

Abstract
The role of haem oxygenase (HO) in the hepatic accumulation of leukocytes in mice during the initiation of remote organ injury following normotensive limb ischaemia-reperfusion (I-R) was investigated. Remote organ injury was initiated by 1 h bilateral hindlimb ischaemia followed by either 1 or 1.5 h reperfusion (I-R) in male C57BL/6 mice. Mice were randomly assigned to either sham (no I-R, n = 4), I-R (n = 4 for both time points), I-R plus chromium mesoporphyrin (CrMP, n = 4) to inhibit HO or I-R plus haemin (n = 4) to increase HO. Leukocyte accumulation and leukocyte-endothelial interaction were directly measured using fluorescence intravital microscopy. Leukocytes were labelled via an injection of rhodamine 6G. In sinusoids the total number and the number of stationary leukocytes were assessed. In postsinusoidal venules the number of adherent and rolling leukocytes and the velocities of both red blood cells and leukocytes were measured. The total number of leukocytes increased in sinusoids of I-R mice reaching a plateau within 1 h compared with sham animals, while the number of stationary leukocytes progressively increased over the entire study period. Stationary leukocytes in sinusoids increased after 1 and 1.5 h of I-R following CrMP, while they were significantly reduced following haemin treatment compared to animals treated with I-R only. In postsinusoidal venules a progressive increase in adherent leukocytes also occurred. As observed in sinusoids, CrMP significantly increased, while haemin significantly reduced leukocyte adhesion. The number of rolling leukocytes increased after CrMP in both I-R groups (1 and 1.5 h). The velocities of rolling leukocytes declined following 1.5 h of I-R compared with sham. Haemin treatment of 1.5 h I-R animals restored the velocities back to sham levels. The calculated wall shear rates in postsinusoidal venules were significantly lower in all I-R groups in comparison to sham animals. Combination of 1.5 h I-R with CrMP resulted in the lowest shear rates of all I-R groups. The number of stationary leukocytes within sinusoids and adherent leukocytes in postsinusoidal venules were correlated to the corresponding alanine aminotransferase (ALT) levels. In conclusion, endogenous HO reduces leukocyte-endothelial interactions within the liver. Thus, endogenous HO activity provides an important mechanism controlling the hepatic inflammatory response during the initiation of remote organ injury following normotensive limb ischaemia-reperfusion.
Although the consequences of trauma or infection are normally controlled at the site of injury, loss of local control may lead to a whole body response, which has been identified clinically as the systemic inflammatory response syndrome (SIRS). If this systemic inflammatory process involves whole body infection, the condition is termed sepsis. In its worst case, SIRS results in multiple organ dysfunction occurring in approximately 30 % of septic patients, in 24 % of patients suffering from pancreatitis, in over 30 % of trauma and in 40 % of burn patients (Beal & Cerra, 1994; Davies & Hagen, 1997).
Liver injury and dysfunction occurring during SIRS has often been overshadowed by concerns regarding cardiac, renal and respiratory function. The fact that liver dysfunction plays a central role in remote injury during SIRS has been supported by studies identifying liver dysfunction as one of the major factors contributing to the mortality of surgical patients suffering from multiple organ dysfunction following infrarenal aortic reconstruction (Huber et al. 1995; Maziak et al. 1998). Currently, pharmacologic protocols and mechanical devices are available to support most vital organ functions but none exists for the liver.
Leukocyte accumulation has been linked to organ injury during SIRS (Stephens et al. 1988; Wakefield et al. 1993; Rosenbloom et al. 1995). The microvascular location of such accumulation may differ between organs. For example, inflammation within skeletal muscle and mesentery, results in leukocyte-endothelial cell interactions within postcapillary venules. Leukocyte accumulation within the liver occurs not only in the collecting venules (post-sinusoidal venules), but also in the hepatic sinusoids in models of inflammation (Ferguson et al. 1993; Vollmar et al. 1994) and infection (Vollmar et al. 1993). However, to our knowledge there is no information regarding the onset of leukocyte accumulation within the liver during the early stages of remote organ injury.
Recent evidence suggests that the activity of haem oxygenase (HO), the rate-limiting enzyme in the degradation of haem into carbon monoxide and bilirubin, may possess anti-inflammatory properties. HO activity suppresses the complement-dependent inflammatory response in the brain and spleen of rats, while a reduction in HO activity shows the reverse effect (Appleton et al. 1999). In addition, recent studies suggest that HO activity may be capable of reducing monocyte chemotaxis (Ishikawa et al. 1997) and altering the expression of adhesion molecules such as ICAM-1 on endothelial cells (Wagener et al. 1999) and P-selectin in the liver (Vachharajani et al. 2000). It has recently been shown that the induction of HO activity, via the application of haemin, resulted in reduced leukocyte adhesion elicited by oxidative stress in the mesenteric microcirculation (Hayashi et al. 1999). Although the liver is one of the most abundant sources of endogenous HO activity, there is little information regarding its role in leukocyte accumulation during the initiation of remote organ injury.
Ischaemia-reperfusion (I-R) of skeletal muscle has been shown to induce both local and remote organ injury (Harris et al. 1986; Sexton et al. 1990). While models of limb I-R associated with hypotension have shown evidence of remote injury in the lung (Welbourn et al. 1991), heart and kidney (Walker, 1991), recent studies using a normotensive model of limb I-R have shown remote injury in the intestine (Corson et al. 1992) and most recently within the liver (Brock et al. 1999, 2001).
The aim of the present study was to test the hypothesis that leukocyte accumulation in the hepatic microcirculation is controlled, at least in part, by endogenous HO activity during the early stages of remote organ injury. To address this aim, we employed an established normotensive model of limb I-R (Nie et al. 2000), thereby eliminating the confounding effects of hypotension, while mimicking the normotensive condition of fluid resuscitated patients who develop remote injury following trauma or vascular surgery. We applied fluorescence intravital videomicroscopy to directly assess leukocyte behaviour within the hepatic microvasculature. Using this approach, we provide direct evidence that HO activity is an important mechanism controlling leukocyte accumulation, particularly within postsinusoidal venules during the early stages of remote injury.
METHODS
Animal description and care
All experimental protocols were in compliance with the guidelines of the Committee on the Care and Use of Laboratory Animals of the Institute of Laboratory Animal Resources, National Research Council, Canada. The mice were fed a standard laboratory diet and water ad libitum. The Council on Animal Care at the University of Western Ontario, Canada approved the experimental protocol.
Experimental protocol
Anaesthesia was induced in male C57BL/6 mice (22–28 g) by inhalation of isoflurane (5 % induction, 2 % surgery, 1–1.5 % maintenance; Abbott Laboratories, Missisauga, ON, Canada) in a 60 % oxygen-40 % nitrogen mixture. The left carotid artery was cannulated (PE-10 polyethylene tubing; Becton-Dickenson, USA) under sterile conditions for the continuous measurement of systemic arterial blood pressure and heart rate. The carotid artery cannula was also used for administration of drugs and fluorescent vital dyes. The tubing was tunnelled to the back of the neck and connected to a swivel (Fluid Swivel, Stoelting, IL, USA) to allow unlimited movement during recovery. Normal saline was administered at a rate of 0.4 ml h−1 throughout the experiment to maintain normal mean arterial pressure.
The animals were randomly assigned to either a sham (no limb ischaemia) or limb ischaemia-reperfusion (I-R) group and then further randomized to be examined at either 1 or 1.5 h after the onset of limb reperfusion. Animals in the I-R group (n = 4 for both reperfusion times) were treated with 60 min bilateral hindlimb ischaemia induced by tightening a tourniquet located above the greater trochanter of each leg. Sham animals (n = 4) were not subjected to ischaemia, but remained anaesthetized for the same period of time as the length of ischaemia. The tourniquets were removed just prior to recovery from anaesthesia. The animals were awake during the reperfusion period and reanaesthetized for the microscopy procedure.
To assess the influence of HO activity on leukocytes in the liver microvasculature, the animals were randomly allocated to groups treated either with chromium mesoporphyrin (CrMP; 40 μmol kg−1, i.p., Porphyrin Products Inc., Logan, UT, USA; n = 4 for both reperfusion times), a competitive inhibitor of HO activity or with haemin (10 mg kg−1, i.p., Porphyrin Products; n = 4 at 1.5 h reperfusion only), a stimulus for the induction of HO activity. Both haemin and CrMP were injected directly following the onset of limb ischaemia. Following intravital microscopy, the animals were killed by arterial exsanguination and the blood was collected for later analysis. The liver was removed and immediately frozen in liquid nitrogen for the analysis of HO protein.
Intravital microscopy
A small transverse epigastric laparotomy with protection of the left superior epigastric artery allowed the exposure of the liver and preserved a dry operative field. The left lobe of the liver was reflected onto the stage of an inverted microscope (Diaphot-TMD, Nikon, Japan). The surface of the organ was moistened with 0.9 % saline and covered by plastic film (Saran, Dow Chemical, Canada). Infrared heat lamps preserved body and organ temperatures (36–37 °C).
Leukocyte parameters
All animals received an intra-arterial injection of rhodamine 6G (0.3 mg kg−1, Sigma, St Louis, MO, USA). The dye was administered immediately after the tourniquets were removed (or at the corresponding time point in the sham group). Rhodamine 6G fluorescence was visualized by epi-illumination at 510 to 560 nm, using a 590 nm emission filter.
After preparing the liver for intravital microscopy, 10 fields of view were randomly chosen and viewed with a × 40 objective (CF Achromat 40/0.4; Nikon). Within each field of view at least one postsinusoidal venule and between 8 and 10 sinusoids were observed. Images were projected onto a charge-coupled device (CCD) video camera (Dage-MTI) and each field of view was recorded for 30 s using an SVHS videotape (Maxell, Concord, ON, Canada) and a video recorder (Panasonic PV-S 4480) for later analysis. Views were always completed 5–10 mm from the edge of the organ.
Quantification of leukocytes in sinusoids and postsinusoidal venules was performed off-line by frame-by-frame analysis of the videotaped images. Tapes were analysed by a single observer in a blinded fashion. Leukocytes were considered adherent in postsinusoidal venules if they remained stationary for 30 s. The number of adherent leukocytes in postsinusoidal venules is given per square millimetre of endothelial surface, calculated from the diameter and length of the postsinusoidal vessel segment, assuming cylindrical geometry. Leukocytes in postsinusoidal venules were considered to be rolling if their velocity was less than 250 μm s−1 (Zeintl et al. 1989) and expressed as the percentage of total non-adherent (all leukocytes passing through the vessel during the observation period of 30 s) leukocytes. Centreline red blood cell velocity and leukocyte velocity were analysed and wall shear rate calculated based on the Newtonian definition (γ = 8 × V/D where V is mean velocity (centreline velocity/1.6) and D is diameter of the individual microvessel). The total number of leukocytes in sinusoids is given as the number of cells per field including all leukocytes seen during the observation period of 30 s. Leukocytes were considered stationary in sinusoids if they were not moving during the observation period of 30 s.
Alanine aminotransferase
Blood samples obtained by cardiac puncture of the left ventricle were used to determine serum levels of alanine aminotransferase (ALT). The samples were stored in serum tubes (Capiject, Terumo Medical Corporation, USA) and immediately centrifuged at 6500 g for 5 min. The ALT within the serum samples was determined at 37 °C by means of standard enzymatic techniques.
HO protein via Western Blot
HO protein was measured in liver microsomes. Briefly, the frozen liver tissue samples were homogenized in homogenizing buffer (50 mm Tris, 1.15 % KCl, 1 mm EDTA, pH 7.4) using a Brinkmann Polytron homogenizer. The homogenate was centrifuged at 10 000 g for 20 min at 4 °C, and the resulting supernatant was further centrifuged for 60 min at 105 000 g at 4 °C. The microsomal pellets were resuspended in freezing buffer (0.1 m potassium phosphate buffer (KPBS), pH 7.5). Protein concentrations were estimated using the Bradford assay, with bovine serum albumin (BSA) as standard. Liver microsomes (80 μg) were electrophoretically resolved on a 12.5 % polyacrylamide gel, and transferred to nitrocellulose membranes, which were then blocked in 5 % non-fat dry milk (NFDM) with 0.1 % Tween-20 for 1 h at room temperature. The membranes were then incubated with HO-1 monoclonal antibody (1:1000 dilution in 5 % NFDM, StressGen Corp., Victoria, BC, Canada) for 2 h at room temperature. The membranes were washed with Tween-phosphate-buffered saline, and incubated in HRP-conjugated mouse IgG (1:1000 dilution in 5 % NFDM) for 1 h at room temperature. Blots were developed by enhanced chemiluminescence (0.4 nm coumaric acid, 2.5 mm luminol in 10 mm Tris, pH 8.5 added 1:1 to a solution of 0.2 % H2O2 in 100 mm Tris, pH 8.5 exposed to blots for 1 min) and visualized on X-ray film. Bands were analysed using image densitometry and expressed as the relative optical density per pixel (OD pixel−1).
Measurement of HO activity
HO activity was measured in the liver microsomes as described previously (Trakshel et al. 1986). Briefly, microsomes from harvested tissues were added to a reaction mixture containing 0.1 m KPBS (pH 7.4), haemin (25 μm) and mouse liver cytosol prepared from 105 000 g supernatant as a source of biliverdin reductase. The reaction was initiated by addition of NADPH (0.4 mm) to the samples, while the same volume of 0.1 m KPBS was added to the blanks. The reaction was conducted in triplicate in the dark, in a shaker water bath at 37 °C for 30 min, and terminated by placing the samples on ice. Bilirubin concentration was calculated as the difference in absorbance between 470 and 530 nm, using an extinction coefficient of 40 mm cm−2. HO activity was then expressed as production of bilirubin in picomoles per milligram of protein per hour.
Statistical analysis
The data were analysed on SigmaStat software (SPSS, Chicago, IL, USA) using ANOVA and Student's t test with Bonferroni's correction for multiple comparisons. If the criteria for parametric tests were not met, comparisons were made using Kruskal-Wallis ANOVA on ranks. The correlation between variables was tested with the Pearson product moment correlation. Criterion for significance was P < 0.05 in all experiments. The data are presented as means ± s.e.m.
RESULTS
In all experimental groups mean arterial blood pressure (MAP) remained normotensive (> 80 mmHg) throughout the study period. Although MAP in the sham group was significantly higher than in all other groups, MAP did not differ between the various I-R groups throughout the study period (data not shown).
Leukocytes within sinusoids
The total number of leukocytes (stationary + moving) significantly increased 1 h following limb ischaemia compared with sham mice, with no further increase occurring following 1.5 h of reperfusion (Fig. 1). No significant difference in the total number of leukocytes was found between all I-R treatment groups. Thus, the 1.5 h group treated with CrMP showed the same total number of leukocytes within sinusoids as mice treated with haemin.
Figure 1. Leukocytes within sinusoids.
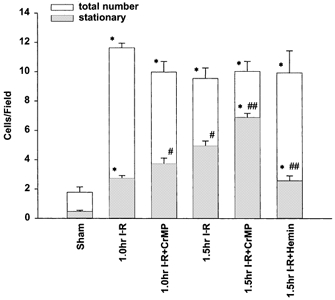
The total number of leukocytes (stationary + moving; open bars) within liver sinusoids significantly increased within 1 h compared to sham and remained at this level in all I-R treatment groups. The number of stationary leukocytes (grey bars) progressively increased from 1 h to 1.5 h compared to sham. Inhibition of HO via CrMP significantly increased, while haemin decreased leukocyte adhesion. *P < 0.05 compared to sham; #P < 0.05 compared to 1 h I-R; ##P < 0.05 compared to 1.5 h I-R.
The number of stationary leukocytes within sinusoids increased significantly following 1 h of I-R compared to sham animals. After 1.5 h of I-R, the number of stationary leukocytes continued to increase (Fig. 1). The total number of leukocytes remained unchanged compared to 1 h of I-R. Application of CrMP resulted in a further significant increase in stationary leukocytes at both the 1 h and 1.5 h time points, while haemin caused a significant reduction in stationary leukocytes at 1.5 h. Although haemin was effective in significantly reducing the number of stationary leukocytes compared to the untreated 1.5 h I-R group, sinusoids in the haemin-treated animals retained significantly increased numbers of stationary leukocytes compared to sham.
Leukocytes in postsinusoidal venules
The velocities of rolling leukocytes progressively declined compared to sham, becoming significantly lower following 1.5 h of I-R (Table 1 and Fig. 2). Rolling velocities at 1 h of I-R became significantly different from sham only after the application of CrMP. Interestingly, CrMP did not result in a further decline following 1.5 h, but the application of haemin significantly increased rolling velocity back to sham levels.
Table 1.
Leukocyte flow and microcirculatory data in postsinusoidal venules
| Sham | 1 h I–R | 1 h I–R + CrMP | 1.5 h I–R | 1.5 h I–R + CrMP | 1.5 h I–R + haemin | |
|---|---|---|---|---|---|---|
| Number of leukocytes analysed | 81 | 311 | 372 | 366 | 386 | 403 |
| Rolling leukocyte velocity (μm s−1) | 188.8 ± 11.7 | 165.6 ± 4.4 | 146.7 ± 6.1* | 131.9 ± 3.5† | 121.2 ± 3.7 | 183.8 ± 3.6‡ |
| Centreline RBC velocity (μm s−1) | 819.7 ± 32.7 | 634.8 ± 25.1* | 537.2 ± 20* | 593.8 ± 32.1* | 449.1 ± 18.1*†‡ | 569.8 ± 12.8* |
| Diameter of vessel (μm) | 50.8 ± 4.7 | 52.3 ± 2.4 | 48.9 ± 2.1 | 47.1 ± 2.8 | 45.1 ± 1.9 | 51.6 ± 2.3 |
| Wall shear rate (l s−1) | 84.9 ± 4.3 | 62.8 ± 4.7* | 55.7 ± 1.8* | 65.8 ± 1.9* | 48.2 ± 3.4*†‡ | 62.9 ± 3.1* |
Mean velocity (μm s−1) of rolling leukocytes was significantly lower than sham following 1.5 h. Inhibition of HO activity significantly reduced while induction of HO activity restored mean velocity of rolling leukocytes. Microhaemodynamics showed decreased centreline velocities in I–R-treated animals compared to sham animals. Inhibition of HO activity with CrMP in animals treated with 1.5 h of I–R caused a significantly lower centreline velocity and wall shear rate compared to only I–R-treated animals.
P < 0.05 compared to sham;
P < 0.05 compared to 1 h I–R;
P < 0.05 compared to 1.5 h I–R.
Figure 2. Distribution of leukocyte velocity over increments of 50 μm s−1 in postsinusoidal venules.
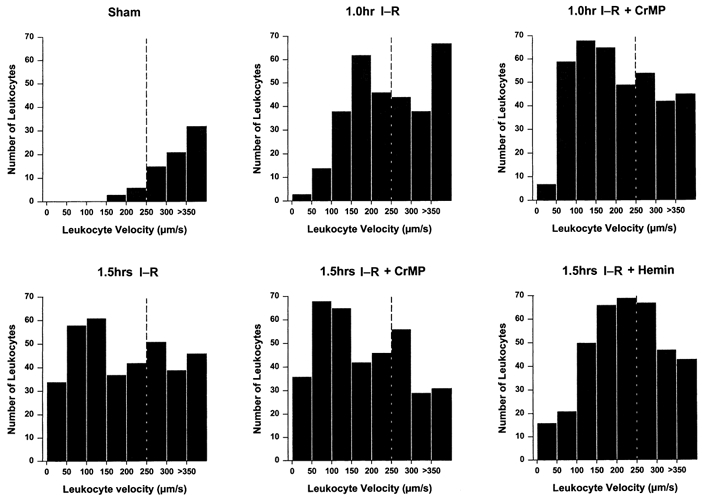
All leukocytes with velocities < 250 μm s−1 were considered rolling (dashed vertical line). Following 1.5 h I-R there was a significant reduction of rolling leukocyte velocities compared to sham. Inhibition of HO further reduced rolling leukocyte velocities, while application of haemin restored rolling velocity.
The proportion of rolling leukocytes in the 1 h I-R group was not significantly different compared to sham animals. However, a significant increase in leukocyte rolling occurred after 1.5 h (Fig. 3). Inhibition of HO activity following 1 h resulted in a significant increase in rolling leukocytes, compared to the untreated group at the 1 h time point. Likewise, application of CrMP caused a further significant increase in rolling leukocytes at 1.5 h. Application of haemin at 1.5 h caused no significant change in the incidence of rolling leukocytes compared with time-matched untreated I-R animals.
Figure 3. Number of rolling leukocytes in postsinusoidal venules.
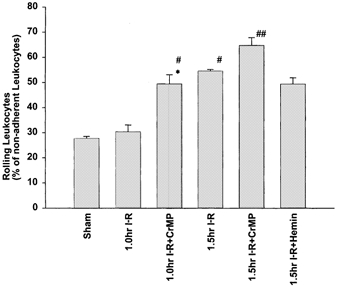
The number of rolling leukocytes within postsinusoidal venules (expressed as a percentage of non-adherent leukocytes) significantly increased following 1.5 h compared to sham. Inhibition of HO activity via CrMP resulted in a significant increase in the incidence of rolling leukocytes at both time points, but application of haemin had no effect. *P < 0.05 compared to sham; #P < 0.05 compared to 1 h I-R; ##P < 0.05 compared to 1.5 h I-R.
Microhaemodynamics in postsinusoidal venules
The red blood cell (RBC) centreline velocity in postsinusoidal venules was significantly lower in all I-R groups in comparison to sham animals (Table 1). Administration of CrMP resulted in a further significant reduction in RBC velocity in the 1.5 h I-R group only, while treatment with haemin did not restore the RBC velocity beyond I-R-treated levels. No differences were detectable in the diameters of the postsinusoidal venules between the different groups. Thus, the calculated wall shear rates showed the same course as RBC velocities. All treatment groups showed lower shear rates than sham animals. The 1.5 h I-R group treated with CrMP resulted in the lowest shear rates of all I-R groups.
The number of adherent leukocytes within postsinusoidal venules significantly increased following 1 h of limb ischaemia compared to sham animals with the number progressively increasing at 1.5 h (Fig. 4). The application of CrMP resulted in a significant increase in adherent leukocytes in both the 1 and 1.5 h I-R groups. Application of haemin significantly reduced the number of adherent leukocytes compared to untreated animals (154.1 ± 12.2), but postsinusoidal venules retained significant adhesion compared to sham animals.
Figure 4. Number of adherent leukocytes in postsinusoidal venules.
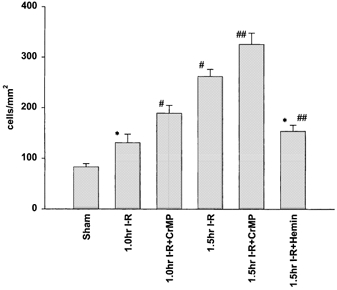
Number of adherent leukocytes (cells mm−2) in postsinusoidal venules progressively increased over 1.5 h compared to sham. Inhibition of HO activity via CrMP significantly increased leukocyte adhesion, while application of haemin significantly reduced the number of adherent leukocytes. *P < 0.05 compared to sham; #P < 0.05 compared to 1 h I-R; ##P < 0.05 compared to 1.5 h I-R.
Alanine aminotransferase
A trend toward progressively increased ALT was measured with increasing time following limb ischaemia (Fig. 5). The removal of HO activity via application of CrMP resulted in an increase in ALT-levels at all times. The administration of haemin caused a significant decrease in ALT-levels, which approached levels measured in sham animals. Interestingly, the number of stationary leukocytes within sinusoids and the number of adherent leukocytes in postsinusoidal venules were correlated to the corresponding ALT-levels (r = 0.633; P < 0.001 and r = 0.619; P < 0.001, respectively).
Figure 5. Liver injury estimated from serum alanine aminotransferase (ALT) levels.
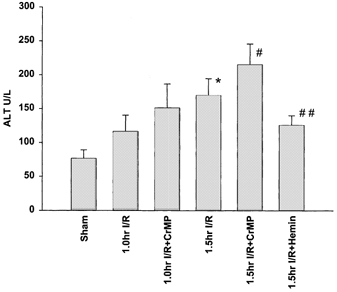
Plasma activities of ALT were higher in all I-R-treated animals compared to sham. Inhibition of HO activity with CrMP caused an increase in ALT levels, while induction of HO activity with haemin resulted in ALT levels nearly similar to sham animals. *P < 0.05 compared to sham; #P < 0.05 compared to 1 h I-R; ##P < 0.05 compared to 1.5 h I-R.
Induction of HO protein and HO activity
No significant induction of HO-1 protein was measured after 1.5 h of I-R (1.8 ± 0.3 OD pixel−1) compared to sham animals (1.4 ± 0.4 OD pixel−1). However, HO protein increased significantly following the application of haemin (2.9 ± 0.4 OD pixel−1), while remaining unchanged following CrMP (0.8 ± 0.10 OD pixel−1). The HO activity (expressed as production of bilirubin in picomoles per milligram of protein per hour) in the 1.5 h I-R group treated with haemin was significantly higher (4810 ± 1228 pmol (mg protein)−1 h−1) compared with sham animals (1259 ± 544 pmol (mg protein)−1 h−1) and compared with the untreated 1.5 h I-R group (1298 ± 528 pmol (mg protein)−1 h−1), while animals treated with 1.5 h of I-R plus CrMP showed almost no HO activity (8 ± 7 pmol (mg protein)−1 h−1).
DISCUSSION
The current study demonstrates that the total number of leukocytes within liver sinusoids increased as early as 1 h following limb ischaemia-reperfusion. No further increase in the total leukocyte numbers occurred following this initial response, at least up to the 1.5 h study period. In spite of such stable leukocyte numbers, an increase in leukocyte-endothelial cell interaction was evident in both sinusoids and postsinusoidal venules. Although induction of HO protein did not occur over the study period, it appears that endogenous HO activity plays an important role in controlling leukocyte behaviour within both sinusoids and postsinusoidal venules.
The incidence of leukocyte adhesion in postsinusoidal venules appeared similar to the incidence of stationary leukocytes in sinusoids. That is, a progressive increase in leukocyte adhesion occurred over 1.5 h. Inhibition of HO activity exacerbated, while haemin reduced, the incidence of leukocyte adhesion. Since the interaction of leukocytes with the endothelial cell depends on both the shear acting upon the leukocyte and the expression of adhesion molecules, it is likely that one or both of these factors are involved during the early stages of remote liver injury following limb I-R.
Although the average number of leukocytes rolling within postsinusoidal venules remained unchanged at 1 h of I-R compared to the sham group, there was an increased incidence of leukocyte adhesion. It has been shown that reduced shear is often associated with increased leukocyte-endothelial cell interaction, which may result in firm adhesion when the appropriate molecules are expressed on the leukocyte and endothelial surfaces (i.e. CD18 and ICAM-1) (Walpola et al. 1995; Mohan et al. 1999). Our data showed that the shear rate within postsinusoidal venules following 1 h of I-R was significantly lower compared to sham animals. Although we did not directly test the role of adhesion molecules, such results strongly suggest that the increased interaction between leukocytes and the vessel wall in this group was significantly influenced by the reduced shear rate. However, the shear rate did not significantly change in the 1.5 h I-R group compared to 1 h I-R, nor was there induction of HO protein, yet the incidence of leukocyte adhesion significantly increased, shown by an increase of leukocytes rolling slower than 50 μm s−1(Fig. 2). Such a result could be due to increased expression of adhesion molecules.
The effect of reducing HO activity via the application of CrMP, resulted in a significant increase in leukocyte adhesion within postsinusoidal venules in both the 1 and 1.5 h I-R groups, yet the shear rate was significantly lower in the 1.5 h I-R group only. Such results suggest that the reduction in flow, and thus shear, may have been a significant influence on the increased leukocyte adhesion in the latter group, but would not have accounted for the increased adhesion in the 1 h I-R group. Thus, increased adhesion in the 1 h I-R group, treated with CrMP, is likely to have reflected increased expression of adhesion molecules.
In animals treated with 1.5 h of I-R plus haemin, the velocities of rolling leukocytes in postsinusoidal venules increased compared to the untreated 1.5 h I-R group, yet the shear rate within these vessels was unchanged compared to the 1.5 h I-R group. In addition, the number of adherent leukocytes were significantly reduced compared to the 1.5 h I-R group. In fact, adhesion reached levels indistinguishable from those measured in the 1 h I-R group. Interestingly, the shear rates between the 1 h, the 1.5 h I-R and the 1.5h plus haemin groups were not significantly different. Thus, if the reduction in leukocyte adhesion in the 1.5 h I-R plus haemin group was the result of reduced shear (compared to untreated I-R) only, then one would have expected the number of adherent leukocytes to be similar to that in the untreated 1.5 h I-R group, yet the resulting adhesion reduced to levels measured in the 1 h I-R group. We believe the reduced adhesion following haemin not only reflects the shear acting on the leukocyte, but a reduction in the expression of adhesion molecules. However, whether altered expression of adhesion molecules occurred on both leukocyte and endothelial cell awaits further study.
Within sinusoids, a progressive increase in the number of stationary leukocytes occurred over 1.5 h. Inhibition of endogenous HO activity resulted in a significant increase, while increasing HO resulted in a significant decline in the numbers of stationary leukocytes. Although the design of the present study did not directly test specific mechanisms of HO control on leukocyte-endothelial interactions and accumulation within sinusoids, the reduced RBC velocity within postsinusoidal venules in all I-R groups suggests that sinusoidal flow was likely also reduced, particularly as no change in venular diameter occurred. However, as indicated above, the centreline velocities were unchanged in the 1 and 1.5 h groups in postsinusoidal venules, yet there was a progressive increase in stationary leukocytes within sinusoids. Interestingly, the application of CrMP did not alter RBC velocity in postsinusoidal venules in the 1 h I-R group; yet removing HO activity resulted in a significant increase in stationary sinusoidal leukocytes. Such results suggest that mechanisms other than changes in flow alone operate to restrict leukocyte flow through sinusoids during the remote injury process. Such mechanisms may include, loss of volume control within endothelial cells and/or contraction of Ito cells thereby reducing sinusoidal diameter leading to entrapment of leukocytes (Vollmar et al. 1995), changes in the deformability of leukocytes (Simon et al. 1999) and/or the expression of adhesion molecules (Steinhoff et al. 1993).
HO degrades protohaem IX into carbon monoxide (CO), free divalent iron and biliverdin-IXα. It has been previously shown that CO causes active relaxation of Ito cells thereby reducing resistance and improving sinusoidal perfusion (Suematsu et al. 1995). As some studies suggested that leukocytes become physically trapped within liver sinusoids (Vollmar et al. 1995), it may be possible that increasing HO activity would influence the driving force acting upon leukocytes by increasing sinusoidal blood flow, thereby forcing them through the sinusoidal bed. Although we cannot exclude this possibility, the RBC velocity within postsinusoidal venules did not change following application of haemin, compared to other I-R groups, suggesting that sinusoidal flow probably did not significantly change. An alternative mechanism may involve the interaction of adhesion molecules. Although studies suggest that selectin-mediated rolling is not involved in leukocyte sequestration within liver sinusoids (Wong et al. 1997), cell adhesion per se may be important since expression of ICAM-1 on the surface of hepatocytes, endothelial cells of the sinusoids and Kupffer cells has been demonstrated during various inflammatory conditions (Steinhoff et al. 1993). Although HO activity has been shown to provide anti-inflammatory properties by altering the expression of ICAM-1 (Wagener et al. 1999), the precise mechanism of HO control over leukocyte accumulation within sinusoids during pathologic processes remains an active area of study.
It was recently shown that increased production of bilirubin by the application of haemin prevented oxidant-induced leukocyte adhesion in the mesenteric tissue (Hayashi et al. 1999). In addition, HO appears to regulate the expression of P-selectin in the vascular bed of the liver for up to 4 h after lipopolysaccharide (LPS) treatment. The LPS-induced P-selectin expression was significantly reduced by pretreatment with haemin or biliverdin and increased by zinc protoporphyrin, a potent HO-1 inhibitor (Vachharajani et al. 2000). In human umbilical vein endothelial cells (HUVECs), the administration of haem was associated with an increase of ICAM-1 expression on the endothelial surface. The increase in ICAM-1 expression was detectable at less than 60 min after the administration of haem. Interestingly, pre-induction of HO activity resulted in a marked decrease in haem-induced ICAM-1 expression (Wagener et al. 1999). The ability of HO to generate bilirubin, a potent antioxidant, as well as carbon monoxide and cellular free iron could all be factors that contribute to the alleviation of an inflammatory reaction.
Contradictory results have been reported regarding leukocyte accumulation and the extent of liver injury. Vollmar et al. (1994) showed that in warm liver I-R the number of adherent leukocytes in venules, but not in sinusoids, correlated with the extent of liver injury. On the other hand, it has been shown that in endotoxic shock only neutrophils from the sinusoids actually transmigrated at a time when parenchymal cell injury occurred (Chosay et al. 1997). In the present study, a correlation was found between ALT, an indicator of liver injury, and the number of stationary leukocytes within both sinusoids and postsinusoidal venules. The disparity in results of the various studies may reflect the use of different inflammatory models; nevertheless the results suggest that recruitment of leukocytes into the liver is an important step in the pathogenesis of liver injury (Jaeschke et al. 1996).
In conclusion, the results of the present study provide the first direct evidence that endogenous HO activity within the liver provides an important anti-inflammatory mechanism, which retards the accumulation of leukocytes within the hepatic microvasculature in the early stages of remote organ injury following limb I-R. Although remote injury may occur in any organ, death usually results as a consequence of heart, lung, kidney or liver failure. Even though elaborate protocols exist to support declining function of the heart, lung and kidney, no such supportive approach is clinically available for the liver. Consequently, future studies exploring the role of endogenous protective mechanisms, such as the activity of haem oxygenase, may contribute to the development of a therapeutic approach to protect the liver during conditions leading to remote organ injury.
Acknowledgments
This work was supported in part by grants from the Canadian Institutes of Health Research and London Health Science Centre Research Fund.
REFERENCES
- Appleton SD, Chretien ML, McLaughlin BE, Vreman HJ, Stevenson DK, Brien JF, Nakatsu K, Maurice DH, Marks GS. Selective inhibition of heme oxygenase, without inhibition of nitric oxide synthase or soluble guanylyl cyclase, by metalloporphyrins at low concentrations. Drug Metabolism and Disposition. 1999;27:1214–1219. [PubMed] [Google Scholar]
- Beal AL, Cerra FB. Multiple organ failure syndrome in the 1990s. Systemic inflammatory response and organ dysfunction. Journal of the American Medical Association. 1994;271:226–233. [PubMed] [Google Scholar]
- Brock RW, Lawlor DK, Harris KA, Potter RF. Initiation of remote hepatic injury in the rat: interactions between Kupffer cells, tumor necrosis factor-alpha, and microvascular perfusion. Hepatology. 1999;30:137–142. doi: 10.1002/hep.510300132. [DOI] [PubMed] [Google Scholar]
- Brock RW, Nie RG, Harris KA, Potter RF. Kupffer cell-initiated remote hepatic injury following bilateral hindlimb ischemia is complement dependent. American Journal of Physiology - Gastrointestinal and Liver Physiology. 2001;280:279–284. doi: 10.1152/ajpgi.2001.280.2.G279. [DOI] [PubMed] [Google Scholar]
- Chosay JG, Essani NA, Dunn CJ, Jaeschke H. Neutrophil margination and extravasation in sinusoids and venules of liver during endotoxin-induced injury. American Journal of Physiology. 1997;272:G1195–1200. doi: 10.1152/ajpgi.1997.272.5.G1195. [DOI] [PubMed] [Google Scholar]
- Corson RJ, Paterson IS, O'dwyer ST, Rowland P, Kirkman E, Little RA, McCollum CN. Lower limb ischaemia and reperfusion alters gut permeability. European Journal of Vascular Surgery. 1992;6:158–163. doi: 10.1016/s0950-821x(05)80234-8. [DOI] [PubMed] [Google Scholar]
- Davies MG, Hagen PO. Systemic inflammatory response syndrome. British Journal of Surgery. 1997;84:920–935. doi: 10.1002/bjs.1800840707. [DOI] [PubMed] [Google Scholar]
- Ferguson D, McDonagh PF, Biewer J, Paidas CN, Clemens MG. Spatial relationship between leukocyte accumulation and microvascular injury during reperfusion following hepatic ischemia. International Journal of Microcirculation, Clinical and Experimental. 1993;12:45–60. [PubMed] [Google Scholar]
- Harris K, Walker PM, Mickle DA, Harding R, Gatley R, Wilson GJ, Kuzon B, McKee N, Romaschin AD. Metabolic response of skeletal muscle to ischemia. American Journal of Physiology. 1986;250:H213–220. doi: 10.1152/ajpheart.1986.250.2.H213. [DOI] [PubMed] [Google Scholar]
- Hayashi S, Takamiya R, Yamaguchi T, Matsumoto K, Tojo SJ, Tamatani T, Kitajima M, Makino N, Ishimura Y, Suematsu M. Induction of heme oxygenase-1 suppresses venular leukocyte adhesion elicited by oxidative stress: role of bilirubin generated by the enzyme. Circulation Research. 1999;85:663–671. doi: 10.1161/01.res.85.8.663. [DOI] [PubMed] [Google Scholar]
- Huber TS, Harward TR, Flynn TC, Albright JL, Seeger JM. Operative mortality rates after elective infrarenal aortic reconstructions. Journal of Vascular Surgery. 1995;22:287–293. doi: 10.1016/s0741-5214(95)70143-5. [DOI] [PubMed] [Google Scholar]
- Ishikawa K, Navab M, Leitinger N, Fogelman AM, Lusis AJ. Induction of heme oxygenase-1 inhibits the monocyte transmigration induced by mildly oxidized LDL. Journal of Clinical Investigation. 1997;100:1209–1216. doi: 10.1172/JCI119634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaeschke H, Smith CW, Clemens MG, Ganey PE, Roth RA. Mechanisms of inflammatory liver injury: adhesion molecules and cytotoxicity of neutrophils. Toxicology and Applied Pharmacology. 1996;139:213–226. doi: 10.1006/taap.1996.0160. [DOI] [PubMed] [Google Scholar]
- Maziak DE, Lindsay TF, Marshall JC, Walker PM. The impact of multiple organ dysfunction on mortality following ruptured abdominal aortic aneurysm repair. Annals of Vascular Surgery. 1998;12:93–100. doi: 10.1007/s100169900123. [DOI] [PubMed] [Google Scholar]
- Mohan S, Mohan N, Valente AJ, Sprague EA. Regulation of low shear flow-induced HAEC VCAM-1 expression and monocyte adhesion. American Journal of Physiology. 1999;276:C1100–1107. doi: 10.1152/ajpcell.1999.276.5.C1100. [DOI] [PubMed] [Google Scholar]
- Nie RG, Brock RW, Harris KA, Potter RF. Heme oxygenase in remote liver injury during the systemic inflammatory response syndrome. Federation of American Societies for Experimental Biology Journal. 2000;14:A367. [Google Scholar]
- Rosenbloom AJ, Pinsky MR, Bryant JL, Shin A, Tran T, Whiteside T. Leukocyte activation in the peripheral blood of patients with cirrhosis of the liver and SIRS. Correlation with serum interleukin-6 levels and organ dysfunction. Journal of the American Medical Association. 1995;274:58–65. [PubMed] [Google Scholar]
- Sexton WL, Korthuis RJ, Laughlin MH. Ischemia-reperfusion injury in isolated rat hindquarters. Journal of Applied Physiology. 1990;68:387–392. doi: 10.1152/jappl.1990.68.1.387. [DOI] [PubMed] [Google Scholar]
- Simon SI, Cherapanov V, Nadra I, Waddell TK, Seo SM, Wang Q, Doerschuk CM, Downey GP. Signaling functions of l-selectin in neutrophils: alterations in the cytoskeleton and colocalization with CD18. Journal of Immunology. 1999;163:2891–2901. [PubMed] [Google Scholar]
- Steinhoff G, Behrend M, Schrader B, Pichlmayr R. Intercellular immune adhesion molecules in human liver transplants: overview on expression patterns of leukocyte receptor and ligand molecules. Hepatology. 1993;18:440–453. [PubMed] [Google Scholar]
- Stephens KE, Ishizaka A, Wu ZH, Larrick JW, Raffin TA. Granulocyte depletion prevents tumor necrosis factor-mediated acute lung injury in guinea pigs. American Review of Respiratory Disease. 1988;138:1300–1307. doi: 10.1164/ajrccm/138.5.1300. [DOI] [PubMed] [Google Scholar]
- Suematsu M, Goda N, Sano T, Kashiwagi S, Egawa T, Shinoda Y, Ishimura Y. Carbon monoxide: an endogenous modulator of sinusoidal tone in the perfused rat liver. Journal of Clinical Investigation. 1995;96:2431–2437. doi: 10.1172/JCI118300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trakshel GM, Kutty RK, Maines MD. Purification and characterization of the major constitutive form of testicular heme oxygenase. The noninducible isoform. Journal of Biological Chemistry. 1986;261:11131–11137. [PubMed] [Google Scholar]
- Vachharajani TJ, Work J, Issekutz AC, Granger DN. Heme oxygenase modulates selectin expression in different regional vascular beds. American Journal of Physiology - Heart and Circulatory Physiology. 2000;278:1613–1617. doi: 10.1152/ajpheart.2000.278.5.H1613. [DOI] [PubMed] [Google Scholar]
- Vollmar B, Glasz J, Menger MD, Messmer K. Leukocytes contribute to hepatic ischemia/reperfusion injury via intercellular adhesion molecule-1-mediated venular adherence. Surgery. 1995;117:195–200. doi: 10.1016/s0039-6060(05)80085-6. [DOI] [PubMed] [Google Scholar]
- Vollmar B, Glasz J, Senkel A, Menger MD, Messmer K. Role of leukocytes in the initial hepatic microvascular response to endotoxemia. Zentralblatt für Chirurgie. 1993;118:691–696. [PubMed] [Google Scholar]
- Vollmar B, Menger MD, Glasz J, Leiderer R, Messmer K. Impact of leukocyte-endothelial cell interaction in hepatic ischemia-reperfusion injury. American Journal of Physiology. 1994;267:786–793. doi: 10.1152/ajpgi.1994.267.5.G786. [DOI] [PubMed] [Google Scholar]
- Wagener FA, Da Silva JL, Farley T, De Witte T, Kappas A, Abraham NG. Differential effects of heme oxygenase isoforms on heme mediation of endothelial intracellular adhesion molecule 1 expression. Journal of Pharmacology and Experimental Therapeutics. 1999;291:416–423. [PubMed] [Google Scholar]
- Wakefield CH, Carey PD, Foulds S, Monson JR, Guillou PJ. Polymorphonuclear leukocyte activation. An early marker of the postsurgical sepsis response. Archives of Surgery. 1993;128:390–395. doi: 10.1001/archsurg.1993.01420160028003. [DOI] [PubMed] [Google Scholar]
- Walker PM. Ischemia/reperfusion injury in skeletal muscle. Annals of Vascular Surgery. 1991;5:399–402. doi: 10.1007/BF02015307. [DOI] [PubMed] [Google Scholar]
- Walpola PL, Gotlieb AI, Cybulsky MI, Langille BL. Expression of ICAM-1 and VCAM-1 and monocyte adherence in arteries exposed to altered shear stress. Arteriosclerosis, Thrombosis, and Vascular Biology. 1995;15:2–10. doi: 10.1161/01.atv.15.1.2. [DOI] [PubMed] [Google Scholar]
- Welbourn R, Goldman G, O'riordain M, Lindsay TF, Paterson IS, Kobzik L, Valeri CR, Shepro D, Hechtman HB. Role for tumor necrosis factor as mediator of lung injury following lower torso ischemia. Journal of Applied Physiology. 1991;70:2645–2649. doi: 10.1152/jappl.1991.70.6.2645. [DOI] [PubMed] [Google Scholar]
- Wong J, Johnston B, Lee SS, Bullard DC, Smith CW, Beaudet AL, Kubes P. A minimal role for selectins in the recruitment of leukocytes into the inflamed liver microvasculature. Journal of Clinical Investigation. 1997;99:2782–2790. doi: 10.1172/JCI119468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeintl H, Sack FU, Intaglietta M, Messmer K. Computer assisted leukocyte adhesion measurement in intravital microscopy. International Journal of Microcirculation, Clinical and Experimental. 1989;8:293–302. [PubMed] [Google Scholar]