechanism.
METHODS
Acute dissociation procedure
PFC neurons from young adult (3–5 weeks postnatal) rats were acutely dissociated using procedures similar to those described previously (Yan & Surmeier, 1996). All experiments were carried out with the approval of the State University of New York at Buffalo Animal Care Committee. In brief, rats were anaesthetized by inhaling 2-bromo-2-chloro-1,1,1-trifluoroethane (1 ml (100 g)−1, Sigma Chemical Co., St Louis, MO, USA) and decapitated; brains were quickly removed, iced and then blocked for slicing. The blocked tissue was cut in 400 μm slices with a Vibrotome while bathed in a low Ca2+ (100 μm), Hepes-buffered salt solution (mm: 140 sodium isethionate, 2 KCl, 4 MgCl2, 0.1 CaCl2, 23 glucose, 15 Hepes, 1 kynurenic acid, pH 7.4, 300–305 mosmol l−1). Slices were then incubated for 1–6 h at room temperature (20–22 °C) in a NaHCO3-buffered saline bubbled with 95 % O2, 5 % CO2; composition (mm): 126 NaCl, 2.5 KCl, 2 CaCl2, 2 MgCl2, 26 NaHCO3, 1.25 NaH2PO4, 10 glucose, 1 pyruvic acid, 0.05 gluta-thione, 0.1 NG-nitro-l-arginine, 1 kynurenic acid, pH 7.4, 300–305 mosmol l−1). All reagents were obtained from Sigma Chemical Co.
Slices were then removed into the low Ca2+ buffer and regions of the PFC were dissected and placed in an oxygenated Cell-Stir chamber (Wheaton, Inc., Millville, NJ, USA) containing pronase (1–3 mg ml−1) in Hepes-buffered Hanks' balanced salt solution (HBSS, Sigma Chemical Co.) at 35 °C. After 30 min of enzyme digestion, tissue was rinsed three times in the low Ca2+, Hepes-buffered saline and mechanically dissociated with a graded series of fire-polished Pasteur pipettes. The cell suspension was then plated into a 35 mm Lux Petri dish which was then placed on the stage of a Nikon inverted microscope.
Whole-cell recordings
Whole-cell recordings of currents employed standard voltage clamp techniques (Hamill et al. 1981; Yan & Surmeier, 1997; Yan et al. 1999). Electrodes were pulled from Corning 7052 glass and fire-polished prior to use. The internal solution (Yan & Surmeier, 1997) consisted of (mm): 180 N-methyl-d-glucamine (NMG), 40 Hepes, 4 MgCl2, 5 BAPTA, 12 phosphocreatine, 2 Na2ATP, 0.2 Na3GTP, 0.1 leupeptin, pH7.2–7.3, 265–270 mosmol l−1. The external solution consisted of (mm): 135 NaCl, 20 CsCl, 1 MgCl2, 10 Hepes, 0.001 TTX, 5 BaCl2, 10 glucose, pH 7.3, 300–305 mosmol l−1.
Recordings were obtained with an Axon Instruments 200B patch clamp amplifier that was controlled and monitored with an IBM PC running pCLAMP (v. 8) with a DigiData 1320 series interface (Axon instruments, Union City, CA, USA). Electrode resistances were typically 2–4 MΩ in the bath. After seal rupture, series resistance (4–10 MΩ) was compensated (70–90 %) and periodically monitored. Care was exercised to monitor the constancy of the series resistance, and recordings were terminated whenever a significant increase (> 20 %) occurred. The cell membrane potential was held at 0 mV. The application of GABA (100 μm) evoked a partially desensitizing outward current with the decay rate fitted by a single or double exponential. Peak values were measured for generating the plot as a function of time and drug application. GABA was applied for 2 s every minute to minimize desensitization-induced decrease of current amplitude. Drugs were applied with a gravity-fed ‘sewer pipe’ system. The array of application capillaries (ca 150 μm i.d.) was positioned a few hundred micrometres from the cell under study. Solution changes were effected by the SF-77B fast-step solution stimulus delivery device (Warner Instrument Co., Hamden, CT, USA).
Serotonin receptor ligands 5-methoxytryptamine, SDZ205557 (Sigma/RBI), RS67506, RS23597–190 and 2-[1-(4-piperonyl) piperaziny]benzothiazole (Tocris, Ballwin, MO, USA), as well as second messenger reagents sp-cAMPS, rp-cAMPS, okadaic acid (Sigma/RBI), cpt-cAMP, cAMP and PKI[5–24] (Calbiochem, San Diego, CA, USA) were made up as concentrated stocks in water or DMSO and stored at −20 °C. Stocks were thawed and diluted immediately prior to use. The amino acid sequence for the PKA-anchoring inhibitory peptide H31 is:
DLIEEAASRIVDAVIEQVKAAY.
The amino acid sequence for the scrambled peptide sH31 is:
QDISAEAEIVAYAVIRDKVELA.
Data analyses were performed with AxoGraph (Axon Instruments), Kaleidagraph (Albeck Software, Reading, PA, USA) and StatView (SAS Institute Inc., Cary, NC, USA). Box plots were used for graphic presentation of the data because of the small sample sizes (Tukey, 1977). The box plot represents the distribution as a box with the median as a central line and the hinges as the edges of the box (the hinges divide the upper and lower distributions in half). The inner fences (shown as a line originating from the edges of the box) run to the limits of the distribution (i.e. the minimal value of the lower half distribution and the maximal value of the upper half distribution) excluding outliers (defined as points that are > 1.5 times the interquartile range beyond the interquartiles). In all the box plots, the positive values of percentage modulation mean enhancement and negative values mean reduction. For analysis of statistical significance, Mann-Whitney U tests were performed to compare the current amplitudes in the presence or absence of various agonists. Student's unpaired t test was performed to compare the differential degrees of current modulation between groups subjected to different treatment.
Electrophysiological recordings in slices
To evaluate the regulation of spontaneous inhibitory synaptic currents (IPSCs) by 5-HT4 receptors in PFC slices, the whole-cell patch technique was used for voltage clamp and current clamp recordings using patch electrodes (5–9 MΩ) filled with the following internal solution (mm): 60 K2SO4, 60 NMG, 40 Hepes, 4 MgCl2, 5 BAPTA, 12 phosphocreatine, 2 Na2ATP, 0.2 Na3GTP, 0.1 leupeptin, pH 7.2–7.3, 265–270 mosmol l−1. The slice (300 μm) was placed in a perfusion chamber attached to the fixed-stage of an upright microscope (Olympus) and submerged in continuously flowing oxygenated artificial cerebrospinal fluid (ACSF). Cells were visualized with a × 40 water-immersion lens and illuminated with near infrared (IR) light and the image was detected with an IR-sensitive CCD camera. A Multiclamp 700A amplifier was used for these recordings. Tight seals (2–10 GΩ) from visualized pyramidal neurons were obtained by applying negative pressure. The membrane was disrupted with additional suction and the whole-cell configuration was obtained. The access resistances ranged from 20 to 35 MΩ and were compensated 50–70 %. Cells were held at 0 or 10 mV for the recording of spontaneous IPSCs. Mini Analysis Program (Synaptosoft, Leonia, NJ, USA) was used to analyse synaptic activity. IPSCs of 1 min (200–1000 events) under each different treatment were used for analysis. Statistical comparisons of the synaptic currents were made using the Kolmogorov-Smirnov (K–S) test.
Single-neuron mRNA profiling
Detection of mRNAs for 5-HT4 receptors and GABAA receptor β1–3 subunits in PFC pyramidal neurons used the single-cell RT-PCR technique similar to that described previously (Yan & Surmeier, 1996, 1997). A patch electrode was used to lift a dissociated neuron into a stream of control solution and then the neuron was aspirated into the electrode by applying negative pressure. After aspiration, the electrode was broken and the contents ejected into a 0.5 ml Eppendorf tube containing 5 μl diethyl pyrocarbonate (DEPC)-treated water, 0.5 μl RNAsin (28 U μl−1) and 0.5 μl dithiothreitol (DTT) (0.1 m) and 1 μl Oligo(dT) primer (0.5 μg μl−1). The mixture was heated to 70 °C for 10 min and then incubated on ice for more than 1 min. Single strand cDNA was synthesized from the cellular mRNA by adding SuperScript II reverse transcriptase (1 μl, 200 U μl−1) and buffer (4 μl, 5 × first strand buffer (mm): 250 Tris-HCl, 375 KCl, 15 MgCl2), RNAsin (0.5 μl, 28 U μl−1), DTT (1.5 μl, 0.1 m) and mixed dNTPs (1 μl, 10 mm). The reaction mixture (20 μl) was incubated at 42 °C for 50 min. The reaction was terminated by heating the mixture to 70 °C for 15 min and then icing. The RNA strand in the RNA-DNA hybrid was then removed by adding 1 μl RNase H (2 U μl−1) and incubating for 20 min at 37 °C. All reagents were obtained from GIBCO BRL (Grand Island, NY, USA).
The cDNA from the reverse transcription (RT) of RNA in single PFC neurons was amplified with the polymerase chain reaction (PCR), which was carried out with a thermal cycler (MJ Research, Inc., Wate rtown, MA, USA) in thin-walled plastic tubes. Reaction mixtures contained 2.5 mm MgCl2, 0.5 mm of each of the dNTPs, 0.4 μm primers, 2.5 U Taq DNA polymerase (Promega, Madison, WI, USA), 5 μl 10 × PCR buffer (Promega) and one-fourth (5 μl) of the cDNA template made from the single-cell RT reaction. Two rounds of amplification were performed to detect GABAA receptor β1–3 subunits. The thermal cycling programme for the first-round amplification was: 94 °C for 1 min, 52 °C for 1 min, 72 °C for 1.5 min for 30 cycles. Two microlitres of the first-round PCR product was used as the cDNA template for the second-round amplification: 94 °C for 1 min, 56 °C for 1 min, 72 °C for 1.5 min for 45 cycles. The PCR primers for GABAA receptor β1–3 subunits were the same as previously described (Yan & Surmeier, 1997). One-round 45-cycle PCR amplification was carried out to detect 5-HT4 receptors. The primers for 5-HT4 receptors were
5′-ACAAGATGACCCCTCTAC
and
5′-TAGCGCTCATCATCACAG.
PCR products were separated by electrophoresis in ethidium bromide-stained 1.5 % agarose gels. Negative controls for contamination from extraneous and genomic DNA were run for every batch of neurons.
PKA assay
After PFC slices were incubated in NaHCO3-buffered saline for 1 h, half of them were transferred to the high KCl (25 mm) saline solution and incubated for 15–30 min. Following the treatment, slices were transferred to ice cold extraction buffer (5 mm EDTA, 50 mm Tris, pH 7.5, with various protease inhibitors) and homogenized immediately on ice. Insoluble material was removed by centrifugation (13 000 g for 15 min at 4 °C). Assays of PKA enzymatic activity in the supernatant fractions were carried out using the PKA assay system from Life Technology (Rockville, MD, USA). Briefly, samples were aliquoted into four tubes (a-d). PKA activity was measure by phosphorylation of the Kemptide (LRRASLG) in the absence or presence of activator (cAMP) and/or inhibitor (PKI). Tube (a) contained samples only, while tube (b) also contained PKI. Only cAMP was added to samples in tube (c), while both cAMP and PKI were added to tube (d). After 5 min incubation at 30 °C, samples were spotted on phosphocellulose discs, which were then washed in 1 % phosphoric acid and measured in a liquid scintillation counter. The difference of counts (a − b) in tubes (a) and (b) reflects the amount of activated PKA in the sample, while (c − d) reflects the amount of total PKA. The ratio (a − b)/(c − d) represents the level of PKA activation, which is indicative of the intracellular cAMP level. Percentages of PKA activation in response to different stimuli were normalized against the control. Data were represented as means ± s.e.m. of five experiments, and were analysed with an unpaired t test.
Immunocytochemistry
Freshly dissociated neurons were precipitated on poly-lysine-coated coverslips. After 10 min, they were fixed in 4 % paraformaldehyde in PBS for 20 min and permeabilized with 0.3 % Triton X-100 for 5 min. Following 1 h incubation with 10 % bovine serum albumin (BSA) to block non-specific staining, the cells were incubated overnight with the phospho-CREB (Ser133) antibody (Cell Signaling Technology Inc., Beverly, MA, USA, 1:1000) at 4 °C. After washing off the primary antibody, the cells were incubated with a fluorescein-conjugated secondary antibody (Sigma, 1:200) for 50 min at room temperature. Rhodamine-phalloidin (Molecular Probes, Eugene, OR, USA 1:2000) was then added to the cells which were incubated for 20 min. After washing in PBS three times, the coverslips were mounted on slides with VECTASHIELD mounting media (Vector Laboratories Inc., Burlingame, CA, USA). Fluorescent images were obtained using a Bio-Rad confocal microscope with a × 100 oil lens. To quantify nuclear p-CREB, the nucleus of each neuron examined was manually outlined, and then the intensity of immunoreactivity for p-CREB within the outlined nuclei was quantified using Universal Imaging Matamorph software (Universal Imaging Co., Downingtown, PA, USA).
RESULTS
Activation of 5-HT4 receptors has a dual effect on GABAA currents in PFC pyramidal neurons
In rats, the prelimbic, infralimbic and ventral anterior cingulate cortex represent the major subdivisions of the PFC (Groenewegen, 1988). Based on their patterns of neural connectivity, these regions are thought to be functionally related to PFC in the primate (Kolb, 1984; Uylings & van Eden, 1990; Conde et al. 1995). To test the potential impact of 5-HT4 receptors on the activation of postsynaptic GABAA receptors in PFC, we examined the effect of 5-HT4 receptor agonist 5-methoxytryptamine (5-MT) on GABAA receptor-mediated currents in dissociated pyramidal neurons located in the intermediate and deep layers (III-VI) of the rat PFC. Acutely isolated PFC pyramidal neurons were readily distinguished from GABAergic interneurons by their distinct morphological features: a pyramidal-shaped soma and a prominent apical dendrite. Neurons with similar soma sizes and dendritic arborizations were selected to minimize the heterogeneity of cells. The expression of glutamic acid decarboxylase mRNA was consistently negative in the harvested neurons (data not shown), confirming that they are not GABAergic interneurons.
In the 154 PFC pyramidal neurons we tested, bath application of 5-MT (20 μm) had an effect on GABA-evoked currents in 81 neurons (52.6 %), consistent with the presence of 5-HT4 receptors in about 60 % of the PFC pyramidal neurons detected with the single-cell mRNA profiling method (Feng et al. 2001). Among the 81 responsive cells, 5-MT caused an enhancement of GABAA currents in 49 cells and a reduction in the other 32 cells. The dual effect of 5-MT in PFC pyramidal neurons is illustrated in Fig. 1 (enhancement: Fig. 1A and B; reduction: Fig. 1C and D). The bidirectional modulation of GABAA currents by 5-MT was reversible, and had slow onset kinetics, taking 2–4 min to stabilize. Following recovery from the first application, a second application of 5-MT resulted in a similar response with the same direction (enhancement: 90.1 ± 9.3 % of first response, n = 5; reduction: 93.7 ± 7.5 % of first response, n = 6). The desensitization kinetics of the GABAA current were not significantly altered by 5-MT (current decay rate fitted by a single exponential: τ = 1.15 ± 0.19 s in the absence of 5-MT, τ = 1.17 ± 0.22 s in the presence of 5-MT, n = 12, P > 0.1, unpaired t test). The enhancement of peak GABAA currents by 5-MT was 17.8 ± 9.7 % (mean ± s.d., n = 49, P < 0.01, Mann-Whitney U test), and the reduction of peak GABAA currents by 5-MT was 13.6 ± 4.9 % (n = 32, P < 0.01). Similar modulation was observed when different concentrations (25 μm, 100 μm and 1 mm) of GABA were applied or membrane potentials were held at different levels (−40 mV, −20 mV and 0 mV) (data not shown).
Figure 1. Application of 5-HT4 agonists caused an enhancement or a reduction of GABAA receptor currents in PFC pyramidal neurons.
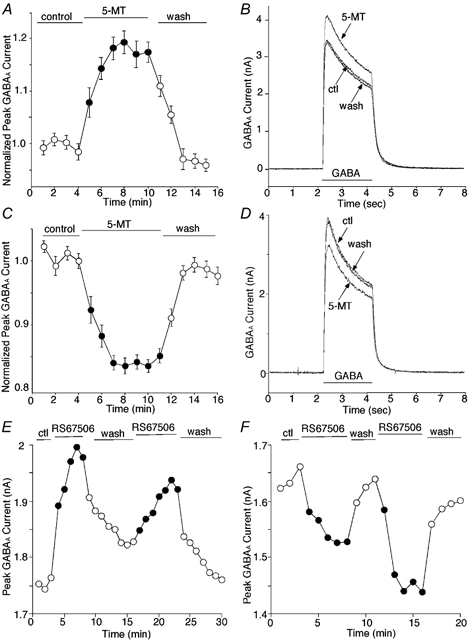
A and C, plot of mean ± s.e.m. normalized peak GABAA current of all PFC pyramidal neurons that responded to 5-MT (n = 81) as a function of time and agonist application. The 5-HT4 agonist 5-MT (20 μm) reversibly enhanced (A, n = 42) or reduced (C, n = 39) GABA (100 μm)-evoked currents in these cells. B and D, current traces taken from representative cells in which enhancement (B) or reduction (D) was induced by 5-MT. E and F, plot of peak GABAA current as a function of time and agonist application. The selective 5-HT4 agonist RS67506 (20 μm) reversibly enhanced (E) or reduced (F) GABA (50 μm)-evoked currents in PFC pyramidal neurons.
We also tested the impact of another structurally unrelated, selective 5-HT4 agonist RS67506 (Eglen et al. 1995a) on GABAA currents in PFC pyramidal neurons. Two representative examples are shown in Fig. 1. Similar to 5-MT, RS67506 enhanced GABAA currents in some cells (Fig. 1E), and reduced them in others (Fig. 1F). Consecutive applications of RS67506 produced comparable responses. The enhancement of peak GABAA currents by RS67506 was 18.1 ± 8.6 % (mean ± s.d., n = 9, P < 0.01, Mann-Whitney U test), and the reduction of peak GABAA currents by RS67506 was 13.7 ± 5.8 % (n = 11, P < 0.01). Another 5-HT4 receptor agonist 2-[1-(4-piperonyl)piperaziny] benzothiazole (20 μm, Monge et al. 1994) gave similar effects: enhancing (n = 4) or reducing GABAA currents (n = 5) by amounts similar to those found with 5-MT treatment (data not shown).
To further confirm that 5-HT4 receptors mediate the effect of 5-MT on GABAA currents, the ability of the selective 5-HT4 receptor antagonist SDZ205557 to prevent the action of 5-MT was examined. As shown in Fig. 2A and B, 5-MT reversibly enhanced GABAA currents in the PFC pyramidal neuron, and this effect was abolished in the presence of SDZ205557 (20 μm). Figure 2C shows results for another PFC pyramidal neuron. 5-MT reduced GABAA currents in this cell, and co-application of SDZ205557 blocked this effect. Removing the antagonist restored the ability of 5-MT to reduce GABAA currents.
Figure 2. Selective 5-HT4 receptor antagonists blocked the dual effect of 5-HT4 agonists on GABAA currents.
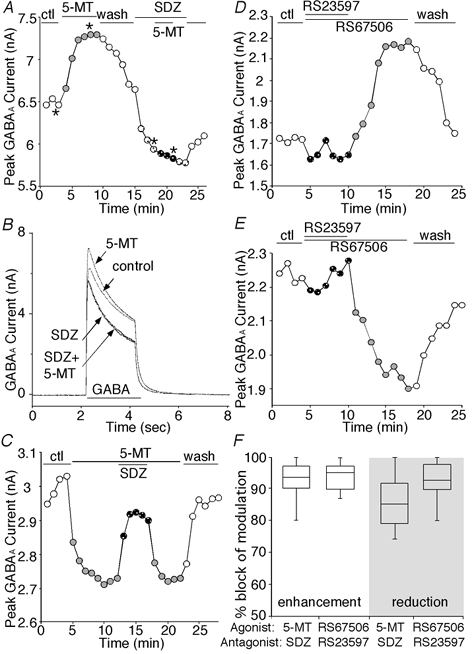
A, plot of peak GABAA current as a function of time and ligand application. 5-MT (20 μm) reversibly enhanced GABAA currents in the PFC pyramidal neuron, and this effect was eliminated in the presence of the 5-HT4 antagonist SDZ205557 (20 μm). B, representative current traces taken from the records used to construct A (at time points denoted by *). C, plot of peak GABAA current as a function of time and ligand application. 5-MT (20 μm) reduced GABAA currents in the PFC pyramidal neuron, and co-application of SDZ205557 (20 μm) abolished this effect. Washing off the antagonist led to recovery of the 5-MT inhibition. D and E, plot of peak GABAA current as a function of time and ligand application. In the presence of the 5-HT4 antagonist RS23597-190, the 5-HT4 agonist RS67506 had little effect, and washing off the antagonist led to emergence of RS67506-induced enhancement (D) or reduction (E) of GABAA currents. F, box plots showing the percentage block of agonist (5-MT, 20 μm or RS67506, 20 μm) effect on GABAA currents by 5-HT4 antagonist (SDZ205557, 20 μm or RS23597-190, 20 μm). Note that SDZ205557 significantly blocked 5-MT-induced enhancement (n = 5) or reduction (n = 9) of GABAA currents, whereas RS23597-190 significantly blocked RS67506-induced enhancement (n = 6) or reduction (n = 5) of GABAA currents.
We also examined the ability of another selective 5-HT4 receptor antagonist RS23597–190 (Eglen et al. 1993) to prevent the action of the selective 5-HT4 receptor agonist RS67506 on GABAA currents in PFC pyramidal neurons. As shown in Fig. 2D and E, RS67506 had little effect on GABAA currents in the presence of RS23597-190. After washing off the antagonist, application of RS67506 induced a strong enhancement (Fig. 2D) or reduction (Fig. 2E) of GABAA currents. A summary of the percentage block of agonist effect on GABAA currents by 5-HT4 antagonists in PFC pyramidal neurons is shown in Fig. 2F. SDZ205557 abolished 92.4 ± 7.8 % (mean ± s.d., n = 5) of 5-MT-induced enhancement of GABAA currents and 85.7 ± 9.3 % (n = 9) of 5-MT-induced reduction of GABAA currents (P < 0.001, 5-MT effect in the absence vs. presence of SDZ205557, unpaired t test). Likewise, RS23597-190 abolished 96.7 ± 8.0 % (n = 6) of RS67506-induced enhancement of GABAA currents and 92.8 ± 8.3 % (n = 5) of RS67506-induced reduction of GABAA currents (P < 0.001, RS67506 effect in the absence vs. presence of RS23597-190, unpaired t test). These results indicate that the dual effect on GABAA currents is mediated by 5-HT4 receptors.
Our previous work has shown that 5-HT, by activating 5-HT2 receptors, inhibits GABAA currents through anchored PKC (Feng et al. 2001). Because of the abundant expression of 5-HT2 receptors in almost all PFC pyramidal neurons, the 5-HT2-mediated effect seems to play a dominant role in serotonergic regulation of the postsynaptic GABA response. To reveal the role of 5-HT4 receptors in serotonin signalling, we have tested the impact of serotonin on GABAA currents in the presence of the specific 5-HT2 antagonist ketanserin (2 μm). A bidirectional modulation was observed with serotonin when 5-HT2 receptors were selectively blocked (enhancement: n = 5; reduction: n = 6, data not shown), suggesting that by activating 5-HT4 receptors, 5-HT exerts a dual effect on GABAA channels in PFC neurons.
The dual effect of 5-HT4 receptors on GABAA currents in PFC neurons is mediated by anchored PKA
We next examined the signal transduction pathways mediating the bidirectional modulation of GABAA currents by 5-HT4 receptors. GABAA channels are thought to be heteropentameric structures, composed of different subunits (Macdonald & Olsen 1994). PKA phosphorylation of GABAA receptor subunits exerts a powerful impact on recombinant and native GABAA channels (Porter et al. 1990; Moss et al. 1992a, b; Kapur & Macdonald, 1996). Activation of 5-HT4 receptors can couple to Gs proteins to stimulate adenylyl cyclase and cAMP production (Monferini et al. 1993). This led us to speculate that the 5-HT4 regulation of GABAA currents is through the PKA-mediated pathway. To test this, we first applied selective PKA activators. Application of the membrane-permeant cAMP analogue cpt-cAMP (200 μm) mimicked the dual effect of the 5-HT4 agonist 5-MT. In the PFC neurons in which 5-MT enhanced GABAA currents, subsequent application of cpt-cAMP after currents had returned to the basal level following washing off of the 5-MT, also caused an enhancement of GABAA currents (Fig. 3A). Similarly, in the PFC neurons in which 5-MT reduced GABAA currents, subsequent application of cpt-cAMP after currents returned to their basal level following washing off of the 5-MT, also caused a reduction of GABAA currents (Fig. 3B). Another PKA activator, sp-cAMPS (50 μm), gave similar results in PFC neurons, mimicking the dual effect of 5-MT on GABAA currents (data not shown). Bath application of the membrane-impermeant cAMP (200 μm) had no effect on GABAA currents in 5-MT-responsive neurons (n = 8; data not shown). Figure 3C shows a summary comparing the effects of PKA activators (cpt-cAMP and sp-cAMPS) and the 5-HT4 agonist 5-MT. The enhancement by PKA activators (17.4 ± 2.1 %, mean ± s.d., n = 7) and reduction by PKA activators (16.8 ± 1.8 %, n = 10) was similar to that seen with 5-MT (enhancement: 15.7 ± 1.3 %, n = 7; reduction: 13.9 ± 1.9 %, n = 10; P > 0.05, PKA activators vs. 5-MT, unpaired t test).
Figure 3. The dual effect of 5-MT was mimicked by PKA activators and blocked by PKA inhibition.
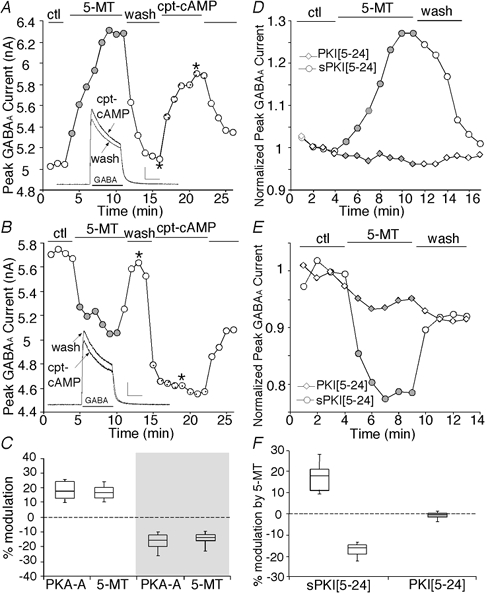
A and B, plot of peak GABAA current as a function of time and drug application. The 5-HT4 agonist 5-MT (20 μm) reversibly enhanced (A) or reduced (B) GABAA currents in the PFC neuron. Following recovery, application of the membrane-permeant cAMP analogue cpt-cAMP (200 μm) produced an effect that was similar to 5-MT, enhancing (A) or reducing (B) GABAA currents. Inset: representative current traces taken from the records used to construct A or B (at time points denoted by *). Scale: 1 nA, 1 s C, box plot summary of the percentage modulation of GABAA currents by PKA activators (PKA-A) or 5-MT. Note that these PKA activators (cpt-cAMP or sp-cAMPS) mimicked both the enhancement (n = 7) and reduction (n = 10) of GABAA currents caused by 5-MT. D and E, plot of peak GABAA current as a function of time and drug application in neurons dialysed with PKI[5–24] or sPKI[5–24]. The specific PKA inhibitory peptide PKI[5–24] (20 μm), but not the scrambled control peptide sPKI[5–24] (20 μm), eliminated 5-MT-induced enhancement (D) or reduction (E) of GABAA currents. F, box plot summary of the percentage modulation of GABAA currents by 5-MT in 5-HT4 mRNA-positive neurons dialysed with PKI[5–24] (n = 10) or sPKI[5–24] (enhancement: n = 5; reduction: n = 6).
To confirm the involvement of PKA in 5-MT modulation of GABAA currents, we dialysed neurons with the specific PKA inhibitory peptide PKI[5–24] (Knighton et al. 1991) and then examined the effect of 5-MT; this was followed by single-cell RT-PCR for the detection of 5-HT4 receptor mRNA in the harvested cells. Representative cells with positive expression of 5-HT4 mRNA are shown in Figs 3D and E. 5-MT-induced enhancement or reduction was abolished in neurons dialysed with PKI[5–24] (20 μm), while the dual effect was intact in neurons dialysed with the scrambled control peptide sPKI[5–24] (20 μm). In the 10 PKI[5–24]-loaded neurons that were 5-HT4 mRNA positive (total cells tested: n = 20), 5-MT-induced modulation of GABAA currents was significantly less than the effect of 5-MT in the 11 sPKI[5–24]-loaded neurons that were 5-HT4 mRNA positive (total cells tested: n = 20) (Fig. 3F; P < 0.01, PKI vs. sPKI, unpaired t test). Taken together, these results indicate that PKA is mediating the dual effect of 5-MT on GABAA currents.
Emerging evidence has shown that PKA, a kinase with broad substrate selectivity, achieves the efficacy and specificity of signal transduction through anchoring protein-mediated subcellular targeting to its substrates in central neurons (Colledge & Scott, 1999). We next examined whether the PKA-anchoring proteins AKAPs (A kinase anchoring proteins) are involved in 5-HT4 modulation of GABAA channels in PFC pyramidal neurons. If AKAPs are responsible for targeting PKA to GABAA receptor channels and allowing PKA to effectively phosphorylate these substrates, then blocking the interaction of PKAQ-AKAPs should lead to the removal of PKA from the vicinity of GABAA receptors, thereby attenuating PKA regulation of these channels. To test this hypothesis, we dialysed neurons with the PKA-anchoring inhibitory peptide H31 (Rosenmund et al. 1994) and examined the effect of 5-HT4 receptors on GABAA currents. As shown in Fig. 4A, dialysis with H31 eliminated the ability of 5-MT to modulate GABAA currents, even though 5-HT4 receptors were expressed in this neuron (Fig. 4A, inset). In contrast, a control peptide with scrambled amino acid sequence, sH31, had no effect on 5-HT4 regulation of GABAA currents (Fig. 4B). In the 11 5-HT4 mRNA-positive PFC neurons dialysed with the peptide H31, 5-MT enhanced GABAA currents by 3.4 ± 1.5 % (mean ± s.d., n = 6, P > 0.05, Mann-Whitney U test, Fig. 4C) and reduced GABAA currents by 3.3 ± 2 % (n = 5, P > 0.05, Fig. 4C). On the other hand, in the 13 5-HT4 mRNA-positive PFC neurons dialysed with the control peptide sH31, 5-MT enhanced GABAA currents by 15.8 ± 7.2 % (n = 7, P < 0.01, Fig. 4C) and reduced GABAA currents by 15.3 ± 3.5 % (n = 6, P < 0.01, Fig. 4C). Thus, the dual effect of 5-MT on GABAA currents was significantly attenuated by the PKAanchoring inhibitory peptide H31 (P < 0.01, H31 vs. sH31, unpaired t test).
Figure 4. 5-HT4 modulation of GABAA receptor function required anchoring of PKA to the channel by AKAPs.
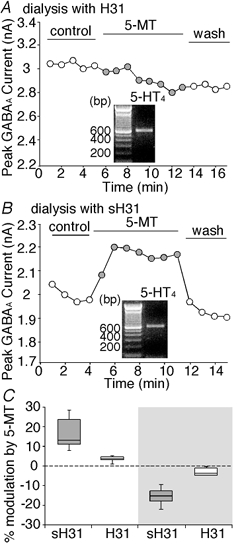
A and B, plot of peak GABAA current as a function of time and agonist application with the PKA-anchoring inhibitory peptide H31 (5 μm) (A) or the control peptide sH31 (5 μm) (B) in the recording pipette. Inset: expression profile of 5-HT4 receptor mRNA in the same PFC neuron harvested after recording. C, box plot summary of the percentage modulation of GABAA currents by 5-MT (20 μm) in the presence of H31 peptide (enhancement: n = 6; reduction: n = 5) or the scrambled peptide sH31 (enhancement: n = 7; reduction: n = 6). Note that the dual effect of 5-MT on GABAA currents was significantly attenuated by the PKA-anchoring inhibitory peptide H31.
The 5-HT4-induced bidirectional regulation of GABAA currents in different PFC neurons cannot be attributed to differential expression of GABAA receptor β1 and β3 subunits
Multiple PKA phosphorylation sites have been identified in GABAA receptor β1 and β3 subunits (Moss et al. 1992a, b). In recombinant systems, PKA phosphorylation of β3 subunit-containing receptors at S408 and S409 enhanced GABAA currents, whereas PKA phosphorylation of β1 subunit-containing receptors solely on S409 reduced GABAA currents (McDonald et al. 1998). To reveal the potential mechanisms underlying 5-MT-induced bidirectional regulation of GABAA currents in PFC pyramidal neurons, we first examined the expression of GABAA receptor β subunits in these neurons. We hypothesize that PFC pyramidal neurons may express different GABAA receptor β subunits, and differential expression of β subunits may explain the different effects of the 5-HT4/PKA signalling on GABAA currents.
To test this idea, the single-cell mRNA profiling technique (Yan & Surmeier, 1996, 1997) was used to detect the coordinated expression of GABAA receptor β1–3 subunits in individual PFC pyramidal neurons. Figure 5A shows two representative examples, with one PFC pyramidal neuron showing the co-expression of β1 and β3 subunits and another neuron showing the co-expression of all three β subunits (β1-β3). The coordinated expression of GABAA receptor β subunits in a sample of 12 PFC pyramidal neurons is summarized in Fig. 5B. β1 and β3 subunits were consistently co-expressed at similar levels in all the cells we tested. The co-expression of GABAA receptor β1 and β3 subunits in PFC pyramidal neurons suggests that these cells have a mixture of β1 subunit-containing and β3 subunit-containing GABAA receptors. The 5-MT-induced enhancement or reduction of GABAA currents in PFC neurons may therefore be caused by factors other than the differential expression of GABAA receptor β subunits in these cells.
Figure 5. Both β1 and β3 GABAA receptor subunits were co-expressed in single PFC pyramidal neurons.
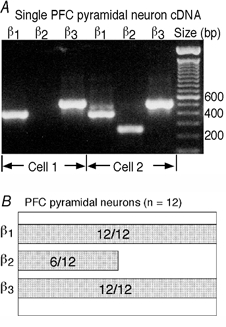
A, expression profile of GABAA receptor β1–3 subunit mRNAs in two representative PFC pyramidal neurons. In one neuron, β1 and β3 were co-expressed, while in the other neuron, all three β subunits were co-expressed. B, bar plot showing the coordinated expression of GABAA receptor β1–3 subunit mRNAs in a sample of 12 PFC pyramidal neurons. The extent of co-expression is indicated by the overlap of the bars.
The direction of the effect of 5-HT4 on GABAA currents can be converted by changing PKA activation levels
Since the direction of the effect of 5-MT on GABAA currents cannot be attributed to differential expression of GABAA receptor β subunits in PFC pyramidal neurons, we hypothesize that the dual effect of 5-MT on GABAA currents may be dependent on the basal phosphorylation states of GABAA receptors. When the basal PKA level is low, 5-MT may cause an enhancement of GABAA currents primarily due to β3 subunit phosphorylation. On the other hand, when the basal PKA level is high, 5-MT may cause a reduction of GABAA currents primarily due to β1 subunit phosphorylation. If this hypothesis is true, then changing PKA activation levels and the ensuing phosphorylation state of GABAA receptors should be able to alter the direction of the effect of 5-MT on GABAA currents in the same cell.
To test this, we transiently applied the selective PKA activator sp-cAMPS or PKA inhibitor rp-cAMPS, both of which are resistant to hydrolysis by cyclic nucleotide phosphodiesterases, to adjust the basal phosphorylation states of GABAA receptors. Consistent with our hypothesis, the direction of the effect of 5-MT on GABAA currents was changed by these reagents. As shown in Fig. 6A, 5-MT slightly reduced GABAA currents in this PFC pyramidal neuron (presumably due to its high basal level of PKA activation). After inhibiting PKA with rp-cAMPS, reapplication of 5-MT caused a significant enhancement of GABAA currents. Figure 6B shows another case; 5-MT enhanced GABAA currents in this PFC pyramidal neuron (presumably due to its low basal level of PKA activation). After activating PKA with sp-cAMPS, reapplication of 5-MT caused a reduction of GABAA currents. In agreement with this, a transient application of 3-isobutyl-1-methylxanthine (IBMX; Sigma Chemical Co.), which inhibits cAMP phosphodiesterase (PDE) and thus raises cAMP levels to activate PKA, also switched the effect of 5-MT from enhancement to reduction (Fig. 6C). Furthermore, when the basal phosphorylation state of GABAA receptors was elevated by inhibiting protein phosphatase 1/2A with okadaic acid (OA), the 5-MT-induced enhancement of GABAA currents was converted to reduction. After washing off okadaic acid to lower the basal phosphorylation state of GABAA receptors, the 5-MT-induced enhancement of GABAA currents recovered (Fig. 6D). Figure 6E summarizes the results, showing that 5-MT-induced reduction of GABAA currents was converted to enhancement following rp-cAMPS treatment (n = 9), while 5-MT-induced enhancement of GABAA currents was converted to reduction by sp-cAMPS (n = 5), IBMX (n = 5) or OA (n = 5).
Figure 6. The direction of effect of 5-MT on GABAA currents was reversed by changing PKA activation levels.
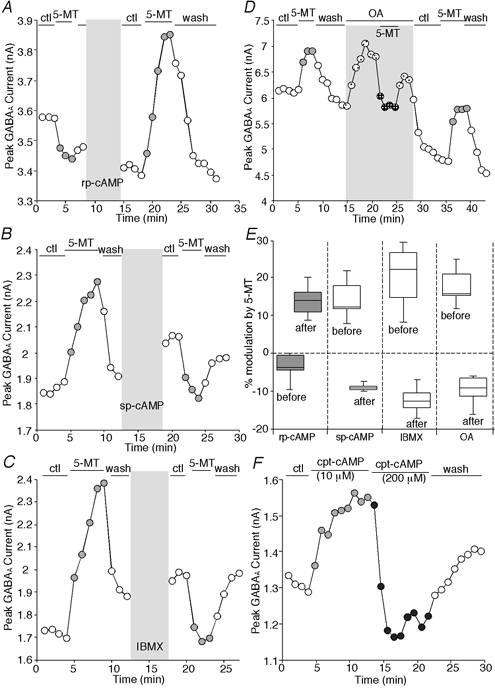
A–D, plots of peak GABAA current as a function of time and drug application. Transient treatment with the PKA inhibitor rp-cAMPS (50 μm) converted the effect of 5-MT (20 μm) from reduction to enhancement of GABAA currents (A). Short treatment with the PKA activator sp-cAMPS (50 μm) (B) or PDE inhibitor IBMX (1 mm) (C) converted the effect of 5-MT (20 μm) from enhancement to reduction. Application of the protein phosphatase 1/2A inhibitor okadaic acid (OA, 0.5 μm) also converted the effect of 5-MT (20 μm) from enhancement to reduction (D). E, box plot summary showing that the direction of 5-MT effect on GABAA currents in PFC neurons was reversed by rp-cAMPS (n = 9), sp-cAMPS (n = 5), IBMX (n = 5) or OA (n = 5). F, plot of peak GABAA current as a function of time and drug application. Following PKA activation by a low concentration (10 μm) of cpt-cAMP, application of a high concentration (200 μm) of cpt-cAMP converted the enhancing effect to a depressing one.
To provide further evidence that the polarity of the 5-MT/PKA effect is dependent on the basal phosphorylation level of GABAA receptors, we consecutively applied different concentrations of cpt-cAMP. As shown in Fig. 6F, application of a low concentration (10 μm) of cpt-cAMP caused an enhancement of GABAA currents in the PFC pyramidal neuron. On the basis of elevated PKA and enhanced GABAA currents, subsequent application of a high concentration (200 μm) of cpt-cAMP induced a depressing effect. In the PFC neurons we tested, cpt-cAMP (10 μm) enhanced GABAA currents by 15.6 ± 4.5 % (mean ± s.d., n = 7, P < 0.01, baseline: original basal current) and subsequent application of cpt-cAMP (200 μm) reduced GABAA currents by 16.5 ± 6.9 % (n = 7, P < 0.01, baseline: cpt-cAMP (10 μm)-elevated current).
Neuronal activity determines basal PKA activation levels and the direction of the effect of 5-HT4 on GABAA currents
Since 5-MT can enhance or reduce GABAA currents in PFC pyramidal neurons depending on their basal PKA activation levels and the phosphorylation states of GABAA receptors, we hypothesize that different levels of neuronal activity may determine the basal PKA activation levels and therefore the direction of the effect of 5-MT on GABAA currents in PFC neurons. If this is the case, then changing neuronal activity should be able to alter the direction of the effect of 5-MT on GABAA currents. To test this hypothesis, we incubated PFC slices with a solution containing a high concentration (25 mm) of KCl to increase the basal neuronal activity, this was followed by examination of the effect of 5-MT on GABAA currents in PFC neurons acutely dissociated from these slices. To determine whether high K+-induced membrane depolarization could increase PKA activity in PFC neurons, we first compared the levels of PKA activation in non-treated and KCl-treated slices using a PKA assay system. As shown in Fig. 7A, high KCl (25 mm) treatment increased PKA activity 2.3 ± 0.3-fold (n = 5), while forskolin increased it 5.2 ± 0.4-fold.
Figure 7. Neuronal activity determined basal PKA activation levels and the direction of 5-MT effect on GABAA currents.
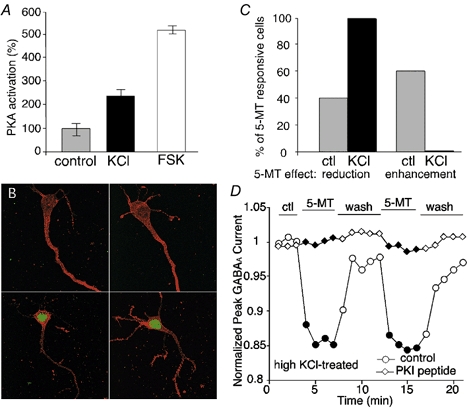
A, activation of PKA in response to a high concentration of KCl or forskolin (FSK). PFC slices were treated with KCl (25 mm) or forskolin (10 μm). Levels of PKA activation were measured and normalized to the control (n = 5). B, confocal images of double immunostaining with rhodamine-phalloidin (red) and a phosph-CREB (Ser133) antibody (green) in acutely dissociated PFC pyramidal neurons from non-treated (upper panel) vs. KCl-treated (lower panel) slices. Two representative cells are shown in each case. C, histogram summary showing the percentage of neurons in which 5-MT caused a reduction or an enhancement of GABAA currents under control and KCl-treated conditions. In non-treated neurons which showed responses to 5-MT (n = 81), 5-MT-induced reduction of GABAA currents was found in about 40 % of them (32/81) and enhancement was found in about 60 % of them (49/81). In KCl-treated neurons which showed responses to 5-MT (n = 12), 5-MT-induced reduction of GABAA currents was found in all of them (12/12) and no enhancement was found (0/12). D, plot of peak GABAA current as a function of time and agonist application in high KCl-treated PFC neurons dialysed with or without PKI[5–24] (20 μm). In the absence of PKI[5–24], 5-MT (20 μm) reversibly reduced GABAA currents.
To provide additional evidence showing the different PKA activation levels in isolated PFC pyramidal neurons from non-treated vs. KCl-treated slices, we next examined the phosphorylation of cAMP response element-binding protein (CREB) in these cells. CREB is a plasticity-associated transcription factor that is phosphorylated at Ser-133 by multiple protein kinases, including PKA and Ca2+/calmodulin-dependent protein kinases (for review see Shaywitz & Greenberg, 1999). Once CREB is phosphorylated, it is translocated from the cytosol to the nucleus. Confocal images of double labelling with phospho-CREB (Ser133) and F-actin in dissociated PFC pyramidal neurons are shown in Fig. 7B. In the neurons from non-treated slices (upper panel), p-CREB signals were barely visible, while in the neurons from KCl-treated slices (lower panel), strong p-CREB signals in the nucleus were evident. Confocal quantification of nuclear p-CREB immunofluorescence confirmed that KCl treatment induced a significant increase of CREB phosphorylation (mean arbitrary fluorescence units: 82 (n = 10, non-treated), 224 (n = 10, KCl-treated), P < 0.001, unpaired t test). These results suggest that our dissociation procedure did not cause a massive increase in the intracellular cAMP level, while KCl-induced increase of PKA activity could directly lead to the elevation of p-CREB. Alternatively, p-CREB is stimulated by an activity-dependent increase of cytosolic Ca2+ following KCl treatment, which also requires PKA activation (Impey et al. 1998).
We then examined the effect of 5-MT on GABAA currents in KCl-treated PFC neurons. For the 23 KCl-treated neurons that were responsive to 5-MT (n = 12), only a reduction of GABAA currents was observed in response to 5-MT (12/12); no enhancement was found (0/12; Fig. 7C). In contrast, for the 154 non-treated PFC neurons that were responsive to 5-MT (n = 81), 5-MT caused a reduction of GABAA currents in about 40 % (32/81) and an enhancement of GABAA currents in about 60 % of them (49/81; Fig. 7C). These results indicate that increased neuronal activity can elevate the basal level of PKA activation, switching the effect of 5-MT on GABAA currents from enhancement to reduction. A representative example showing the modulation of 5-MT on GABAA currents in a high KCl-treated PFC neuron is illustrated in Fig. 7D. Repeated application of 5-MT caused a reversible reduction of GABAA currents in this cell that had been subjected to membrane depolarization and high synaptic activity. On the contrary, in another KCl-treated neuron that was dialysed with PKI[5–24], 5-MT had little effect on GABAA currents (Fig. 7D). Single-cell mRNA profiling after recording showed that 5-HT4 receptor mRNA was expressed in this cell (data not shown). In KCl-treated 5-HT4 mRNA-positive neurons loaded with the control internal solution, 5-MT reduced GABAA currents by 18.2 ± 6.3 % (mean ± s.d., n = 12, P < 0.01, Mann-Whitney U test), while in KCl-treated 5-HT4 mRNA-positive neurons loaded with PKI[5–24], 5-MT reduced GABAA currents by 1.6 ± 1.0 % (n = 8; P > 0.1), which was significantly smaller (P < 0.001, control vs. PKI[5–24], unpaired t test). These results suggest that the effect of KCl is not due to factors other than PKA.
To understand the impact of 5-HT4 receptors on GABAergic synaptic transmission, we further examined the effect of 5-MT on GABAA receptor-mediated inhibitory synaptic currents (IPSCs) in PFC slices. Spontaneous IPSCs (sIPSCs), which were sensitive to the GABAA receptor antagonist bicuculline (30 μm), were recorded in PFC pyramidal neurons. Among the 14 responsive neurons (total n = 21), 5-MT showed bidirectional regulation of the amplitude of sIPSCs (enhancement: 21.2 ± 7.3 %, mean ± s.e.m., n = 8, P < 0.001, K-S test; reduction: 23.3 ± 6.2 %, n = 6, P < 0.001). The frequency of sIPSCs was not significantly changed by 5-MT in most of the 14 responsive cells (6.6 ± 5.3 %, mean ± s.e.m., n = 12, P > 0.05, K-S test), and only two cells showed an 5-MT-induced increase in the frequency of sIPSC (24.6 ± 5.0 %, P < 0.01, K-S test). These results suggest that presynaptic function or spontaneous firing of the presynaptic neuron was not significantly altered by 5-HT4 receptors in most cases. A representative example is illustrated in Fig. 8. In the same neuron, 5-MT initially increased the sIPSC amplitude reversibly (mean ± s.e.m.: 14.8 ± 0.3 pA (control), 18.8 ± 0.4 pA (5-MT), P < 0.001, K-S test, Fig. 8A and B). Bath perfusion of KCl (15 mm) induced a strong depolarization and triggered trains of action potentials (Fig. 8C and D). Following the KCl-treatment, reapplication of 5-MT reversibly decreased the sIPSC amplitude (mean ± s.e.m.: 24.3 ± 0.6 pA (control), 13.1 ± 0.5 pA (5-MT), P < 0.001, Fig. 8E and F). The KCl-induced switch of the effect of 5-MT on sIPSC amplitude from enhancement to reduction was observed in four other pyramidal neurons we tested. Overall, our results suggest that the direction of 5-HT4 modulation of GABAA receptor channels is determined by intracellular PKA activation levels, which in turn are controlled by neuronal activity.
Figure 8. Changing neuronal activity switched the effect of 5-MT on the amplitude of spontaneous inhibitory synaptic currents in PFC slices.
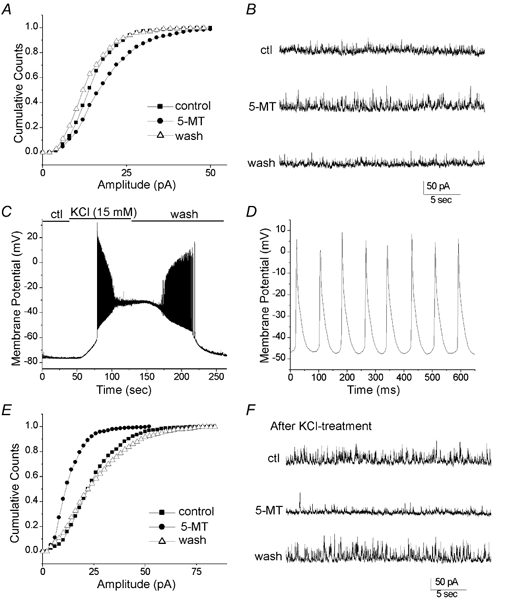
A, cumulative plots indicating that the distribution of sIPSC amplitude was reversibly increased by 5-MT (20 μm). B, sIPSCs recorded under control conditions, during bath application of 5-MT and after washing off the agonist. C, Current clamp recording showing the changes of membrane potentials under control conditions, during bath application of KCl (15 mm) and after washing off KCl. KCl-induced depolarization triggered trains of action potentials in the recorded neuron. D, expanded view of AP spikes during KCl treatment. E, cumulative plots after KCl treatment indicating that the distribution of sIPSC amplitude was reversibly decreased by 5-MT (20 μm). F, sIPSCs recorded after KCl treatment under control conditions, during bath application of 5-MT and after washing off the agonist.
DISCUSSION
Though highly enriched in the limbic system, including basal ganglia, hippocampus and frontal cortex (Domenech et al. 1994), 5-HT4 receptors selectively facilitate the release of ACh in the frontal cortex (Consolo et al. 1994), suggesting that frontal cortical 5-HT4 receptors are not tonically activated (Eglen et al. 1995b). Consistent with a potential role in cognition, 5-HT4 receptors decline significantly in the hippocampus and frontal cortex of patients with Alzheimer's disease (Reynolds et al. 1995). The regulation of synaptic plasticity, which is fundamental in learning and memory, involves modulation of ion channel activity. Previous studies have found that 5-HT4 receptor activation exerts an excitatory impact on neurons by regulating different voltage-dependent ion channels (Bobker & Williams, 1989; Ansanay et al. 1995; Torres et al. 1995; Cardenas et al. 1997). In this study, we identified the GABAA receptor channel as a molecular target of 5-HT4 receptors in PFC.
Our experiments showed that application of 5-HT4 receptor agonists caused a bidirectional modulation of GABAA currents in PFC pyramidal neurons and the dual effect was were mediated by PKA. Among all the 5-HT receptors, only 5-HT4, 5-HT6 and 5-HT7 link to Gs proteins to stimulate PKA. Our single-cell mRNA profiling experiments have shown that 5-HT6 and 5-HT7 were rarely found in PFC pyramidal neurons, whereas 5-HT4 mRNA was detected in ∼60 % of these cells (Feng et al. 2001), consistent with the present finding of a response to 5-MT in 50–60 % of PFC pyramidal neurons. Antagonist blockade and PKA dependence of the dual effect of 5-HT4 agonists further corroborate the mediation by 5-HT4 receptors. Because of the broad substrate selectivity of PKA, subcellular targeting through association with anchoring proteins (AKAPs) has emerged as an important mechanism by which PKA achieves precise substrate recognition and enhanced efficacy of signal transduction (Rosenmund et al. 1994; Gao et al. 1997; Colledge & Scott, 1999). Our results with the PKA-anchoring inhibitory peptide suggest that the 5-HT4 modulation of GABAA receptor channels is a highly localized event that requires the fraction of PKA that is anchored on AKAPs, rather than PKA that is freely floating in the cytosol.
Both PKA-induced enhancement and reduction of GABAA currents have been reported in recombinant systems and native neurons (Moss et al. 1992b; Kapur & Macdonald, 1996). In heterologous expression systems, it has been show that the differential effects of PKA can be attributed to phosphorylation of different GABAA receptor β subunits (McDonald et al. 1998). However, the co-expression of both β1 and β3 subunits in PFC pyramidal neurons led us to search for other reasons underlying the dual effect of 5-HT4 receptors on GABAA currents. Our next set of experiments suggests that the dual effect of 5-MT on GABAA currents is dependent on the basal phosphorylation states of GABAA receptors. In the same neuron, when PKA activation levels were changed by PKA activators or inhibitors, the direction of 5-HT4 modulation of GABAA currents was reversed. Decreasing PKA activation levels switched 5-HT4-induced reduction to enhancement, while increasing PKA activation levels switched 5-HT4-induced enhancement to reduction. One potential mechanism underlying the changes in the polarity of 5-MT effect is the differential phosphorylation of different β subunits of GABAA receptors. When the basal PKA level is relatively low, β3 subunits may first get phosphorylated after exposure to a 5-HT4 agonist, leading to current enhancement. When the basal PKA level is relatively high, 5-HT4 receptor activation may cause the phosphorylation of both β1 and β3 subunits, leading to current reduction due to the predominant role of β1 subunits. Of course, we cannot rule out the possibility that in addition to GABAA receptor β subunits, a further regulatory protein could also be the site of PKA action.
To understand the underlying reasons for the varied basal PKA activation levels in different PFC pyramidal neurons under physiological conditions, we tested the role of neuronal activity in this process. When membrane depolarization was induced in the PFC network, PKA activity was elevated. Presumably, KCl depolarization activated voltage-gated Ca2+ channels, and Ca2+ that entered through these channels stimulated Ca2+/calmodulin-regulated adenylyl cyclases (Hanoune & Defer, 2001). The direction of 5-HT4 regulation is likely to be activity dependent for the following reasons. First, only reduction of GABAA currents by 5-MT was observed in neurons isolated from PFC slices with high neuronal activity and PKA activity (Fig. 7). Second, 5-MT enhancement of sIPSC amplitude in PFC pyramidal neurons in slices was converted to reduction after KCl-induced elevation of excitability (Fig. 8). Because the effect of 5-MT is mediated by PKA (Fig. 3) and the direction of 5-MT modulation can be converted by changing PKA activation levels (Fig. 6), the switch of 5-MT effect from enhancement to reduction in KCl-treated neurons is likely to be attributable to their elevated PKA activation levels.
We notice that the activity-dependent bidirectional regulation of GABAergic signalling by 5-HT4 receptors could potentially form a neural computational module, namely, ‘Activity-Locked Loop (ALL)’ (Fig. 9), which could be simplified to resemble digital circuits controlled by logic gates. For those PFC pyramidal neurons that are at the ‘high activity’ state (logic ‘l’ state), activation of 5-HT4 receptors (logic ‘l’ state) will reduce their postsynaptic responses to GABA (logic ‘0’ state) through a ‘NAND’ gate controlled by high PKA activity. A ‘NAND’ gate performs the logic operation in which two inputs of ‘l’ give an output of ‘0‘. The reduced GABAergic inhibition on these cells will reinforce their ‘high activity’ state through the ‘NOT’ gate afforded by the intrinsic Cl− channel of GABAA receptors (High Activity-Locked Loop). A ‘NOT’ gate performs the logic operation in which one input of ‘0’ gives an output of ‘1’ and vice versa. On the other hand, for those PFC pyramidal neurons that are at the ‘low activity’ state (logic ‘0’ state), activation of 5-HT4 receptors (logic ‘l’ state) will enhance their postsynaptic response to GABA (logic ‘l’ state) through an ‘OR’ gate controlled by low PKA activity. An ‘OR’ gate performs the logic operation in which one input of ‘l’ and one input of ‘0’ gives an output of ‘1’. The enhanced GABAergic inhibition of these cells will reinforce their ‘low activity’ state (Low Activity-Locked Loop).
Figure 9. A neural computational module for 5-HT4 action in PFC pyramidal neurons (see note in Discussion about use of quotes).
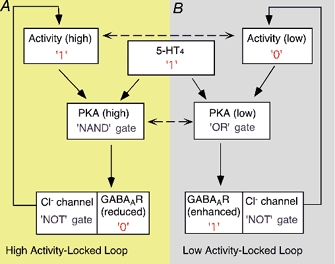
For those cells at the ‘high activity’ state, the concomitant activation of PKA to a high level by 5-HT4 receptors and high neuronal activity leads to a reduction of postsynaptic GABAA receptor functions (‘NAND’ gate), and a reduction of GABA-mediated inhibitory synaptic transmission, which causes the neurons to be locked at the ‘high activity’ state. On the other hand, for those cells at the ‘low activity’ state, the concomitant activation of PKA to a low level by 5-HT4 receptors and low neuronal activity leads to an enhancement of postsynaptic GABAA receptor functions (‘OR’ gate), and an enhancement of GABA-mediated inhibitory synaptic transmission, which causes the neurons to be locked at the ‘low activity’ state. ‘0’, logic 0 state; ‘1’, logic 1 state. ‘NAND’ gate, a gate performing the ‘NOT AND’ function, i.e. the output is ‘0’ only when two inputs are both ‘l’. ‘OR’ gate, a gate performing the logic that the output is ‘l’ as long as there is at least one input at ‘1’. ‘NOT’ gate, a gate performing the logic that the output is ‘l’ when the input is ‘0’ and vice versa.
This ALL module would allow serotonin, by acting on 5-HT4 receptors, to use key molecules within a single neuron (possibly even a single synapse) to perform complex neural computation to maintain the activity state of a neuron (or a synapse). The underlying principle may offer insights into the computational mechanisms of serotonin functions in cognition and emotion. One can envisage that non-physiological, activity-uncoupled changes in the activation level of PKA caused by various drugs of abuse (e.g. cocaine) could disrupt the normal reinforcement of this ALL module, which may lead to altered output that could be manifested behaviourally. Similarly, perturbations of this module at other inputs (e.g. convulsants on GABAAR and antidepressants on 5-HT4R) could also have dramatic behavioural consequences.
Functional implications
This study mechanistically links together two important neurotransmitter systems, serotonin and GABA, both of which have been implicated in neuropsychiatric disorders, such as depression and anxiety (Griebel 1995; Benes et al. 1996; Abi-Dargham et al. 1997; Stockmeier 1997; Ohnuma et al. 1999; Lewis, 2000). The 5-HT4 receptor-mediated modulation of GABAA receptor currents provides a cellular mechanism for the functional roles of serotonin in PFC neurons. Emerging evidence suggests that imbalances in serotonergic neurotransmission and the ensuing dysregulation of GABAergic signalling may contribute significantly to the pathogenesis of mental diseases (Dean, 2001). A novel feature of this modulation is that it is bidirectional depending on neuronal activity, and PKA provides the cellular mechanism for a sliding threshold of current modification between suppression and potentiation. This explains from a unique angle why many neuromodulators, like dopamine and serotonin, can have either excitatory or inhibitory functions in the central nervous system (Andrade, 1998; Nicola et al. 2000). These neuromodulators often have dual roles not only because they can act on a variety of different targets, but also because they can act differently on the same target under different physiological conditions. This mechanism ensures that the modulation is flexible, accurate and dynamic.
Acknowledgments
This work was supported by the start-up packages from SUNY-Buffalo, NARSAD Young Investigator Award (Z.Y.), NSF grant IBN-0117026 (Z.Y.), NIH grant MH63128 (Z.Y.) and NIH grant NS41722 (J.F.).
REFERENCES
- Abi-Dargham A, Laruelle M, Aghajanian GK, Charney D, Krystal J. The role of serotonin in the pathophysiology and treatment of schizophrenia. Journal of Neuropsychiatry and Clinical Neurosciences. 1997;9:1–17. doi: 10.1176/jnp.9.1.1. [DOI] [PubMed] [Google Scholar]
- Andrade R. Regulation of membrane excitability in the central nervous system by serotonin receptor subtypes. Annals of the New York Academy of Sciences. 1998;861:190–203. doi: 10.1111/j.1749-6632.1998.tb10191.x. [DOI] [PubMed] [Google Scholar]
- Ansanay H, Dumuis A, Sebben M, Bockaert J, Fagni L. cAMP-dependent, long-lasting inhibition of a K+ current in mammalian neurons. Proceeding of the National Academy of Sciences of the USA. 1995;92:6635–6639. doi: 10.1073/pnas.92.14.6635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benes FM, Vincent SL, Marie A, Khan Y. Up-regulation of GABAA receptor binding on neurons of the prefrontal cortex in schizophrenic subjects. Neuroscience. 1996;75:1021–1031. doi: 10.1016/0306-4522(96)00328-4. [DOI] [PubMed] [Google Scholar]
- Bobker DH, Williams JT. Serotonin augments the cationic current Ih in central neurons. Neuron. 1989;2:1535–1540. doi: 10.1016/0896-6273(89)90041-x. [DOI] [PubMed] [Google Scholar]
- Breier A. Serotonin, schizophrenia and antipsychotic drug action. Schizophrenia Research. 1995;14:187–202. doi: 10.1016/0920-9964(94)00043-8. [DOI] [PubMed] [Google Scholar]
- Buhot MC. Serotonin receptors in cognitive behaviors. Current Opinion in Neurobiology. 1997;7:243–254. doi: 10.1016/s0959-4388(97)80013-x. [DOI] [PubMed] [Google Scholar]
- Bunin MA, Wightman RM. Paracrine neurotransmission in the CNS: involvement of 5-HT. Trends in Neurosciences. 1999;22:377–382. doi: 10.1016/s0166-2236(99)01410-1. [DOI] [PubMed] [Google Scholar]
- Cardenas CG, Del Mar LP, Cooper BY, Scroggs RS. 5HT4 receptors couple positively to tetrodotoxin-insensitive sodium channels in a subpopulation of capsaicin-sensitive rat sensory neurons. Journal of Neuroscience. 1997;17:7181–7189. doi: 10.1523/JNEUROSCI.17-19-07181.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colledge M, Scott JD. AKAPs: from structure to function. Trends in Cell Biology. 1999;9:216–221. doi: 10.1016/s0962-8924(99)01558-5. [DOI] [PubMed] [Google Scholar]
- Conde F, Marie-lepoivre E, Audinat E, Crepel F. Afferent connections of the medial frontal cortex of the rat. II. Cortical and subcortical afferents. The Journal of Comparative Neurology. 1995;352:567–593. doi: 10.1002/cne.903520407. [DOI] [PubMed] [Google Scholar]
- Consolo S, Arnaboldi S, Giorgi S, Russi G, Ladinsky H. 5-HT4 receptor stimulation facilitates acetylcholine release in rat frontal cortex. Neuroreport. 1994;5:1230–1232. doi: 10.1097/00001756-199406020-00018. [DOI] [PubMed] [Google Scholar]
- Costall B, Naylor RJ. The pharmacology of the 5-HT4 receptor. International Clinical Psychopharmacology. 1993;8(Suppl. 2):11–18. doi: 10.1097/00004850-199311002-00002. [DOI] [PubMed] [Google Scholar]
- Davidson RJ, Putnam KM, Larson CL. Dysfunction in the neural circuitry of emotion regulation - a possible prelude to violence. Science. 2000;289:591–594. doi: 10.1126/science.289.5479.591. [DOI] [PubMed] [Google Scholar]
- Deakin JF. The role of serotonin in panic, anxiety and depression. International Clinical Psychopharmacology. 1998;13(Suppl. 4):1–5. doi: 10.1097/00004850-199804004-00001. [DOI] [PubMed] [Google Scholar]
- Dean B. A predicted cortical serotonergic/cholinergic/GABAergic interface as a site of pathology in schizophrenia. Clinical and Experimental Pharmacology and Physiology. 2001;28:74–78. doi: 10.1046/j.1440-1681.2001.03401.x. [DOI] [PubMed] [Google Scholar]
- Domenech T, Beleta J, Fernandez AG, Gristwood RW, Cruz Sanchez F, Tolosa E, Palacios JM. Identification and characterization of serotonin 5-HT4 receptor binding sites in human brain: comparison with other mammalian species. Brain Research. Molecular Brain Research. 1994;21:176–180. doi: 10.1016/0169-328x(94)90392-1. [DOI] [PubMed] [Google Scholar]
- Dubovsky SL, Thomas M. Serotonergic mechanisms and current and future psychiatric practice. Journal of Clinical Psychiatry. 1995;56(Suppl. 2):38–48. [PubMed] [Google Scholar]
- Eglen RM, Bley K, Bonhaus DW, Clark RD, Hegde SS, Johnson LG, Leung E, Wong EH. RS 23597-190: a potent and selective 5-HT4 receptor antagonist. British Journal of Pharmacology. 1993;110:119–126. doi: 10.1111/j.1476-5381.1993.tb13780.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eglen RM, Bonhaus DW, Johnson LG, Leung E, Clark RD. Pharmacological characterization of two novel and potent 5-HT4 receptor agonists, RS 67333 and RS 67506, in vitro and in vivo. British Journal of Pharmacology. 1995a;115:1387–1392. doi: 10.1111/j.1476-5381.1995.tb16628.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eglen RM, Wong EHF, Dumuis A, Bockaert J. Central 5-HT4 receptors. Trends in Pharmacological Sciences. 1995b;16:391–398. doi: 10.1016/s0165-6147(00)89081-1. [DOI] [PubMed] [Google Scholar]
- Feng J, Cai X, Zhao JH, Yan Z. Serotonin receptors modulate GABAA receptor channels through activation of anchored protein kinase C in prefrontal cortical neurons. Journal of Neuroscience. 2001;21:6502–6511. doi: 10.1523/JNEUROSCI.21-17-06502.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao T, Yatani A, Dell'Acqua ML, Sako H, Green SA, Dascal N, Scott JD, Hosey MM. cAMP-dependent regulation of cardiac L-type Ca2+ channels requires membrane targeting of PKA and phosphorylation of channel subunits. Neuron. 1997;19:185–196. doi: 10.1016/s0896-6273(00)80358-x. [DOI] [PubMed] [Google Scholar]
- Goldman-Rakic PS. Cellular basis of working memory. Neuron. 1995;14:477–485. doi: 10.1016/0896-6273(95)90304-6. [DOI] [PubMed] [Google Scholar]
- Griebel G. 5-Hydroxytryptamine-interacting drugs in animal models of anxiety disorders: more than 30 years of research. Pharmacology and Therapeutics. 1995;65:319–395. doi: 10.1016/0163-7258(95)98597-j. [DOI] [PubMed] [Google Scholar]
- Groenewegen HJ. Organization of the afferent connections of the mediodorsal thalamic nucleus in the rat, related to the mediodorsal-prefrontal topography. Neuroscience. 1988;24:379–431. doi: 10.1016/0306-4522(88)90339-9. [DOI] [PubMed] [Google Scholar]
- Gurevich EV, Joyce JN. Alterations in the cortical serotonergic system in schizophrenia: a postmortem study. Biological Psychiatry. 1997;42:529–545. doi: 10.1016/S0006-3223(97)00321-1. [DOI] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high resolution current recording from cells and cell-free membrane patches. Pflügers Archiv. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Hanoune J, Defer N. Regulation and role of adenylyl cyclase isoforms. Annual Review of Pharmacology and Toxicology. 2001;41:145–174. doi: 10.1146/annurev.pharmtox.41.1.145. [DOI] [PubMed] [Google Scholar]
- Impey S, Obrietan K, Wong ST, Poser S, Yano S, Wayman G, Deloulme JC, Chan G, Storm DR. Cross talk between ERK and PKA is required for Ca2+ stimulation of CREB-dependent transcription and ERK nuclear translocation. Neuron. 1998;21:869–883. doi: 10.1016/s0896-6273(00)80602-9. [DOI] [PubMed] [Google Scholar]
- Jaffe EH, De Frias V, Ibarra C. Changes in basal and stimulated release of endogenous serotonin from different nuclei of rats subjected to two models of depression. Neuroscience Letters. 1993;162:157–160. doi: 10.1016/0304-3940(93)90584-8. [DOI] [PubMed] [Google Scholar]
- Kapur J, Macdonald RL. Cyclic AMP-dependent protein kinase enhances hippocampal dentate granule cell GABAA receptor currents. Journal of Neurophysiology. 1996;76:2626–2634. doi: 10.1152/jn.1996.76.4.2626. [DOI] [PubMed] [Google Scholar]
- Knighton DR, Zheng JH, Ten Eyck LF, Xuong NH, Taylor SS, Sowadski JM. Structure of a peptide inhibitor bound to the catalytic subunit of cyclic adenosine monophosphate-dependent protein kinase. Science. 1991;253:414–420. doi: 10.1126/science.1862343. [DOI] [PubMed] [Google Scholar]
- Kolb B. Functions of the frontal cortex of the rat: a comparative review. Brain Research. 1984;320:65–98. doi: 10.1016/0165-0173(84)90018-3. [DOI] [PubMed] [Google Scholar]
- Lewis DA. GABAergic local circuit neurons and prefrontal cortical dysfunction in schizophrenia. Brain Research. Brain Research Reviews. 2000;31:270–276. doi: 10.1016/s0165-0173(99)00042-9. [DOI] [PubMed] [Google Scholar]
- Macdonald RL, Olsen RW. GABAA receptor channels. Annual Review of Neuroscience. 1994;17:569–602. doi: 10.1146/annurev.ne.17.030194.003033. [DOI] [PubMed] [Google Scholar]
- McDonald BJ, Amato A, Connolly CN, Benke D, Moss SJ, Smart TG. Adjacent phosphorylation sites on GABAA receptor beta subunits determine regulation by cAMP-dependent protein kinase. Nature Neuroscience. 1998;1:23–28. doi: 10.1038/223. [DOI] [PubMed] [Google Scholar]
- Monferini E, Gaetani P, Rodriguez Y, Baena R, Giraldo E, Parenti M, Zocchetti A, Rizzi CA. Pharmacological characterization of the 5-hydroxytryptamine receptor coupled to adenylyl cyclase stimulation in human brain. Life Science. 1993;52:61–65. doi: 10.1016/0024-3205(93)90083-f. [DOI] [PubMed] [Google Scholar]
- Monge A, Pena MC, Palop JA, Caldero JM, Roca J, Garcia E, Romero G, Del Rio J. Synthesis of 2-piperazinylbenzothiazole and 2-piperazinylbenzoxazole derivatives with 5-HT3 antagonist and 5-HT4 agonist properties. Journal of Medical Chemistry. 1994;37:1320–1325. doi: 10.1021/jm00035a012. [DOI] [PubMed] [Google Scholar]
- Moss SJ, Doherty CA, Huganir RL. Identification of the cAMP-dependent protein kinase and protein kinase C phosphorylation sites within the major intracellular domains of the beta 1, gamma 2S, and gamma 2L subunits of the gamma-aminobutyric acid type A receptor. Journal of Biological Chemistry. 1992a;267:14470–14476. [PubMed] [Google Scholar]
- Moss SJ, Smart TG, Blackstone CD, Huganir RL. Functional modulation of GABAA receptors by cAMP-dependent protein phosphorylation. Science. 1992b;257:661–665. doi: 10.1126/science.1323140. [DOI] [PubMed] [Google Scholar]
- Nicola SM, Surmeier J, Malenka RC. Dopaminergic modulation of neuronal excitability in the striatum and nucleus accumbens. Annual Review of Neuroscience. 2000;23:185–215. doi: 10.1146/annurev.neuro.23.1.185. [DOI] [PubMed] [Google Scholar]
- Ohnuma T, Augood SJ, Arai H, McKenna PJ, Emson PC. Measurement of GABAergic parameters in the prefrontal cortex in schizophrenia: focus on GABA content, GABAA receptor alpha-1 subunit messenger RNA and human GABA transporter-1 (HGAT-1) messenger RNA expression. Neuroscience. 1999;93:441–448. doi: 10.1016/s0306-4522(99)00189-x. [DOI] [PubMed] [Google Scholar]
- Porter NM, Twyman RE, Uhler MD, Macdonald RL. Cyclic AMP-dependent protein kinase decreases GABAA receptor current in mouse spinal neurons. Neuron. 1990;5:789–796. doi: 10.1016/0896-6273(90)90338-g. [DOI] [PubMed] [Google Scholar]
- Reynolds GP, Mason SL, Meldrum A, De Keczer S, Parnes H, Eglen RM, Wong EH. 5-Hydroxytryptamine 5-HT4 receptors in post mortem human brain tissue: distribution, pharmacology and effects of neurodegenerative diseases. British Journal of Pharmacology. 1995;114:993–998. doi: 10.1111/j.1476-5381.1995.tb13303.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenmund C, Carr DW, Bergeson SE, Nilaver G, Scott JD, Westbrook GL. Anchoring of protein kinase A is required for modulation of AMPA/kainate receptors on hippocampal neurons. Nature. 1994;368:853–856. doi: 10.1038/368853a0. [DOI] [PubMed] [Google Scholar]
- Shaywitz AJ, Greenberg ME. CREB: a stimulus-induced transcription factor activated by a diverse array of extracellular signals. Annual Review of Biochemistry. 1999;68:821–861. doi: 10.1146/annurev.biochem.68.1.821. [DOI] [PubMed] [Google Scholar]
- Smiley JF, Goldman-Rakic PS. Serotonergic axons in monkey prefrontal cerebral cortex synapse predominantly on interneurons as demonstrated by serial section electron microscopy. Journal of Comparative Neurology. 1996;367:431–443. doi: 10.1002/(SICI)1096-9861(19960408)367:3<431::AID-CNE8>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Somogyi P, Kisvardy ZF, Martin KAC, Whitteridge D. Synaptic connections of morphologically identified and physiologically characterized basket cells in the striate cortex of the cat. Neuroscience. 1983;10:261–294. doi: 10.1016/0306-4522(83)90133-1. [DOI] [PubMed] [Google Scholar]
- Stockmeier CA. Neurobiology of serotonin in depression and suicide. Annals of the New York Academy of Sciences. 1997;836:220–232. doi: 10.1111/j.1749-6632.1997.tb52362.x. [DOI] [PubMed] [Google Scholar]
- Torres GE, Chaput Y, Andrade R. Cyclic AMP and protein kinase A mediate 5-hydroxytryptamine type 4 receptor regulation of calcium-activated potassium current in adult hippocampal neurons. Molecular Pharmacology. 1995;47:191–197. [PubMed] [Google Scholar]
- Tukey JW. Exploratory Data Analysis. Menlo Park, CA, USA: Addison-Weley; 1977. [Google Scholar]
- Uylings HBM, Van Eden CG. Qualitative and quantitative comparison of the prefrontal cortex in rat and in primates, including humans. Progress in Brain Research. 1990;85:31–61. doi: 10.1016/s0079-6123(08)62675-8. [DOI] [PubMed] [Google Scholar]
- Yan Z, Hsieh-Wilson L, Feng J, Tomizawa K, Allen PB, Fienberg AA, Nairn AC, Greengard P. Protein phosphatase 1 modulation of neostriatal AMPA channels:regulation by DARPP-32 and spinophilin. Nature Neuroscience. 1999;2:13–17. doi: 10.1038/4516. [DOI] [PubMed] [Google Scholar]
- Yan Z, Surmeier DJ. Muscarinic (m2/m4) receptors reduce N- and P-type Ca2+ currents in rat neostriatal cholinergic interneurons through a fast, membrane-delimited, G-protein pathway. The Journal of Neuroscience. 1996;16:2592–2604. doi: 10.1523/JNEUROSCI.16-08-02592.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Z, Surmeier DJ. D5 dopamine receptors enhance Zn2+-sensitive GABAA currents in striatal cholinergic interneurons through a PKA/PP1 cascade. Neuron. 1997;19:1115–1126. doi: 10.1016/s0896-6273(00)80402-x. [DOI] [PubMed] [Google Scholar]