

Abstract
We tested the hypotheses that: (i) exercise with low muscle glycogen would reduce pyruvate flux through the alanine aminotransferase (AAT) reaction and attenuate the increase in tricarboxylic acid (TCA) cycle intermediates, and (ii) attenuation of tricarboxylic acid cycle intermediate (TCAI) pool expansion would limit TCA cycle flux, thereby accelerating phosphocreatine (PCr) degradation. Eight men cycled for 10 min at 70% of their VO2,max on two occasions: (i) following their normal diet (CON) and (ii) after cycling to exhaustion and consuming a low carbohydrate diet for ∼2 days (LG). Biopsies (m. vastus lateralis) confirmed that [glycogen] was lower in LG vs. CON at rest (257 ± 18 vs. 611 ± 54 mmol (kg dry mass)−1; P ≤ 0.05); however, net glycogenolysis was not different after 1 or 10 min of exercise. PCr degradation from rest to 1 min was ∼26% higher in LG vs. CON (38 ± 4 vs. 28 ± 4 mmol (kg dry mass)−1; P ≤ 0.05). The sum of five measured TCAIs (∼90 % of total pool) was not different between trials at rest and after 1 min, but was higher after 10 min in LG vs. CON (5.51 ± 0.43 vs. 4.45 ± 0.49 mmol (kg dry mass)−1; P ≤ 0.05). Pyruvate dehydrogenase complex (PDC) activity was lower during exercise in LG vs. CON (2.2 ± 0.2 vs. 1.4 ± 0.2 mmol min−1 (kg wet weight)−1 after 10 min; P ≤ 0.05), and acetylcarnitine was ∼threefold less, implying increased pyruvate availability for flux through AAT. Resting muscle [glutamate] was higher in LG vs. CON (16.1 ± 0.8 vs. 11.8 ± 0.4 mmol (kg dry mass)−1; P ≤ 0.05) and the net decrease in [glutamate] during exercise was ∼30 % greater in LG vs. CON. These findings suggest that: (i) contrary to our hypotheses, LG increased anaplerosis by decreasing PDC flux and/or increasing the conversion of glutamate carbon to TCAIs, and (ii) accelerating the rate of muscle TCAI expansion did not affect oxidative energy provision during the initial phase of contraction, since changes in [TCAI] were not temporally related to PCr degradation.
The reaction catalysed by alanine aminotransferase (AAT; pyruvate + glutamate ⇌ 2-oxoglutarate + alanine) appears to be primarily responsible for the rapid increase in muscle tricarboxylic acid cycle intermediates (TCAIs) which occurs during the initial minutes of moderate to intense exercise in humans (Sahlin et al. 1990; Gibala et al. 1997). While there are other potential anaplerotic pathways in muscle (for review, see Graham & Gibala, 1998; Constantin-Teodosiu & Greenhaff, 1999), it is believed that the initial expansion of the TCAI pool occurs when the rate of pyruvate formation from glycolysis transiently exceeds its rate of oxidation in the pyruvate dehydrogenase complex (PDC) reaction and causes a rightward shift of the near-equilibrium AAT reaction. This interpretation is supported by the fact that the concentration of muscle glutamate–the co-substrate for the AAT reaction–decreases rapidly during the initial minutes of exercise (Bergström et al. 1985; Gibala et al. 1997), and the concentration of muscle alanine increases in a near-stoichiometric manner equivalent to the change in TCAIs (Gibala et al. 1997). While it is the carbon skeleton from glutamate which is converted to 2-oxoglutarate (and subsequently other TCAIs), an increase in pyruvate flux appears crucial in order to drive the AAT reaction to the right (Constantin-Teodosiu et al. 1999).
The mechanistic explanation for the increase in muscle TCAIs at the start of exercise is generally accepted; however, the physiological significance of this phenomenon remains controversial. Several investigators have suggested that the increase in TCAIs is necessary in order to attain high rates of aerobic energy provision, presumably by activating various near-equilibrium reactions in the TCA cycle (Sahlin et al. 1990; Wagenmakers, 1998). However, we have argued that the increase in TCAIs during exercise primarily reflects a mass action phenomenon, and is of little functional importance for the regulation of oxidative energy provision (Gibala et al. 1998; Constantin-Teodosiu et al. 1999). Before this debate can be resolved, it will be necessary to conduct studies which manipulate the concentrations of TCAIs and determine what effect–if any–this has on skeletal muscle metabolism and exercise performance. In this regard, two recent investigations from our laboratories demonstrated that administration of the pharmacological agent dichloroacetate (DCA) reduced the concentrations of TCAIs in resting skeletal muscle (Constantin-Teodosiu et al. 1999; Gibala & Saltin, 1999)–presumably by increasing the oxidative disposal of pyruvate and diverting substrate away from anaplerotic pathways–but DCA did not alter the concentrations of muscle TCAIs during exercise (Gibala & Saltin, 1999). Bruce et al. (2001) recently reported that glutamine ingestion at rest resulted in a higher concentration of TCAIs after 10 min of exercise compared to a placebo trial, presumably by increasing the availability of glutamate for entry into the cycle at the level of 2-oxoglutarate. Importantly, this study also demonstrated a clear dissociation between TCAI expansion and oxidative energy delivery during the initial phase of exercise, since there was no difference between trials in the extent of muscle phosphocreatine (PCr) degradation after 10 min of exercise.
Another potential strategy to manipulate muscle TCAIs during exercise would be to reduce muscle glycogen concentration and therefore the availability of pyruvate for the AAT reaction. In the present study, we had subjects perform cycle exercise to exhaustion and then consume a diet low in carbohydrate (CHO) for ∼2 days in order to reduce the resting concentration of muscle glycogen prior to a subsequent bout of exercise. We hypothesized that this manipulation would reduce the anaplerotic flux of pyruvate through the AAT reaction during exercise and attenuate the increase in TCAIs, compared to a control bout which was performed with normal resting glycogen. A previous study (Spencer et al. 1992) suggested that low resting muscle glycogen reduced the concentrations of TCAIs during moderate-intensity exercise; however measurements were only made at rest and after prolonged cycling to fatigue (∼40 min during the low carbohydrate trial). In the present study, we specifically examined the effect of low glycogen on muscle TCAI expansion during the initial period of contraction, since the increase in TCAI pool size has been shown to peak within the first 5–10 min of exercise (Gibala et al. 1997). We also sought to determine whether altering the rate of TCAI expansion affected the rate of oxidative energy provision at the onset of exercise and, if so, we expected to observe an accelerated rate of PCr degradation during the carbohydrate-restricted trial.
METHODS
Subjects
Eight healthy men, with a mean (± s.e.m.) age, height and body mass of 22 ± 1 year, 184 ± 1 cm and 77 ± 2 kg, respectively, took part in the investigation. The subjects were recreationally active individuals recruited from the undergraduate student population at the University of Nottingham. The experimental protocol was approved by the University of Nottingham Ethics Committee, and all subjects provided written, informed consent. All studies were performed according to the Declaration of Helsinki. At least 2 weeks prior to the start of the experiment, subjects performed a progressive exercise test on an electrically braked cycle ergometer (Lode, The Netherlands) in order to determine their maximal oxygen uptake (VO2,max), which was confirmed by a second test 3 days later. The mean VO2,max for the group was 52 ± 2 ml kg−1 min−1.
Experimental protocol
Subjects performed a 10 min bout of cycle exercise on two occasions, separated by ∼48 h, under two different pre-exercise dietary conditions: (1) following their normal, mixed diet (control, CON) and (2) after cycling to exhaustion and then consuming a diet low in carbohydrate for ∼2 days (low glycogen, LG). An ordered design was employed, such that the CON trial was always performed first. Upon arrival at the laboratory on the first day of the experiment, subjects rested in the supine position while the area over the lateral aspect of one thigh was anaesthetized (1 % w/v lignocaine hydrochloride, Antigen Pharmaceuticals Ltd, Ireland) and prepared for the extraction of needle biopsy samples from the vastus lateralis muscle. A resting needle biopsy sample was obtained ∼15 min prior to the start of exercise. Subjects then cycled on the electrically-braked ergometer at a workload corresponding to ∼70 % VO2,max (210 ± 10 W) for 10 min. Needle biopsy samples were obtained after 1 and 10 min of exercise–from the same leg as for the resting sample–and expired gas measurements were made throughout exercise using an on-line gas analysis system (SensorMedics BV, UK). Exercise was performed in an air conditioned laboratory at a temperature of 20 °C. Approximately 15 min after completion of the first exercise bout, the subject remounted the ergometer and cycled at the same work intensity (∼70 % VO2,max) until volitional exhaustion (∼90 min; defined as the point at which the pedal cadence fell below 50 revs min−1). Subjects ingested 200 ml of water every 15 min during this second ride. After a short period of recovery (∼10 min), during which only water was ingested, subjects were required to recommence cycling for a third time and again continue until the point of volitional fatigue (≤5 min). Following the final ride, subjects were instructed to consume a diet low in carbohydrate for the next ∼45 h in order to ensure that the concentration of muscle glycogen was substantially reduced prior to the second exercise bout (LG trial). Subjects were provided with a list of acceptable food choices designed to maintain dietary carbohydrate intake at ≤10 % of total energy intake. Subjects reported back to the laboratory ∼48 h following the start of the first exercise trial in order to perform the second 10 min exercise bout at the same work intensity. Needle biopsy samples and cardiorespiratory measurements were obtained as described for the first trial, except that the opposite leg was used for muscle sampling.
Muscle analyses
Needle biopsy samples were immediately frozen in liquid nitrogen. A 10–20 mg piece of frozen muscle was subsequently removed from each sample under liquid nitrogen and used for the determination of the active fraction of pyruvate dehydrogenase complex (PDCa) using the method of Constantin-Teodosiu et al. 1991b. The remaining portion of each biopsy sample was freeze-dried, powdered, dissected free of non-muscle elements and stored at −80 °C. A ∼10 mg portion of freeze-dried muscle was extracted with 0.5 m perchloric acid (containing 1 mm EDTA), neutralized with 2.2 m KHCO3, and assayed for creatine, PCr, ATP, lactate, citrate, isocitrate, succinate, fumarate, malate, alanine and glutamate using enzymatic methods (Bergmeyer, 1974; Harris et al. 1974; Passoneau & Lowry, 1993) adapted for fluorometry (Hitachi F-2000 fluorescence spectrophotometer, Hitachi Instruments, Japan). Acetylcarnitine and carnitine were determined using a radioisotopic assay (Cederblad et al. 1990). In order to correct for differences in blood or connective tissue between samples, muscle metabolites were corrected to the highest total creatine value obtained in all biopsy samples for a given subject.
Statistical analyses
Muscle metabolite and enzyme activity data were analysed using a two-factor (time × condition) repeated measures analysis of variance (ANOVA). Significant interactions and main effects were subsequently analysed using Tukey's honestly significant difference post hoc test. Net changes in metabolite concentrations during exercise were examined using Student's paired t test. Statistical significance for all analyses was accepted as P ≤ 0.05. All data are expressed as mean ± s.e.m.
RESULTS
Cardiorespiratory data
Pulmonary VO2, expired minute ventilation, and respiratory exchange ratio data during exercise are summarized in Table 1.
Table 1.
Cardiorespiratory data during exercise
| Control | Low glycogen | |
|---|---|---|
| V˙o2(ml kg min−1) | 40.9 ± 1.5 | 42.7 ± 1.5 |
| V˙E(BTPS) (l min−1) | 61.5 ± 2.6 | 59.8 ± 2.7 |
| RER | 0.90 ± 0.02 | 0.78 ± 0.01* |
Values are means ± s.e.m., n = 8. Data were obtained during the final 3 min of each 10 min bout of exercise with normal (control) and low muscle glycogen. V˙o2, pulmonary oxygen uptake; V˙E(BTPS), expired minute ventilation corrected for body temperature and pressure, saturated with water vapour; RER, respiratory exchange ratio.
P ≤ 0.05 between groups.
Muscle glycogen
The pre-exercise dietary protocol was successful at reducing the concentration of muscle glycogen prior to exercise in the LG trial, and glycogen was lower (P ≤ 0.05) throughout exercise compared to CON (main effect for time, P ≤ 0.05; Fig. 1). There was no difference in muscle glycogen utilization between trials from rest to 1 min of exercise (9.5 ± 5.8 vs. 8.4 ± 7.8 mmol (kg dry mass)−1 for CON and LG, respectively; P > 0.05). From 1 to 10 min of exercise, glycogenolytic rate was ∼30 % lower during LG vs. CON (5.9 ± 1.1 vs. 8.5 ± 1.7 mmol min−1 (kg dry mass)−1); however, this difference was not statistically significant.
Figure 1. Muscle glycogen content at rest and during exercise in control and low glycogen conditions.

Prior to experimental trials, subjects consumed either a normal mixed diet (Control) or performed prolonged cycle exercise to exhaustion and then consumed a low carbohydrate diet for 2 days (Low Glycogen). Values are means ± s.e.m., n = 8. † Main effect for condition (P ≤ 0.05), low glycogen < control.
Muscle TCAIs
The total concentration of the five measured TCAIs (ΣTCAIs) was similar in both groups at rest, as was the net increase in ΣTCAIs during the first minute of exercise (Fig. 2). However, from 1 to 10 min, the net increase in ΣTCAIs was ∼36 % higher (P ≤ 0.05) in the LG trial vs. CON (1.76 ± 0.61 vs. 1.12 ± 0.42 mmol (kg dry mass)−1), such that ΣTCAIs after 10 min of exercise was higher (P ≤ 0.05) in the LG trial vs. CON. With regard to individual TCAIs, the larger pool size after 10 min of exercise during the LG trial was due to larger net increases in succinate, fumarate and malate; there were no differences between trials in citrate or isocitrate during exercise (Table 2).
Figure 2. Total muscle concentration of five measured TCAIs (ΣTCAIs; citrate, isocitrate, succinate, fumarate, malate) at rest and during exercise with normal (control) or low muscle glycogen.
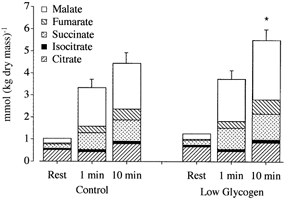
Values are means ± s.e.m. for sum of total TCAI pool, with mean concentrations for individual TCAIs presented within each stacked bar. * P ≤ 0.05vs. control trial at same time point.
Table 2.
Muscle metabolites at rest and during exercise
| Control trial | Low CHO trial | |||||
|---|---|---|---|---|---|---|
| Rest | 1 min | 10 min | Rest | 1 min | 10 min | |
| Citrate | 0.52 ± 0.06 | 0.43 ± 0.03 | 0.80 ± 0.04 | 0.67 ± 0.07 | 0.46 ± 0.04 | 0.86 ± 0.07 |
| Isocitrate | 0.06 ± 0.00 | 0.10 ± 0.01 | 0.11 ± 0.01 | 0.08 ± 0.01 | 0.10 ± 0.01 | 0.13 ± 0.01 |
| Succinate | 0.20 ± 0.02 | 0.76 ± 0.08 | 0.97 ± 0.12 | 0.21 ± 0.02 | 0.96 ± 0.10 | 1.18 ± 0.10 * |
| Fumarate | 0.04 ± 0.00 | 0.30 ± 0.05 | 0.49 ± 0.09 | 0.05 ± 0.00 | 0.30 ± 0.04 | 0.64 ± 0.08 * |
| Malate | 0.21 ± 0.02 | 1.75 ± 0.23 | 2.08 ± 0.29 | 0.25 ± 0.03 | 1.92 ± 0.20 | 2.70 ± 0.26* |
| Alanine † | 7.2 ± 0.6 | 9.6 ± 0.7 | 12.1 ± 1.1 | 4.0 ± 0.2 | 6.7 ± 0.5 | 10.0 ± 0.7 |
| Creatine | 43.5 ± 2.6 | 71.7 ± 3.1 | 80.8 ± 4.5 | 43.3 ± 3.0 | 81.4 ± 4.8* | 89.0 ± 5.5 * |
| ATP | 27.9 ± 0.7 | 27.6 ± 0.6 | 27.9 ± 0.7 | 26.3 ± 0.3 | 26.2 ± 0.5 | 25.7 ± 0.8 |
| Lactate | 3.0 ± 0.3 | 21.3 ± 4.1 | 24.9 ± 6.6 | 1.7 ± 0.2 | 19.2 ± 3.3 | 17.3 ± 4.0 |
| Acetylcarnitine | 6.7 ± 1.1 | 6.1 ± 0.7 | 11.8 ± 1.0 | 8.6 ± 1.5 | 7.1 ± 1.0 | 9.9 ± 1.1 |
| Carnitine | 16.6 ± 1.5 | 16.6 ± 1.0 | 10.8 ± 1.5 | 13.8 ± 1.7 | 15.4 ± 1.5 | 12.4 ± 1.4 |
All values are means ± s.e.m. (n = 8) in mmol (kg dry mass)−1.
P ≤ 0.05vs. control trial at same time point.
P ≤ 0.05 main effect for time (control trial > low CHO trial).
Muscle energy metabolites, acetylcarnitine and carnitine
There were no differences between groups in resting concentrations of PCr, creatine, ATP, lactate, acetylcarnitine or carnitine (Table 2). However, the rate of PCr utilization was ∼26 % higher (P ≤ 0.05) during the first minute of exercise in the LG trial compared to CON (38 ± 4 vs. 28 ± 4 mmol (kg dry mass)−1, respectively), and consequently PCr was lower after 1 and 10 min of exercise in the LG trial vs. CON (Fig. 3). Muscle creatine was higher (P ≥ 0.05) after 1 and 10 min of exercise in the LG trial vs. CON. Notably, the net accumulation in muscle acetylcarnitine during exercise was approximately threefold lower (P ≤ 0.05) during the LG trial compared to CON (Fig. 4). There were no differences between trials in the other metabolites during exercise (Table 2).
Figure 3. Muscle phosphocreatine concentration at rest and during exercise with normal (control) or low muscle glycogen.

Values are means ± s.e.m., n = 8. * P ≤ 0.05 between trials at same time point.
Figure 4. Net change in muscle acetylcarnitine concentration from rest during exercise with normal (control) or low muscle glycogen.

Values are means ± s.e.m., n = 8. * P ≤ 0.05vs. control trial.
Muscle glutamate and alanine
The resting concentration of glutamate was higher (P ≤ 0.05) in the LG trial vs. CON, and the difference persisted after 1 min of exercise (Fig. 5). However, there was no difference between trials in muscle glutamate after 10 min of exercise, such that the net decrease in glutamate from 1 to 10 min of exercise was ∼34 % larger (P ≤ 0.05) during the LG trial vs. CON (10.8 ± 0.7 vs. 7.1 ± 0.6 mmol (kg dry mass)−1). The concentration of muscle alanine was lower throughout the LG trial compared to CON (main effect for time, P ≤ 0.05) (Table 2). Muscle alanine concentration increased during exercise in both trials, and while the net increase in alanine from 1 to 10 min of exercise was ∼30 % larger during the LG trial vs. CON, this difference was not statistically significant.
Figure 5. Muscle glutamate concentration at rest and during exercise with normal (control) or low muscle glycogen.

Values are means ± s.e.m., n = 8. * P ≤ 0.05 between trials at same time point.
Muscle pyruvate dehydrogenase
Resting PDCa was similar between trials (0.2 ± 0.0 vs. 0.3 ± 0.1 mmol min−1 (kg wet weight)−1 in the LG and CON trials, respectively; P ≥ 0.05). However, during exercise the rate of enzyme activation differed between trials such that PDCa was lower (P ≤ 0.05) in the LG trial vs. CON after 1 and 10 min of exercise (Fig. 6).
Figure 6. Measured active fraction of pyruvate dehydrogenase (PDCa) at rest and during exercise with normal (control) or low muscle glycogen.

Values are means ± s.e.m., n = 8. * P ≤ 0.05 between trials at same time point.
DISCUSSION
A major, novel finding from the present study was that the contraction-induced expansion of the TCAI pool was enhanced when subjects began exercise with low muscle glycogen content. The increase in TCAIs normally observed at the start of moderate to intense exercise is most probably due to a rightward shift of the near-equilibrium AAT reaction (pyruvate + glutamate ⇌ 2-oxoglutarate + alanine), subsequent to an increase in pyruvate availability when its rate of formation from glycolysis transiently exceeds its rate of oxidation in the PDC reaction. This causes a decrease in the concentration of muscle glutamate, as the amino group from this amino acid is transferred onto pyruvate, forming alanine, and the carbon skeleton enters the TCA cycle at the level of 2-oxoglutarate. The concentration of 2-oxoglutarate does not increase (Gibala et al. 1998, 1999; Constantin-Teodosiu et al. 1999), but rather it appears that the carbon skeleton is rapidly converted to other intermediates in the ‘second span’ of the cycle, and leads to marked increases in the concentrations of succinate, fumarate and especially malate.
We hypothesized that by reducing the concentration of muscle glycogen at rest, the flux through glycolysis would be reduced during subsequent exercise and thereby would limit the availability of pyruvate for anaplerosis through the AAT reaction. However, we did not observe any significant differences in the glycogenolytic rate during exercise, and muscle lactate accumulation was similar between trials. This suggests that our experimental manipulation may not have been sufficient to impair the rate of pyruvate formation during exercise. In a recent study from our laboratory (Gibala et al. 1999), we observed that the magnitude of TCAI expansion during exercise after a low CHO diet was similar to that observed in a previous group of subjects who performed exercise with no dietary manipulation. Therefore, we concluded that ‘low’ [glycogen] per se–i.e. resting values of ∼200 mmol (kg dry mass)−1–was not sufficient to impair the net increase in TCAIs during exercise, and introduced the concept of a critical minimum concentration of glycogen necessary to provide an adequate pyruvate flux to drive anaplerotic reactions. Nevertheless, prior to the present work, no study had directly compared the effect of low vs. high muscle [glycogen] on TCAI expansion during exercise in the same group of subjects. Interestingly, and contrary to our hypothesis, the present results suggest that low resting [glycogen]–induced by a combination of previous exercise and ingestion of a low CHO diet for ∼2 days–augments the increase in TCAIs during moderate exercise. There are at least two plausible mechanisms to explain these findings, as outlined below.
Muscle PDC, pyruvate availability and TCAI expansion at the onset of exercise
One potential explanation for the larger increase in TCAIs during exercise in the LG trial involves differences in the flux through PDC during exercise. It could be argued that if glycolytic flux, i.e. the rate of glycogen degradation, during exercise was similar between trials, but flux through the PDC reaction was lower in the LG trial vs. CON, the net effect would have been an increase in pyruvate availability for flux through the AAT reaction. In the present study, the measured active fraction of PDC was indeed lower after 1 and 10 min of exercise in the LG trial vs. CON. Furthermore, the extent of acetylcarnitine accumulation (an indicator of the magnitude of flux through the PDC reaction) during 10 min of exercise was also reduced in the LG vs. CON. This is consistent with previous studies which have reported lower activation of PDC during the initial phase of exercise following a CHO-restricted diet (e.g. Putman et al. 1993). It should also be noted that the in vitro measured PDCa fraction only represents the potential for PDC flux, since in vivo the latter is determined by allosteric modulators in addition to covalent modification. Peters et al. (1998) reported that the activity of PDC kinase–the enzyme responsible for phosphorylating PDC to the less active form–is increased in human skeletal muscle at rest after a low CHO diet, and several studies (e.g. Putman et al. 1993) have reported increases in the acetyl CoA/CoA ratio in resting muscle after a low CHO diet, which would function to inhibit PDC flux as classically proposed (Randle, 1995). However, it remains speculative what effect these changes would have on PDC flux during the rest-to-work transition and, indeed, several studies have demonstrated that increases in PDCa during exercise occur independently of changes in the acetyl CoA/CoA ratio (Constantin-Teodosiu et al. 1991a; Dyck et al. 1993; Putman et al. 1993). Irrespective of the mechanism, the lower PDCa and reduced acetylcarnitine accumulation observed in the LG trial suggest that flux through the PDC reaction was attenuated at the onset of exercise. Therefore, it is plausible that lower PDC flux during the initial phase of exercise in the LG trial may have increased pyruvate availability for anaplerosis and contributed to the larger accumulation of TCAIs after 10 min of exercise.
The effect of prior exercise and low carbohydrate availability on resting muscle [glutamate]
A second factor which may have influenced the TCAI response in the present study involves differences in the availability of muscle glutamate, which is the co-substrate for the AAT reaction. Notably, the experimental manipulation of glycogen-depleting exercise followed by the ingestion of a low CHO diet for 2 days led to a significant elevation in the resting concentration of muscle glutamate prior to the LG trial. The precise explanation for this phenomenon–which has been observed by previous investigators (e.g. van Hall et al. 1995)–is difficult to ascertain given the myriad metabolic pathways in which glutamate takes part. One potential source for an increase in muscle glutamate is the catabolism of its amino acid precursor, glutamine, which is catalysed by the enzyme glutaminase (glutamine → glutamate + ammonia). Muscle glutamine was not measured in the present study, however a previous investigation reported a significant decrease in this amino acid following a low carbohydrate diet (Greenhaff et al. 1988). Regardless of the mechanism, the increased glutamate availability may have contributed to a higher flux through the AAT reaction and greater TCAI accumulation during the LG trial. This is supported by recent data from Bruce et al. (2001), who attempted to manipulate glutamate availability during exercise under conditions of reduced muscle glycogen availability. These authors demonstrated that ingestion of a glutamine solution significantly increased the muscle concentration of glutamine at rest, and resulted in a larger increase in muscle TCAIs after 10 min of moderate exercise.
Collectively, these findings suggest that availability of muscle glutamate–provided the flux of pyruvate is sufficient–may influence the net increase in TCAIs during exercise. Alternatively, it can be argued that the larger net decrease in glutamate during the LG trial is also consistent with a reduced flux through PDC (as outlined above) and thus pyruvate availability is the more critical determinant for increases in TCAIs during exercise. This latter view is also supported by the fact that alanine production in skeletal muscle appears linked to the availability of pyruvate above a critical intracellular level (Pardridge & Davidson, 1979), although there is evidence from studies on other tissues that alanine production is also influenced by glutamate availability (e.g. Christie & Butler, 1999). Of note, the concentration of pyruvate in skeletal muscle during exercise tends to fluctuate around the Km of AAT for pyruvate (0.3 mm) whereas the concentration of glutamate tends to be much lower than the Km value (25 mm) and the concentration of this amino acid decreases during exercise (Bergmeyer et al. 1983). Nonetheless, the present data do not permit a definitive conclusion regarding the availability of which substrate–pyruvate or glutamate–is most critical for determining the magnitude of TCAI formation at the onset of exercise, and further studies are warranted in order to resolve this issue.
Significance of changes in TCAIs at the onset of exercise
It has previously been suggested that an increase in TCAIs is necessary in order to attain high rates of TCA cycle flux during strenuous exercise (Sahlin et al. 1990; Wagenmakers et al. 1998). However, until recently no study had directly tested this hypothesis by manipulating TCAIs during the initial phase of exercise and measuring markers of non-oxidative ATP provision such as phosphocreatine degradation (Hultman et al. 1967). Bruce et al. (2001) recently demonstrated that increasing the rate of TCAI expansion during the first 10 min of moderate exercise did not attenuate the extent of muscle PCr degradation, as would be expected if the increase in TCAIs was obligatory for the onset of mitochondrial respiration. These findings are supported by the results of the present study, which showed a clear dissociation between the increase in TCAIs and oxidative ATP provision during exercise. PCr degradation during the first minute of exercise was ∼35 % higher during the LG trial vs. CON despite the similar rate of increase in muscle TCAIs between conditions. Moreover, there was no relationship between these variables from 1 to 10 min of exercise, since the net increase in TCAIs was higher during the LG trial and yet PCr degradation during this time was similar between trials. The present data therefore support the findings of Bruce et al. (2001) and indicate that augmenting the rate of TCAI expansion during the initial phase of exercise does not enhance aerobic energy provision in skeletal muscle. While these data argue against the hypothesis that changes in muscle TCAIs are causally related to mitochondrial respiration, it remains to be determined whether reducing or preventing expansion of the TCAI pool affects oxidative energy production at the onset of exercise. Indeed, the precise factors which regulate aerobic energy provision during the rest-to-work transition are probably complex and this topic remains controversial (Tschakovsky & Hughson, 1999); however, it was recently suggested that the delivery of acetyl groups to the TCA cycle may limit oxidative energy provision at the onset of exercise in skeletal muscle (Timmons et al. 1997, 1998).
The reason for the higher initial rate of PCr degradation during the LG trial in the present study could be related to a lag in PDC flux at the onset of exercise. A brief lag in PDC flux during the rest-to-work transition–for example due to an elevated acetyl CoA/CoA ratio following the low CHO diet–could have transiently delayed the production of pyruvate-derived acetyl CoA and compromised mitochondrial NADH production. This would have led to compensatory increases in the free concentration of ADP and an accelerated initial rate of PCr degradation (Spencer et al. 1992). In support of this, Ren et al. (1990) previously reported that the extent of PCr degradation in the human quadriceps femoris during 30 s of intense electrical stimulation was ∼30 % greater when subjects consumed a low carbohydrate diet vs. a mixed diet prior to exercise.
Conclusion
The results from the present study demonstrate that the normal exercise-induced expansion of the muscle TCAI pool during moderate exercise is enhanced when subjects begin exercise with low resting muscle glycogen content. Notably, the experimental manipulation of exercise to exhaustion followed by the consumption of a low CHO diet for ∼2 days also resulted in a higher concentration of muscle glutamate at rest in the LG trial. Muscle glycogenolyis and lactate accumulation were similar between trials during exercise, but PDCa and muscle acetylcarnitine accumulation were reduced in the LG trial. The net effect of these changes may have been an increased pyruvate flux through the AAT reaction, which consequently led to the larger muscle accumulation of TCAIs observed after 10 min of exercise in the LG trial. The net decrease in glutamate during exercise was larger during the LG trial, which is also consistent with a higher flux through the AAT reaction. Finally, there was a clear dissociation between the initial expansion of the TCAI pool and the rate of PCr utilization, which suggests the increase in TCAIs is not causally related to the onset of aerobic energy production during dynamic exercise in humans.
Acknowledgments
We thank Elizabeth Simpson, John Fox and Dr Julie Lambourne for technical assistance during the experimental trials, and our subjects for their time and effort. M.J.G. was supported by a Postdoctoral Fellowship award from the Natural Sciences and Engineering Research Council of Canada.
REFERENCES
- Bergmeyer HU. Methods of Enzymatic Analysis. 2. New York: Academic Press; 1974. [Google Scholar]
- Bergmeyer HUM, Graßl M, Walter H-E. Section I: Enzymes. In: Bergmeyer HU, editor. Methods of Enzymatic Analysis. 3. Weinheim, Germany: Verlag Chemie; 1983. pp. 136–138. [Google Scholar]
- Bergström J, Fürst P, Hultman E. Free amino acids in muscle tissue and plasma during exercise in man. Clinical Physiology. 1985;5:155–160. doi: 10.1111/j.1475-097x.1985.tb00591.x. [DOI] [PubMed] [Google Scholar]
- Bruce M, Constantin-Teodosiu D, Greenhaff PL, Boobis LH, Williams C, Bowtell JL. Glutamine supplementation promotes anaplerosis but not oxidative energy delivery in human skeletal muscle. American Journal of Physiology - Endocrinology and Metabolism. 2001;280:E669–675. doi: 10.1152/ajpendo.2001.280.4.E669. [DOI] [PubMed] [Google Scholar]
- Cederblad G, Carlin JI, Constantin-Teodosiu D, Harper P, Hultman E. Radioisotopic assays of CoASH and carnitine and their acetylated forms in human skeletal muscle. Analytical Biochemistry. 1990;185:274–278. doi: 10.1016/0003-2697(90)90292-h. [DOI] [PubMed] [Google Scholar]
- Christie A, Butler M. The adaptation of BHK cells to a non-ammoniagenic glutamate-based culture medium. Biotechnology and Bioengineering. 1999;64:298–309. [PubMed] [Google Scholar]
- Constantin-Teodosiu D, Carlin JI, Cederblad G, Harris RC, Hultman E. Acetyl group accumulation and pyruvate dehydrogenase activity in human muscle during incremental exercise. Acta Physiologica Scandinavica. 1991a;143:367–372. doi: 10.1111/j.1748-1716.1991.tb09247.x. [DOI] [PubMed] [Google Scholar]
- Constantin-Teodosiu D, Cederblad G, Hultman E. A sensitive radioisotopic assay of pyruvate dehydrogenase complex in human muscle tissue. Analytical Biochemistry. 1991b;198:347–351. doi: 10.1016/0003-2697(91)90437-x. [DOI] [PubMed] [Google Scholar]
- Constantin-Teodosiu D, Greenhaff PL. The tricarboxylic acid cycle in human skeletal muscle: Is there a role for nutritional intervention? Current Opinions in Clinical Nutrition and Metabolic Care. 1999;2:527–531. doi: 10.1097/00075197-199911000-00017. [DOI] [PubMed] [Google Scholar]
- Constantin-Teodosiu D, Simpson EJ, Greenhaff PL. The importance of pyruvate availability to PDC activation and anaplerosis in human skeletal muscle. American Journal of Physiology. 1999;276:E472–478. doi: 10.1152/ajpendo.1999.276.3.E472. [DOI] [PubMed] [Google Scholar]
- Dyck DJ, Putman CT, Heigenhauser GJF, Hultman E, Spriet LL. Regulation of fat-carbohydrate interaction in skeletal muscle during intense aerobic cycling. American Journal of Physiology. 1993;265:E852–859. doi: 10.1152/ajpendo.1993.265.6.E852. [DOI] [PubMed] [Google Scholar]
- Gibala MJ, Lozej M, Tarnopolsky MA, McLean C, Graham TE. Low glycogen and branched-chain amino acid ingestion do not impair anaplerosis during exercise in humans. Journal of Applied Physiology. 1999;87:1662–1667. doi: 10.1152/jappl.1999.87.5.1662. [DOI] [PubMed] [Google Scholar]
- Gibala MJ, MacLean DA, Graham TE, Saltin B. Anaplerotic processes in human skeletal muscle during brief dynamic exercise. Journal of Physiology. 1997;502:703–713. doi: 10.1111/j.1469-7793.1997.703bj.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibala MJ, MacLean DA, Graham TE, Saltin B. Tricarboxylic acid cycle pool size and estimated cycle flux in human muscle during exercise. American Journal of Physiology. 1998;275:E235–242. doi: 10.1152/ajpendo.1998.275.2.E235. [DOI] [PubMed] [Google Scholar]
- Gibala MJ, Saltin B. PDH activation by dichloroacetate reduces TCA cycle intermediates at rest but not during exercise in humans. American Journal of Physiology. 1999;277:E33–38. doi: 10.1152/ajpendo.1999.277.1.E33. [DOI] [PubMed] [Google Scholar]
- Graham TE, Gibala MJ. Anaplerosis of the tricarboxylic acid cycle during exercise in human skeletal muscle: Magnitude, sources and potential physiological significance. In: Richter EA, Galbo H, Kiens B, Saltin B, editors. Skeletal Muscle Metabolism in Exercise and Diabetes. New York: Plenum Press; 1998. pp. 271–286. [DOI] [PubMed] [Google Scholar]
- Greenhaff PL, Gleeson M, Maughan RJ. The effects of diet on muscle pH and metabolism during high intensity exercise. European Journal of Applied Physiology. 1988;57:531–539. doi: 10.1007/BF00418458. [DOI] [PubMed] [Google Scholar]
- Harris RC, Hultman E, Nordesjo LO. Glycogen, glycolytic intermediates and high-energy phosphates determined in biopsy samples of musculus quadriceps femoris of man at rest. Scandinavian Journal of Clinical and Laboratory Investigation. 1974;33:109–120. [PubMed] [Google Scholar]
- Hultman E, Bergström J, McLeannan Anderson N. Breakdown and resynthesis of phosphocreatine and adenosine triphosphate in connection with muscular work in man. Scandinavian Journal of Clinical and Laboratory Investigation. 1967;19:56–66. doi: 10.3109/00365516709093481. [DOI] [PubMed] [Google Scholar]
- Pardridge WM, Davidson MB. Alanine metabolism in skeletal muscle in tissue culture. Biochimica Biophysica Acta. 1979;585:34–42. doi: 10.1016/0304-4165(79)90322-2. [DOI] [PubMed] [Google Scholar]
- Passoneau JV, Lowry OH. Enzymatic Analysis: A Practical Guide. Totowa, NJ, USA: Humana Press; 1993. [Google Scholar]
- Peters SJ, St Amand TA, Howlett RA, Heigenhauser GJF, Spriet LL. Human skeletal muscle pyruvate dehydrogenase kinase activity increases after a low-carbohydrate diet. American Journal of Physiology. 1998;275:E980–986. doi: 10.1152/ajpendo.1998.275.6.E980. [DOI] [PubMed] [Google Scholar]
- Putman CL, Spriet LL, Hultman E, Lindinger MI, Lands LC, McKelvie RS, Cederblad G, Jones NL, Heigenhauser GJF. Pyruvate dehydrogenase activity and acetyl group accumulation during exercise after different diets. American Journal of Physiology. 1993;265:E752–760. doi: 10.1152/ajpendo.1993.265.5.E752. [DOI] [PubMed] [Google Scholar]
- Randle PJ. Metabolic fuel selection: general integration at the whole-body level. Proceedings of the Nutrition Society. 1995;54:317–327. doi: 10.1079/pns19950057. [DOI] [PubMed] [Google Scholar]
- Ren JM, Broberg S, Sahlin K, Hultman E. Influence of reduced glycogen level on glycogenolysis during short-term stimulation in man. Acta Physiologica Scandinavica. 1990;139:467–474. doi: 10.1111/j.1748-1716.1990.tb08948.x. [DOI] [PubMed] [Google Scholar]
- Sahlin K, Katz A, Broberg S. Tricarboxylic acid cycle intermediates in human muscle during prolonged exercise. American Journal of Physiology. 1990;259:C834–841. doi: 10.1152/ajpcell.1990.259.5.C834. [DOI] [PubMed] [Google Scholar]
- Snell K. Muscle alanine synthesis and hepatic gluconeogenesis. Biochemical Society Transactions. 1980;8:205–213. doi: 10.1042/bst0080205. [DOI] [PubMed] [Google Scholar]
- Spencer MK, Zhen Y, Katz A. Effect of low glycogen on carbohydrate and energy metabolism in human muscle during exercise. American Journal of Physiology. 1992;262:C975–979. doi: 10.1152/ajpcell.1992.262.4.C975. [DOI] [PubMed] [Google Scholar]
- Tschakovksy ME, Hughson RL. Interaction of factors determining oxygen uptake at the onset of exercise. Journal of Applied Physiology. 1999;86:1101–1113. doi: 10.1152/jappl.1999.86.4.1101. [DOI] [PubMed] [Google Scholar]
- Timmons JA, Gustafsson T, Sundberg CJ, Jansson E, Greenhaff PL. Muscle acetyl group availability is a major determinant of oxygen deficit in humans during submaximal exercise. American Journal of Physiology. 1998;274:E377–380. doi: 10.1152/ajpendo.1998.274.2.E377. [DOI] [PubMed] [Google Scholar]
- Timmons JA, Poucher SM, Constantin-Teodosiu D, MacDonald I, Greenhaff PL. The metabolic responses from rest to steady-state determine contractile function in ischemic canine skeletal muscle. American Journal of Physiology. 1997;273:E233–238. doi: 10.1152/ajpendo.1997.273.2.E233. [DOI] [PubMed] [Google Scholar]
- van Hall G, Saltin B, van der Vusse GJ, Söderland K, Wagenmakers AJM. Deamination of amino acids as a source for ammonia production in human skeletal muscle during prolonged exercise. Journal of Physiology. 1995;489:251–261. doi: 10.1113/jphysiol.1995.sp021047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagenmakers AJM. Muscle amino acid metabolism at rest and during exercise: Role in human physiology and metabolism. In: Holloszy JO, editor. Exercise and Sports Science Reviews. Baltimore, MD, USA: Williams and Wilkins; 1998. pp. 287–314. [PubMed] [Google Scholar]