

Abstract
The heart is a dynamic organ capable of adapting its size and architecture in response to alterations in workload associated with developmental maturation, physiological stimulation and pathological diseases. Such alterations in heart size typically result from the hypertrophic growth of individual myocytes, but not myocyte cellular proliferation. In recent years, a great deal of investigation has gone toward elucidating the molecular signalling machinery that underlies the hypertrophic response and manner in which increased cardiac load promotes alterations in gene expression. To this end, the Ca2+-calmodulin-activated phosphatase calcineurin has been proposed as a necessary component of the multi-pathway hypertrophy program in the heart. Despite initial controversy over this hypothesis due to disparate results from pharmacological inhibitory studies in animal models of hypertrophy, compelling data from genetic models with calcineurin inhibition now exist. This review will summarize many of these studies and will attempt to address a number of unanswered issues. In particular, specific downstream mediators of calcineurin signalling will be discussed, as well as the need to identify calcineurin's temporal activation profile, transcriptional targets and cross-communication with other reactive signalling pathways in the heart. Finally, we will present evidence suggesting that calcineurin, as a Ca2+-responsive enzyme, may function as an internal load sensor in cardiac myocytes, matching output demands to hypertrophic growth.
Cardiac hypertrophy is defined by an increase in heart size and/or myofibrillar volume without a change in myocyte number, which occurs in response to both physiological and pathophysiological stimulation. Hypertrophy allows the myocardium to adapt functional performance to alterations in workload associated with developmental maturation, physiological challenge, or injury. While cardiac hypertrophy is typically viewed as a compensatory response that normalizes ventricular wall stress, sustained hypertrophy is correlated with an increase in both the incidence of and mortality from cardiovascular disease (Levy et al. 1990), and often is a first step in the progression to congestive heart failure. Cardiac hypertrophy is also a risk factor for arrhythmia and sudden cardiac death due to prolongation of the myocyte action potential (Kääb et al. 1998). While the clinical consequences of cardiac hypertrophy have been known for some time, only recently have significant inroads been made into our understanding of the molecular underpinnings of this response. A large body of literature has emerged describing the intracellular signalling pathways that transduce hypertrophic stimulation into alterations in gene expression, which include the mitogen-activated protein kinases (MAPKs), protein kinase C (PKC), PI3K-Akt and calcineurin-nuclear factor of activated T-cells (NFAT) (McKinsey & Olson, 1999; Steinberg, 2000; Molkentin & Dorn, 2001).
The Ca2+-calmodulin-activated phosphatase calcineurin and its downstream transcriptional effector NFAT have been implicated as critical transducers of the hypertrophic response that uniquely link alterations in intracellular calcium handling in a myocyte to the hypertrophic growth response. The initial description of calcineurin and NFAT as hypertrophic transducers involved transgenic overexpression of each factor in the heart, which promoted a dramatic hypertrophy response that quickly transitioned to heart failure and death in the mouse (Molkentin et al. 1998). However, controversy quickly followed as pharmacological studies employing the calcineurin inhibitors cyclosporin A or FK506 in different rodent models of heart disease produced largely equivocal results (Olson & Molkentin, 1999; Molkentin, 2000). More recently, data generated by loss-of-function approaches in genetically modified mouse models have revitalized the hypothesis that calcineurin and NFAT function as critical transducers of the hypertrophic response. This review will largely focus on recent studies using genetic manipulations in the mouse to address the importance of calcineurin signalling in hypertrophy, and will conclude with a discussion of important questions that remain to be answered.
Calcineurin as a sufficient and necessary mediator of cardiac hypertrophy
Calcineurin was initially described as a Ca2+-activated, cyclosporin A (CsA)/FK506-inhibited phosphatase that, upon activation, catalysed the dephosphorylation and nuclear accumulation of cytoplasmic NFAT transcription factors (Flanagan et al. 1991; Clipstone & Crabtree, 1992; Shibasaki et al. 1996) (Fig. 1). In the heart, nuclear NFATs bind to the transcription factor GATA-4 and activate transcription of hypertrophic genes. Cardiac overexpression of the constitutively active calcineurin catalytic subunit or a constitutively nuclear NFATc4 mutant protein each induced massive cardiac hypertrophy that quickly transitioned to heart failure and death (Molkentin et al. 1998).
Figure 1. Calcineurin signalling pathways in cardiac myocytes.
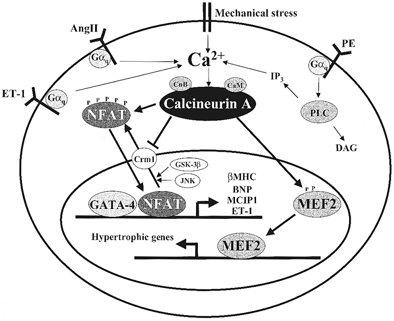
Activation of Gαq-coupled or mechanical stretch receptors leads to an elevation of intracellular Ca2+ and activation of the calmodulin-regulated phosphatase, calcineurin. Calcineurin activation causes nuclear localization of NFAT transcription factors by direct dephosphorylation as well as inhibition of the exportin Crm1 (Zhu & McKeon, 1999). Calcineurin also directly activates nuclear MEF2 factors and probably has other, unknown effectors. These factors, along with GATA-4 and other partners, cooperatively activate transcription of the hypertrophic gene programme. Reductions in [Ca2+]i cause inactivation of calcineurin, de-stimulation of MEF2, and Crm1-mediated nuclear export of NFATs catalysed by NFAT kinases such as GSK-3β and JNK.
Since that initial report, numerous studies have employed CsA or FK506 to determine the necessity of calcineurin signalling in multiple models/experimental systems of hypertrophy. At present count, 22 studies support the hypothesis that calcineurin functions as an important hypertrophic transducing factor in cardiac myocytes (18 in vivo and 4 in vitro) (Mende et al. 1998; Molkentin et al. 1998; Sussman et al. 1998; Meguro et al. 1999; Shimoyama et al. 1999; Hill et al. 2000; Kato et al. 2000; Lim et al. 2000a,b; Murat et al. 2000; Sakata et al. 2000; Shimoyama et al. 2000; Xia et al. 2000; Goldspink et al. 2001; Wang et al. 2001a). In addition, three reports demonstrated electrophysiological changes in two models of hypertrophy that were reversible with CsA administration (Wang et al. 2001a,b; Yatani et al. 2001). However, five studies utilizing these calcineurin inhibitory agents failed to identify a significant attenuation of cardiac hypertrophy in similar rodent models (Luo et al. 1998; Muller et al. 1998; Zhang et al. 1999; Ding et al. 1999; Fatkin et al. 2000). It is likely that experimental variables such as drug dosing and the specifics of each animal model underlie these differing accounts. However, another important issue that should be considered is the specificity of CsA or FK506. For example, high doses of CsA can alter sarcoplasmic reticulum Ca2+ release through the ryanodine receptor, or through a non-specific leakage from the sarcoplasmic reticulum itself (Park et al. 1999; Janssen et al. 2000). CsA also inhibits the Na+,K+-ATPase promoting neurotoxicity and nephrotoxicity, which could lead to a secondary increase in blood pressure and increased load on the heart (reviewed in Klee et al. 1998). Finally, CsA can antagonize apoptosis in certain cell types by binding to mitochondrial cyclophilin D and inhibiting the mitochondrial permeability transition, an effect that is independent of calcineurin activity (Nazareth et al. 1991; Griffiths & Halestrap, 1993, 1995; Halestrap et al. 1997; Lemasters et al. 1997; Molkentin, 2001).
Genetic inhibition of calcineurin
To avoid many of the complications arising from the use of CsA and FK506 as calcineurin inhibitors, recent work has involved the use of genetic models with inhibited calcineurin activity. This has been accomplished by either overexpressing naturally occurring inhibitors of calcineurin (so-called ‘calcipressins’), through dominant-negative strategies, or more recently through gene targeting. The first such approach involved overexpression of the calcineurin inhibitory domain from two proteins, Cabin1/Cain and AKAP79. Cain is a ubiquitously expressed 230 kDa protein containing a C-terminal domain that acts as a non-competitive inhibitor of calcineurin, while AKAP79 is a scaffolding protein that docks calcineurin, PKA and PKC. Using a recombinant adenoviral approach to infect rat neonatal cardiomyocytes, Taigen et al. (2000) demonstrated that the calcineurin inhibitory domain from either Cain or AKAP79 blocked in vitro hypertrophy in response to phenylephrine (PE), angiotensin II (AngII) and 1 % serum. These studies were then extended to the in vivo setting by the creation of Cain and AKAP79 transgenic mice (De Windt et al. 2001). Low-copy Cain or AKAP79 mice displayed normal heart function at baseline and an impaired hypertrophic response to abdominal aortic constriction and angiotensin II infusion. In addition, rats subjected to in vivo cardiac gene transfer with the Cain-expressing adenovirus demonstrated a significant resistance to hypertrophy induced by transverse aortic constriction as compared to controls subjected to an equal transaortic pressure gradient (De Windt et al. 2001) (Table 1).
Table 1.
Summary of genetic models of calcineurin inhibition subjected to hypertrophic stimuli
| Study | Model | Stimulus | % Increase(-inhibitor) | % Increase(+inhibitor) | Relative % decrease |
|---|---|---|---|---|---|
| Bueno et al. 2002 | cnAβ -/- mouse | AngII, 14 days | 14 | −6.5 | 146 |
| Iso, 14 days | 33 | 8 | 75 | ||
| AAC, 14 days | 39 | 13 | 67 | ||
| Hill et al. 2002 | MCIP1 TG mouse | TAC, 21 days | 70 | 40 | 43 |
| De Windt et al. 2001 | ΔCain TG mouse | Iso, 14 days | 24 | 12 | 50 |
| AAC, 14 days | 22 | 5 | 79 | ||
| ΔAKAP TG mouse | Iso, 14 days | 24 | 12 | 50 | |
| AAC, 14 days | 22 | 14 | 38 | ||
| Ad Cain infuse rat | TAC, 7 days | 27 | 16 | 40 | |
| Rothermel et al. 2001 | MCIP1 TG mouse | CnA TG, 12 weeks | 129 | 39 | 70 |
| Iso, 7 days | 23 | 9 | 59 | ||
| Exercise, 28 days | 29 | 12 | 58 | ||
| Zou et al. 2001a | dnCnA TG mouse | AAC, 21 days | 58 | 26 | 56 |
Relative percent decrease for all models was determined to be statistically significant in the original publications. All data represent percent increase and decrease in heart-to-body weight ratio, except Bueno et al. which utilizes heart weight-to-tibia length ratio. Abbreviations: TG, transgenic; AngII, angiotensin II; Iso, isoproterenol; AAC, abdominal aortic constriction; CnA, calcineurin transgenic; TAC, transverse aortic constriction.
A second approach used to genetically inhibit calcineurin involved transgenic overexpression of the calcineurin inhibitory protein MCIP1 (myocyte-enriched calcineurin-interacting protein-1) in the heart. MCIP1 was first identified as DSCR1 (Down syndrome critical region-1), a member of a family of calcineurin inhibitors conserved from yeast to humans (Görlach et al. 2000). MCIP1 is highly enriched in cardiac and slow skeletal muscle and is overexpressed in Down syndrome (potentially lending to the cognitive and cardiac defects observed in the syndrome) (Fuentes et al. 2000; Rothermel et al. 2000; Casas et al. 2001). Interestingly, MCIP1 expression is transcriptionally regulated by calcineurin and NFAT, such that the third intron of the human MCIP1 gene contains a cluster of 15 consensus NFAT binding sites, making it the first known feedback regulator of calcineurin activity (Yang et al. 2000). In accordance with these functions, cardiac-specific MCIP1 overexpressing transgenic mice demonstrated impaired hypertrophy to isoproterenol infusion, treadmill running, pressure overload, and the activated calcineurin catalytic transgene (Rothermel et al. 2001; Hill et al. 2002) (Table 1).
The third transgenic approach used to inhibit cardiac calcineurin activity employed a dominant-negative mutant of calcineurin (Zou et al. 2001a). Dominant negative calcineurin expressing transgenic mice had normal hearts at baseline, but demonstrated an attenuation of cardiac hypertrophy induced by abdominal aortic banding (Table 1). Finally, calcineurin Aβ gene targeted mice were also recently generated and shown to have reduced cardiac hypertrophy in response to aortic banding, isoproterenol infusion, and angiotensin II infusion (Bueno et al. 2002) (Table 1) (see paragraph below). Taken together, four separate transgenic approaches (Cain, AKAP79, MCIP1 and dnCnA) and one gene targeted mouse model (CnAβ nulls) have demonstrated reduced cardiac hypertrophy to a wide variety of acute stimuli in vivo, unequivocally implicating calcineurin as an important signalling constituent in the heart.
Mediators of calcineurin hypertrophic signalling
While the transgenic approaches discussed above have provided a convincing data set, a number of critical questions remain unanswered. For example, the identity of the downstream transcriptional effectors that mediate calcineurin-induced hypertrophy in vivo remains uncharacterised. In addition, the molecular identity of the calcineurin isoform that regulates the hypertrophic response is uncertain (three calcineurin catalytic genes have been identified: CnAα, CnAβ, and CnAγ). Our laboratory has attempted to address each of these issues using gene-targeted mice. Previous in vitro studies indicated that of the three CnA isoforms, only CnAα and CnAβ are present in cardiomyocytes, and interestingly, CnAβ mRNA and protein levels are upregulated in cardiac myocytes undergoing hypertrophy (Taigen et al. 2000). Accordingly, the CnAβ gene was recently targeted in embryonic stem cells and used to generate homozygous null mice. CnAβ null mice are viable, fertile and overtly normal. CnAβ null mice had reduced cardiac calcineurin activity and demonstrated impaired hypertrophy in response to AngII infusion, Iso infusion, or abdominal aortic constriction (Bueno et al. 2002) (Table 1). These data not only further extend the transgenic approaches discussed above, but they more specifically implicate the CnAβ gene in regulating the hypertrophic response.
Finally, the downstream transcriptional mechanisms whereby calcineurin might function in vivo remain largely uncharacterised. However, both NFAT and MEF2 transcriptional regulators are directly regulated by calcineurin, suggesting obvious candidates for genetic analysis in the heart. The NFAT family consists of five members, four of which (NFATc1-c4) are partitioned between the cytoplasm and nucleus by calcineurin (Rao et al. 1997). Most of the isoforms are expressed in several tissues at different times in development (Crabtree, 1999). For example, NFATc1-c2 are expressed in T lymphocytes where they regulate mature immune function, while NFATc3 tends to be more highly expressed in developing immune cells (Oukka et al. 1998). While analysis of mRNA levels suggests that multiple NFAT factors are expressed in the heart (Hoey et al. 1995), the lack of good antibodies and the relatively low abundance of NFAT proteins has made it difficult to correlate mRNA expression with protein expression. Despite definitive data identifying which NFAT protein isoforms are present in the heart, gene targeted mice have been described for NFATc1-c4, which might provide additional insight as to the necessary regulators downstream of calcineurin. Indeed, recent investigation has suggested that the NFATc3 gene plays an important role in mediating the cardiac hypertrophic response downstream of calcineurin (B. J. Wilkins & J. D. Molkentin, unpublished observations). Consistent with this observation, NFATc3 protein, but not NFATc4 protein, is detected in the adult myocardium. While it is uncertain if NFATc1 and/or NFATc2 also participate in calcineurin-induced cardiac hypertrophy, a more pervasive genetic analysis employing NFATc1 null mice or simultaneous disruptions of multiple NFAT genes is not possible given embryonic lethality (de la Pompa et al. 1998; Ranger et al. 1998; Graef et al. 2001). Future work along these lines will most likely require cardiac-specific or conditional inactivation of these factors.
The MEF2 family of transcriptional regulators has also been implicated in calcineurin-mediated signalling in a variety of cell types. Specifically, a cardiac-restricted, constitutively active calcineurin transgene was shown to activate expression of a MEF2 reporter construct in vivo (Passier et al. 2000). Studies in skeletal muscle have shown that calcineurin is able to dephosphorylate and directly bind to MEF2A, to synergize with MEF2A-D-dependent transcription, and to strongly activate a MEF2 reporter transgene (Wu et al. 2000, 2001). In T lymphocytes, NFATc2 acts as a synergistic coactivator of MEF2D and facilitates recruitment of the general coactivator p300 to target promoters (Youn et al. 2000). Finally, studies in cerebellar neurons have shown that calcineurin directly dephosphorylates and enhances the DNA binding activity of MEF2A (Mao & Wiedmann, 1999). Given that MEF2C is also a regulator of embryonic and postnatal cardiac growth (Lin et al. 1997; Kolodziejczyk et al. 1999), it will be of great interest to determine the role of MEF2 factors as downstream effectors of the hypertrophic response.
While previous work has reasonably established that calcineurin activation is necessary for hypertrophy, the exact timing of activation is still a matter of debate. Four reports indicate that calcineurin activity is increased in failing human hearts as compared to normal samples, but activity is increased still further in compensated, non-failing hypertrophy (Lim & Molkentin, 1999, 2000; Tsao et al. 2000; Haq et al. 2001). NFAT and MEF2-driven reporter constructs used in vitro and in vivo should allow investigators to temporally track calcineurin activation under different stimuli that are known to activate hypertrophy through different pathways.
Calcineurin cross-talk with other hypertrophic signalling pathways
A common feature of reactive hypertrophic signalling in cardiac myocytes is that multiple pathways cross-talk with one another to orchestrate a productive response. With respect to calcineurin signalling, its activation is associated with the activation of certain PKC isoforms, c-Jun N-terminal kinase (JNK), and Akt (De Windt et al. 2000a,b). Conversely, β-adrenergic-mediated activation of extracellular signal-regulated kinase 1/2 (ERK1/2) signalling and endothelin-1 transcription is blocked by calcineurin inhibition (Morimoto et al. 2001; Zou et al. 2001b). In addition, ras signalling activates NFAT-mediated transcription (Ichida & Finkel, 2001), JNK signalling antagonizes NFAT nuclear accumulation (Chow et al. 1997), and GSK-3β directly phosphorylates NFAT factors preventing nuclear accumulation, DNA binding, and calcineurin-mediated hypertrophy (Graef et al. 1999; Haq et al. 2000; Neal & Clipstone, 2001; Antos et al. 2002). Each of these descriptions suggests cross-talk whereby calcineurin-NFAT-signalling is ‘fine-tuned’ or coordinated within a larger context of signalling during the hypertrophic response.
Role of calcineurin in physiological and developmental hypertrophy
While a convincing data set has emerged implicating calcineurin as a regulator of pathophysiological hypertrophy, less is understood of calcineurin's role in potentially regulating physiological growth of the heart. Indeed, it is often speculated that pathophysiological and physiological hypertrophy utilize similar signalling pathways, although the timing and degree of signalling probably regulate the ultimate phenotype of each response. By definition, physiological hypertrophy generally refers to the clinical phenomenon known as an ‘athletic’ heart. Physiological hypertrophy is induced by regular exercise training that promotes myocyte growth changes in the heart that are reversible and do not progress to decompensation (Oakley, 2001). For example, professional football (soccer) players were suggested to have physiological hypertrophy associated with increased myocardial insulin-like growth factor-1 (IGF-1) and myocardial sympathetic activation (Serneri et al. 2001).
Circumstantial data suggest a potential role for calcineurin signalling in regulating physiological hypertrophy. In vivo calcineurin inhibition by MCIP1 overexpression attenuates exercise-induced hypertrophy (Rothermel et al. 2001). In additon, athletic hypertrophy is associated with increased IGF-1 production, which was previously shown to cause skeletal muscle hypertrophy through a calcineurin-NFAT-GATA pathway in cultured myoblasts (Musarò et al. 1999; Semsarian et al. 1999). Future studies will need to address how hypertrophic signalling pathways are differentially modulated in these two general forms of hypertrophy, and in particular, what role calcineurin activation plays in each.
In the rodent heart, myocytes exit the cell cycle within the first week of postnatal development, yet the heart enlarges substantially afterwards through a process referred to as developmental hypertrophy (Claycomb, 1977). As with physiological hypertrophy induced by exercise training, developmental hypertrophy is also probably regulated by adaptive haemodynamic load, implicating a similar array of signal transduction factors. That calcineurin might regulate developmental hypertrophy, in part, is suggested by a number of observations. First, MEF2C gene targeted mice die during embryonic development with a hypoplastic right ventricle (Lin et al. 1997). Second, targeted disruption of the calcium regulatory genes calreticulin and connexin45 each resulted in embryonic lethality that was associated with defective NFAT-mediated transcription in the heart (Mesaeli et al. 1999; Kumai et al. 2000). Third, deletion of NFATc1 in the mouse resulted in defective cardiac valve formation and embryonic lethality (de la Pompa et al. 1998; Ranger et al. 1998). In the postnatal heart, two studies have suggested a role for calcineurin in regulating hypertrophic maturation. First, high copy number Cain and AKAP79 transgenic mice each perished during with the first 2 weeks of neonatal development with severely atrophic hearts, although low copy number lines were viable (De Windt et al. 2001). Second, while MCIP-1 transgenic mice survive successfully through neonatal development, adult heart size was 5-10 % smaller than wild-types, suggesting an attenuation in developmental hypertrophy (Rothermel et al. 2001). Collectively, these observations suggest that calcineurin transduces, in part, the signals that control physiological and developmental growth of the myocardium.
Is hypertrophy a beneficial response?
Cardiac hypertrophy is generally assumed to be a necessary compensation to injury or increased workload that benefits the heart in the short term. However, a few recent studies have questioned this assumption and demonstrated that acute injury or pressure overload can be dissociated from the hypertrophic response without leading to decompensation. Specifically, analysis of mice with inhibited Gαq, adrenergic, or calcineurin signalling during acute pressure overload stimulation revealed normal cardiac function in conditions of increased wall stress without decompensation (Hill et al. 2000; Esposito et al. 2002; Hill et al. 2002). However, an early study showed that while calcineurin inhibition reduced pressure overload-induced hypertrophy in mice subjected to TAC, it also resulted in increased decompensation and mortality (Meguro et al. 1999). Given these disparate results, more studies are necessary to resolve the question of how to modulate the hypertrophic response for the greatest clinical benefit (Force et al. 1999).
Calcineurin activation and function in an unstimulated heart
While calcineurin probably transduces physiological and pathophysiological growth responses in the heart, its functional role at baseline in an unstimulated heart is unknown. It has been suggested that calcineurin is inactive in an unstimulated heart given the observation that dominant negative calcineurin expressing transgenic mice have similar calcineurin phosphatase activity compared to wild-type controls (Zou et al. 2001a). However, calcineurin is also known to directly sense intracellular calcium levels, which constantly cycle in contracting myocytes. The calcium transient itself may regulate, in part, basal calcineurin activity as a mechanism for adapting cardiac load or inotropy with hypertrophic signalling. Indeed, the steady-state size of the heart directly responds to haemodynamic load in a continuous fashion. For example, it is well established that heart size closely parallels body size (haemodynamic load) in rats and mice and probably all mammals (Goodman et al. 1984; Ernsberger et al. 1996). By a similar mechanism, unloading of the heart results in severe atrophy such that 50 % of cardiac mass is lost after only 14 days of unloading in the rat (Klein et al. 1990). In other cell types, the nature and duration of calcium increase directly modify calcineurin activity in a graded fashion. For example, calcineurin/GSK-3β signalling in T lymphocytes responds to intracellular Ca2+ levels to monitor the duration of antigen receptor occupancy, which activates immune cells in response to antigen ‘load’ (Neilson et al. 2001). Finally, cardiac myocytes isolated from adult mice treated with CsA demonstrated a 10 % reduction in size compared with wild-type controls (Wang et al. 2001b). Collectively, these observations suggest that calcineurin participates in regulating the homeostatic size of the heart in response to load-associated alterations in calcium handling. To definitively address this hypothesis, complete inactivation of calcineurin activity in the heart is required, especially since CsA and FK506 only partially block calcineurin activity. While the complete disruption of all calcineurin activity results in early embryonic lethality (Graef et al. 2001), tools now exist to conditionally inactivate calcineurin specifically within the heart to directly address this intriguing hypothesis (Sohal et al. 2001; Zeng et al. 2001).
Acknowledgments
The authors would like to thank the members of the Molkentin lab for helpful comments on this manuscript. B.J.W. is supported by a MD/PhD scholar award from the University of Cincinnati Physician Scientist Training Program, and J.D.M. is supported by National Institutes of Health (NIH) and a Scholar award from the Pew Foundation.
REFERENCES
- Antos CL, McKinsey TA, Frey N, Kutschke W, McAnally J, Shelton JM, Richardson JA, Hill JA, Olson EN. Activated glycogen synthase-3β suppresses cardiac hypertrophy in vivo. Proceedings of the National Academy of Sciences of the USA. 2002;99:907–912. doi: 10.1073/pnas.231619298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bueno OF, Wilkins BJ, Tymitz KM, Glascock BJ, Kimball TF, Lorenz JN, Molkentin JD. Impaired cardiac hypertrophic response in calcineurin Aβ-deficient mice. Proceedings of the National Academy of Sciences of the USA. 2002;99:4586–4591. doi: 10.1073/pnas.072647999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casas C, Martínez S, Prichard MA, Fuentes JJ, Nadal M, Guimerà J, Arbonés M, Flórez J, Soriano E, Estivill X, Alcántara S. Dscr1, a novel endogenous inhibitor of calcineurin signaling, is expressed in the primitive ventricle of the heart and during neurogenesis. Mechanisms of Development. 2001;101:289–292. doi: 10.1016/s0925-4773(00)00583-9. [DOI] [PubMed] [Google Scholar]
- Chow C-W, Rincón M, Cavanagh J, Dickens M, Davis RJ. Nuclear accumulation of NFAT4 opposed by the JNK signal transduction pathway. Science. 1997;278:1638–1641. doi: 10.1126/science.278.5343.1638. [DOI] [PubMed] [Google Scholar]
- Claycomb WC. Cardiac-muscle hypertrophy. Differentiation and growth of the heart cell during development. Biochemical Journal. 1977;168:599–601. doi: 10.1042/bj1680599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clipstone NA, Crabtree GR. Identification of calcineurin as a key signalling enzyme in T-lymphocyte activation. Nature. 1992;357:695–697. doi: 10.1038/357695a0. [DOI] [PubMed] [Google Scholar]
- Crabtree GR. Generic signals and specific outcomes: signaling through Ca2+, calcineurin, and NF-AT. Cell. 1999;96:611–614. doi: 10.1016/s0092-8674(00)80571-1. [DOI] [PubMed] [Google Scholar]
- De La Pompa JL, Timmerman LA, Takimoto H, Yoshida H, Elia AJ, Samper E, Potter J, Wakeham A, Marengere L, Langille BL, Crabtree GR, Mak TW. Role of the NF-ATc transcription factor in morphogenesis of cardiac valves and septum. Nature. 1998;392:182–186. doi: 10.1038/32419. [DOI] [PubMed] [Google Scholar]
- De Windt LJ, Lim HW, Bueno OF, Liang Q, Delling U, Braz JC, Glascock BJ, Kimball TF, Del Monte F, Hajjar RJ, Molkentin JD. Targeted inhibition of calcineurin attenuates cardiac hypertrophy in vivo. Proceedings of the National Academy of Sciences of the USA. 2001;98:3322–3327. doi: 10.1073/pnas.031371998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Windt LJ, Lim HW, Haq S, Force T, Molkentin JD. Calcineurin promotes protein kinase C and c-jun NH2-terminal kinase activation in the heart. Journal of Biological Chemistry. 2000b;275:13571–13579. doi: 10.1074/jbc.275.18.13571. [DOI] [PubMed] [Google Scholar]
- De Windt LJ, Lim HW, Taigen T, Wencker D, Condorelli G, Dorn GW, II, Kitsis RN, Molkentin JD. Calcineurin-mediated hypertrophy protects cardiomyocytes from apoptosis in vitro and in vivo. Circulation Research. 2000a;86:255–263. doi: 10.1161/01.res.86.3.255. [DOI] [PubMed] [Google Scholar]
- Ding B, Price RL, Borg TK, Weinberg EO, Halloran PF, Lorell BH. Pressure overload induces severe hypertrophy in mice treated with cyclosporine, an inhibitor of calcineurin. Circulation Research. 1999;84:729–734. doi: 10.1161/01.res.84.6.729. [DOI] [PubMed] [Google Scholar]
- Ernsberger P, Koletsky RJ, Baskin JS, Collins LA. Consequences of weight cycling in obese spontaneously hypertensive rats. American Journal of Physiology. 1996;270:R864–872. doi: 10.1152/ajpregu.1996.270.4.R864. [DOI] [PubMed] [Google Scholar]
- Esposito G, Rapacciuolo A, Naga Prasad SV, Takaoka H, Thomas SA, Koch WJ, Rockman HA. Genetic alterations that inhibit in vivo pressure-overload hypertrophy prevent cardiac dysfunction despite increased wall stress. Circulation. 2002;105:85–92. doi: 10.1161/hc0102.101365. [DOI] [PubMed] [Google Scholar]
- Fatkin D, McConnell BK, Mudd JO, Semsarian C, Moskowitz IGP, Schoen FJ, Giewat M, Seidman CE, Seidman JG. An abnormal Ca2+ response in mutant sarcomere protein-mediated familial hypertrophic cardiomyopathy. Journal of Clinical Investigation. 2000;106:1351–1359. doi: 10.1172/JCI11093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flanagan WM, Corthésy B, Bram RJ, Crabtree GR. Nuclear association of a T-cell transcription factor blocked by FK-506 and cyclosporin A. Nature. 1991;352:803–807. doi: 10.1038/352803a0. [DOI] [PubMed] [Google Scholar]
- Force T, Rosenzweig A, Choukroun G, Hajjar R. Calcineurin inhibitors and cardiac hypertrophy. Lancet. 1999;353:1290–1292. doi: 10.1016/S0140-6736(99)90016-8. [DOI] [PubMed] [Google Scholar]
- Fuentes JJ, Genescà L, Kingsbury TJ, Cunningham KW, Pérez-Riba M, Estivill X, De La Luna S. DSCR1, overexpressed in Down syndrome, is an inhibitor of calcineurin-mediated signaling pathways. Human Molecular Genetics. 2000;9:1681–1690. doi: 10.1093/hmg/9.11.1681. [DOI] [PubMed] [Google Scholar]
- Goldspink PH, McKinney RD, Kimball VA, Geenen DL, Buttrick PM. Angiotensin II induced cardiac hypertrophy in vivo is inhibited by cyclosporin A in adult rats. Molecular and Cellular Biochemistry. 2001;226:83–88. doi: 10.1023/a:1012789819926. [DOI] [PubMed] [Google Scholar]
- Goodman MN, Lowell B, Belur E, Ruderman NB. Sites of protein conservation and loss during starvation: influence of adiposity. American Journal of Physiology. 1984;246:E383–390. doi: 10.1152/ajpendo.1984.246.5.E383. [DOI] [PubMed] [Google Scholar]
- Görlach J, Fox DS, Cutler NS, Cox GM, Perfect JR, Heitman J. Identification and characterization of a highly conserved calcineurin binding protein, CBP1/calcipressin. Cryptococcus neoformans. EMBO Journal. 2000;19:3618–3629. doi: 10.1093/emboj/19.14.3618. in. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graef IA, Chen F, Chen L, Kuo A, Crabtree GR. Signals transduced by Ca2+/calcineurin and NFATc3/c4 pattern the developing vasculature. Cell. 2001;105:863–875. doi: 10.1016/s0092-8674(01)00396-8. [DOI] [PubMed] [Google Scholar]
- Graef IA, Mermelstein PG, Stankunas K, Neilson JR, Deisseroth K, Tsien RW, Crabtree GR. L-type calcium channels and GSK-3 regulate the activity of NF-ATc4 in hippocampal neurons. Nature. 1999;401:703–708. doi: 10.1038/44378. [DOI] [PubMed] [Google Scholar]
- Griffiths EJ, Halestrap AP. Protection by cyclosporin A of ischemia/reperfusion-induced damage in isolated rat hearts. Journal of Molecular and Cellular Cardiology. 1993;25:1461–1469. doi: 10.1006/jmcc.1993.1162. [DOI] [PubMed] [Google Scholar]
- Griffiths EJ, Halestrap AP. Mitochondrial non-specific pores remain closed during cardiac ischaemia, but open upon reperfusion. Biochemical Journal. 1995;307:93–98. doi: 10.1042/bj3070093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halestrap AP, Connern CP, Griffiths EJ, Kerr PM. Cyclosporin A binding to mitochondrial cyclophilin inhibits the permeability transition pore and protects hearts from ischaemia/reperfusion injury. Molecular and Cellular Biochemistry. 1997;174:167–172. [PubMed] [Google Scholar]
- Haq S, Choukroun G, Kang ZB, Ranu H, Matsui T, Rosenzweig A, Molkentin JD, Alessandrini A, Woodgett J, Hajjar R, Michael A, Force T. Glycogen synthase kinase-3β is a negative regulator of cardiomyocyte hypertrophy. Journal of Cell Biology. 2000;151:117–129. doi: 10.1083/jcb.151.1.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haq S, Choukroun G, Lim H, Tymitz KM, Del Monte F, Gwathmey J, Grazette L, Michael A, Hajjar R, Force T, Molkentin JD. Differential activation of signal transduction pathways in human hearts with hypertrophy versus advanced heart failure. Circulation. 2001;103:670–677. doi: 10.1161/01.cir.103.5.670. [DOI] [PubMed] [Google Scholar]
- Hill JA, Karimi M, Kutschke W, Davisson RL, Zimmerman K, Wang Z, Kerber RE, Weiss RM. Cardiac hypertrophy is not a required compensatory response to short-term pressure overload. Circulation. 2000;101:2863–2869. doi: 10.1161/01.cir.101.24.2863. [DOI] [PubMed] [Google Scholar]
- Hill JA, Rothermel B, Yoo KD, Cabuay B, Demetroulis E, Weiss RM, Kutschke W, Bassel-Duby R, Williams RS. Targeted inhibition of calcineurin in pressure-overload cardiac hypertrophy: Preservation of systolic function. Journal of Biological Chemistry. 2002;277:10251–10255. doi: 10.1074/jbc.M110722200. [DOI] [PubMed] [Google Scholar]
- Hoey T, Sun Y-L, Williamson K, Xu X. Isolation of two new members of the NF-AT gene family and functional characterization of the NF-AT proteins. Immunity. 1995;2:461–472. doi: 10.1016/1074-7613(95)90027-6. [DOI] [PubMed] [Google Scholar]
- Ichida M, Finkel T. Ras regulates NFAT3 activity in cardiac myocytes. Journal of Biological Chemistry. 2001;276:3524–3530. doi: 10.1074/jbc.M004275200. [DOI] [PubMed] [Google Scholar]
- Janssen PM, Zeitz O, Keweloh B, Siegel U, Maier LS, Barckhausen P, Pieske B, Prestle J, Lehnart SE, Hasenfuss G. Influence of cyclosporine A on contractile function, calcium handling, and energetics in isolated human and rabbit myocardium. Cardiovascular Research. 2000;47:99–107. doi: 10.1016/s0008-6363(00)00052-3. [DOI] [PubMed] [Google Scholar]
- Kääb S, Dixon J, Duc J, Ashen D, Näbauer M, Beuckelmann DJ, Steinbeck G, McKinnon D, Tomaselli GF. Molecular basis of transient outward potassium current downregulation in human heart failure: a decrease in Kv4. 3 mRNA correlates with a reduction in current density. Circulation. 1998;98:1383–1393. doi: 10.1161/01.cir.98.14.1383. [DOI] [PubMed] [Google Scholar]
- Kato T, Sano M, Miyoshi S, Sato T, Hakuno D, Ishida H, Kinoshita-Nakazawa H, Fukuda K, Ogawa S. Calmodulin kinases II and IV and calcineurin are involved in leukemia inhibitory factor-induced cardiac hypertrophy in rats. Circulation Research. 2000;87:937–945. doi: 10.1161/01.res.87.10.937. [DOI] [PubMed] [Google Scholar]
- Klee CB, Ren H, Wang X. Regulation of the calmodulin-stimulated protein phosphatase, calcineurin. Journal of Biological Chemistry. 1998;273:13367–13370. doi: 10.1074/jbc.273.22.13367. [DOI] [PubMed] [Google Scholar]
- Klein I, Hong C, Schreiber SS. Cardiac atrophy in the heterotopically transplanted rat heart: in vitro protein synthesis. Journal of Molecular and Cellular Cardiology. 1990;22:461–468. doi: 10.1016/0022-2828(90)91481-l. [DOI] [PubMed] [Google Scholar]
- Kolodziejczyk SM, Wang L, Balazsi K, Derepentigny Y, Kothary R, Megeney LA. MEF2 is upregulated during cardiac hypertrophy and is required for normal post-natal growth of the myocardium. Current Biology. 1999;9:1203–1206. doi: 10.1016/S0960-9822(00)80027-5. [DOI] [PubMed] [Google Scholar]
- Kumai M, Nishii K, Nakamura K, Takeda N, Suzuki M, Shibata Y. Loss of connexin45 causes a cushion defect in early cardiogenesis. Development. 2000;127:3501–3512. doi: 10.1242/dev.127.16.3501. [DOI] [PubMed] [Google Scholar]
- Lemasters JJ, Nieminen A-L, Qian T, Trost LC, Herman B. The mitochondrial permeability transition in toxic, hypoxic and reperfusion injury. Molecular and Cellular Biochemistry. 1997;174:159–165. [PubMed] [Google Scholar]
- Levy D, Garrison RJ, Savage DD, Kannel WB, Castelli WP. Prognostic implications of echocardiographically determined left ventricular mass in the Framingham Heart Study. New England Journal of Medicine. 1990;322:1561–1566. doi: 10.1056/NEJM199005313222203. [DOI] [PubMed] [Google Scholar]
- Lim HW, De Windt LJ, Mante J, Kimball TR, Witt SA, Sussman MA, Molkentin JD. Reversal of cardiac hypertrophy in transgenic disease models by calcineurin inhibition. Journal of Molecular and Cellular Cardiology. 2000b;32:697–709. doi: 10.1006/jmcc.2000.1113. [DOI] [PubMed] [Google Scholar]
- Lim HW, De Windt LJ, Steinberg L, Taigen T, Witt SA, Kimball TR, Molkentin JD. Calcineurin expression, activation, and function in cardiac pressure-overload hypertrophy. Circulation. 2000a;101:2431–2437. doi: 10.1161/01.cir.101.20.2431. [DOI] [PubMed] [Google Scholar]
- Lim HW, Molkentin JD. Calcineurin and human heart failure. Nature Medicine. 1999;5:246–247. doi: 10.1038/6430. [DOI] [PubMed] [Google Scholar]
- Lim HW, Molkentin JD. Reply to “Revisiting calcineurin and human heart failure”. Nature Medicine. 2000;6:3. doi: 10.1038/71511. [DOI] [PubMed] [Google Scholar]
- Lin Q, Schwartz J, Bucana C, Olson EN. Control of mouse cardiac morphogenesis and myogenesis by transcription factor MEF2C. Science. 1997;276:1404–1407. doi: 10.1126/science.276.5317.1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Z, Shyu K-G, Gualberto A, Walsh K. Calcineurin inhibitors and cardiac hypertrophy. Nature Medicine. 1998;4:1092–1093. doi: 10.1038/2578. [DOI] [PubMed] [Google Scholar]
- McKinsey TA, Olson EN. Cardiac hypertrophy: sorting out the circuitry. Current Opinion in Genetics and Development. 1999;9:267–274. doi: 10.1016/s0959-437x(99)80040-9. [DOI] [PubMed] [Google Scholar]
- Mao Z, Wiedmann M. Calcineurin enhances MEF2 DNA binding activity in calcium-dependent survival of cerebellar granule neurons. Journal of Biological Chemistry. 1999;274:31102–31107. doi: 10.1074/jbc.274.43.31102. [DOI] [PubMed] [Google Scholar]
- Meguro T, Hong C, Asai K, Takagi G, McKinsey TA, Olson EN, Vatner SF. Cyclosporine attenuates pressure-overload hypertrophy in mice while enhancing susceptibility to decompensation and heart failure. Circulation Research. 1999;84:735–740. doi: 10.1161/01.res.84.6.735. [DOI] [PubMed] [Google Scholar]
- Mende U, Kagen A, Cohen A, Aramburu J, Schoen FJ, Neer EJ. Transient cardiac expression of constitutively active Gαq leads to hypertrophy and dilated cardiomyopathy by calcineurin-dependent and independent pathways. Proceedings of the National Academy of Sciences of the USA. 1998;95:13893–13898. doi: 10.1073/pnas.95.23.13893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesaeli N, Nakamura K, Zvaritch E, Dickie P, Dziak E, Krause K-H, Opas M, MacLennan DH, Michalak M. Calreticulin is essential for cardiac development. Journal of Cell Biology. 1999;144:857–868. doi: 10.1083/jcb.144.5.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molkentin JD. Calcineurin and beyond: cardiac hypertrophic signaling. Circulation Research. 2000;87:731–738. doi: 10.1161/01.res.87.9.731. [DOI] [PubMed] [Google Scholar]
- Molkentin JD. Calcineurin, mitochondrial membrane potential, and cardiomyocyte apoptosis. Circulation Research. 2001;88:1220–1222. doi: 10.1161/hh1201.093159. [DOI] [PubMed] [Google Scholar]
- Molkentin JD, Dorn GW., II Cytoplasmic signaling pathways that regulate cardiac hypertrophy. Annual Review of Physiology. 2001;63:391–426. doi: 10.1146/annurev.physiol.63.1.391. [DOI] [PubMed] [Google Scholar]
- Molkentin JD, Lu J-R, Antos CL, Markham B, Richardson J, Robbins J, Grant SR, Olson EN. A calcineurin-dependent transcription pathway for cardiac hypertrophy. Cell. 1998;93:215–228. doi: 10.1016/s0092-8674(00)81573-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morimoto T, Hasegawa K, Wada H, Kakita T, Kaburagi S, Yanazume T, Sasayama S. Calcineurin-GATA4 pathway is involved in β-adrenergic agonist-responsive endothelin-1 transcription in cardiac myocytes. Journal of Biological Chemistry. 2001;276:34983–34989. doi: 10.1074/jbc.M005498200. [DOI] [PubMed] [Google Scholar]
- Muller JG, Nemoto S, Laser M, Carabello BA, Menick DR. Calcineurin inhibition and cardiac hypertrophy. Science. 1998;282:1007. letter. [Google Scholar]
- Murat A, Pellieux C, Brunner H-R, Pedrazzini T. Calcineurin blockade prevents cardiac mitogen-activated protein kinase activation and hypertrophy in renovascular hypertension. Journal of Biological Chemistry. 2000;275:40867–40873. doi: 10.1074/jbc.M008071200. [DOI] [PubMed] [Google Scholar]
- Musarò A, McCullagh KJA, Naya FJ, Olson EN, Rosenthal N. IGF-1 induces skeletal myocyte hypertrophy through calcineurin in association with GATA-2 and NF-ATc1. Nature. 1999;400:581–585. doi: 10.1038/23060. [DOI] [PubMed] [Google Scholar]
- Nazareth W, Yafei N, Crompton M. Inhibition of anoxia-induced injury in heart myocytes by cyclosporin A. Journal of Molecular and Cellular Cardiology. 1991;23:1351–1354. doi: 10.1016/0022-2828(91)90181-k. [DOI] [PubMed] [Google Scholar]
- Neal JW, Clipstone NA. Glycogen synthase kinase-3 inhibits the DNA binding activity of NFATc. Journal of Biological Chemistry. 2001;276:3666–3673. doi: 10.1074/jbc.M004888200. [DOI] [PubMed] [Google Scholar]
- Neilson J, Stankunas K, Crabtree GR. Monitoring the duration of antigen-receptor occupancy by calcineurin/glycogen-synthase-kinase-3 control of NF-AT nuclear shuttling. Current Opinion in Immunology. 2001;13:346–350. doi: 10.1016/s0952-7915(00)00225-9. [DOI] [PubMed] [Google Scholar]
- Oakley D. The athlete's heart. Heart. 2001;86:722–726. doi: 10.1136/heart.86.6.722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson EN, Molkentin JD. Prevention of cardiac hypertrophy by calcineurin inhibition: hope or hype? Circulation Research. 1999;84:623–632. doi: 10.1161/01.res.84.6.623. [DOI] [PubMed] [Google Scholar]
- Oukka M, Ho IC, De La Brousse FC, Hoey T, Grusby MJ, Glimcher GR. The transcription factor NFAT4 is involved in the generation and survival of T cells. Immunity. 1998;9:295–304. doi: 10.1016/s1074-7613(00)80612-3. [DOI] [PubMed] [Google Scholar]
- Park KS, Kim TK, Kim DH. Cyclosporin A treatment alters characteristics of Ca2+-release channel in cardiac sarcoplasmic reticulum. American Journal of Physiology. 1999;45:H865–872. doi: 10.1152/ajpheart.1999.276.3.H865. [DOI] [PubMed] [Google Scholar]
- Passier R, Zeng H, Frey N, Naya FJ, Nicol RL, McKinsey TA, Overbeek P, Richardson JA, Grant SR, Olson EN. CaM kinase signaling induces cardiac hypertrophy and activates the MEF2 transcription factor in vivo. Journal of Clinical Investigation. 2000;105:1395–1406. doi: 10.1172/JCI8551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranger AM, Grusby MJ, Hodge MR, Gravallese EM, De La Brousse FC, Hoey T, Mickanin C, Baldwin HS, Glimcher LH. The transcription factor NF-ATc is essential for cardiac valve formation. Nature. 1998;392:186–190. doi: 10.1038/32426. [DOI] [PubMed] [Google Scholar]
- Rao A, Luo C, Hogan PG. Transcription factors of the NFAT family: regulation and function. Annual Review of Immunology. 1997;15:707–747. doi: 10.1146/annurev.immunol.15.1.707. [DOI] [PubMed] [Google Scholar]
- Rothermel BA, McKinsey TA, Vega RB, Nicol RL, Mammen P, Yang J, Antos CL, Shelton JM, Bassel-Duby R, Olson EN, Williams RS. Myocyte-enriched calcineurin-interacting protein, MCIP1, inhibits cardiac hypertrophy in vivo. Proceedings of the National Academy of Sciences of the USA. 2001;98:3328–3333. doi: 10.1073/pnas.041614798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothermel BA, Vega RB, Yang J, Wu H, Bassel-Duby R, Williams RS. A protein encoded within the Down syndrome critical region is enriched in striated muscles and inhibits calcineurin signaling. Journal of Biological Chemistry. 2000;275:8719–8725. doi: 10.1074/jbc.275.12.8719. [DOI] [PubMed] [Google Scholar]
- Sakata Y, Masuyama T, Yamamoto K, Nishikawa N, Yamamoto H, Kondo H, Ono K, Otsu K, Kuzuya T, Miwa T, Takeda H, Miyamoto E, Hori M. Calcineurin inhibitor attenuates left ventricular hypertrophy, leading to prevention of heart failure in hypertensive rats. Circulation. 2000;102:2269–2275. doi: 10.1161/01.cir.102.18.2269. [DOI] [PubMed] [Google Scholar]
- Semsarian C, Wu M-J, Ju Y-K, Marciniec T, Yeoh T, Allen DG, Harvey RP, Graham RM. Skeletal muscle hypertrophy is mediated by a Ca2+-dependent calcineurin signalling pathway. Nature. 1999;400:576–581. doi: 10.1038/23054. [DOI] [PubMed] [Google Scholar]
- Serneri GGN, Boddi M, Modesti PA, Cecioni I, Coppo M, Padeletti L, Michelucci A, Colella A, Galanti G. Increased cardiac sympathetic activity and insulin-like growth factor-I formation are associated with physiological hypertrophy in athletes. Circulation Research. 2001;89:977–982. doi: 10.1161/hh2301.100982. [DOI] [PubMed] [Google Scholar]
- Shibasaki F, Price ER, Milan D, Mckeon F. Role of kinases and the phosphatase calcineurin in the nuclear shuttling of transcription factor NF-AT4. Nature. 1996;382:370–373. doi: 10.1038/382370a0. [DOI] [PubMed] [Google Scholar]
- Shimoyama M, Hayashi D, Takimoto E, Zou Y, Oka T, Uozumi H, Kudoh S, Shibasaki F, Yazaki Y, Nagai R, Komuro I. Calcineurin plays a critical role in pressure overload-induced cardiac hypertrophy. Circulation. 1999;100:2449–2454. doi: 10.1161/01.cir.100.24.2449. [DOI] [PubMed] [Google Scholar]
- Shimoyama M, Hayashi D, Zou Y, Takimoto E, Mizukami M, Monzen K, Kudoh S, Hiroi Y, Yazaki Y, Nagai R, Komuro I. Calcineurin inhibitor attenuates the development and induces the regression of cardiac hypertrophy in rats with salt-sensitive hypertension. Circulation. 2000;102:1996–2004. doi: 10.1161/01.cir.102.16.1996. [DOI] [PubMed] [Google Scholar]
- Sohal DS, Nghiem M, Crackower MA, Witt SA, Kimball TR, Tymitz KM, Penninger JM, Molkentin JD. Temporally regulated and tissue-specific gene manipulations in the adult and embryonic heart using a tamoxifen-inducible cre protein. Circulation Research. 2001;89:20–25. doi: 10.1161/hh1301.092687. [DOI] [PubMed] [Google Scholar]
- Steinberg SF. Many pathways to cardiac hypertrophy. Journal of Molecular and Cellular Cardiology. 2000;32:1381–1384. doi: 10.1006/jmcc.2000.1202. [DOI] [PubMed] [Google Scholar]
- Sussman MA, Lim HW, Gude N, Taigen T, Olson EN, Robbins J, Colbert MC, Gualberto A, Wieczorek DF, Molkentin JD. Prevention of cardiac hypertrophy in mice by calcineurin inhibition. Science. 1998;281:1690–1693. doi: 10.1126/science.281.5383.1690. [DOI] [PubMed] [Google Scholar]
- Taigen T, De Windt LJ, Lim HW, Molkentin JD. Targeted inhibition of calcineurin prevents agonist-induced cardiomyocyte hypertrophy. Proceedings of the National Academy of Sciences of the USA. 2000;97:1196–1201. doi: 10.1073/pnas.97.3.1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsao L, Neville C, Musaro A, McCullagh KJA, Rosenthal N. Revisiting calcineurin and human heart failure. Nature Medicine. 2000;6:2–3. doi: 10.1038/71478. [DOI] [PubMed] [Google Scholar]
- Wang Z, Kutschke W, Richardson KE, Karimi M, Hill JA. Electrical remodeling in pressure-overload cardiac hypertrophy: role of calcineurin. Circulation. 2001a;104:1657–1663. doi: 10.1161/hc3901.095766. [DOI] [PubMed] [Google Scholar]
- Wang Z, Nolan B, Kutschke W, Hill JA. Na+-Ca2+ exchanger remodeling in pressure overload cardiac hypertrophy. Journal of Biological Chemistry. 2001b;276:17706–17711. doi: 10.1074/jbc.M100544200. [DOI] [PubMed] [Google Scholar]
- Wu H, Naya FJ, McKinsey TA, Mercer B, Shelton JM, Chin ER, Simard AR, Michel RN, Bassel-Duby R, Olson EN, Williams RS. MEF2 responds to multiple calcium-regulated signals in the control of skeletal muscle fiber type. EMBO Journal. 2000;19:1963–1973. doi: 10.1093/emboj/19.9.1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Rothermel B, Kanatous S, Rosenberg P, Naya FJ, Shelton JM, Hutcheson KA, Dimaio JM, Olson EN, Bassel-Duby R, Williams RS. Activation of MEF2 by muscle activity is mediated through a calcineurin-dependent pathway. EMBO Journal. 2001;20:6414–6423. doi: 10.1093/emboj/20.22.6414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Y, McMillin JB, Lewis A, Moore M, Zhu WG, Williams RS, Kellems RE. Electrical stimulation of neonatal cardiac myocytes activates the NFAT3 and GATA4 pathways and up-regulates the adenylosuccinate synthetase 1 gene. Journal of Biological Chemistry. 2000;275:1855–1863. doi: 10.1074/jbc.275.3.1855. [DOI] [PubMed] [Google Scholar]
- Yang J, Rothermel B, Vega RB, Frey N, McKinsey TA, Olson EN, Bassel-Duby R, Williams RS. Independent signals control expression of the calcineurin inhibitory proteins MCIP1 and MCIP2 in striated muscles. Circulation Research. 2000;87:e61–e68. doi: 10.1161/01.res.87.12.e61. [DOI] [PubMed] [Google Scholar]
- Yatani A, Honda R, Tymitz KM, Lalli MJ, Molkentin JD. Enhanced Ca2+ channel currents in cardiac hypertrophy induced by activation of calcineurin-dependent pathway. Journal of Molecular and Cellular Cardiology. 2001;33:249–259. doi: 10.1006/jmcc.2000.1296. [DOI] [PubMed] [Google Scholar]
- Youn H-D, Chatila TA, Liu JO. Integration of calcineurin and MEF2 signals by the coactivator p300 during T-cell apoptosis. EMBO Journal. 2000;19:4323–4331. doi: 10.1093/emboj/19.16.4323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng H, Chattarji S, Barbarosie M, Rondi-Reig L, Philpot BD, Miyakawa T, Bear MF, Tonegawa S. Forebrain-specific calcineurin knockout selectively impairs bidirectinal synaptic plasticity and working/episodic-like memory. Cell. 2001;107:617–629. doi: 10.1016/s0092-8674(01)00585-2. [DOI] [PubMed] [Google Scholar]
- Zhang W, Kowal RC, Rusnak F, Sikkink RA, Olson EN, Victor RG. Failure of calcineurin inhibitors to prevent pressure-overload left ventricular hypertrophy in rats. Circulation Research. 1999;84:722–728. doi: 10.1161/01.res.84.6.722. [DOI] [PubMed] [Google Scholar]
- Zhu J, McKeon F. NF-AT activation requires suppression of Crm1-dependent export by calcineurin. Nature. 1999;398:256–260. doi: 10.1038/18473. [DOI] [PubMed] [Google Scholar]
- Zou Y, Hiroi Y, Uozumi H, Takimoto E, Toko H, Zhu W, Kudoh S, Mizukami M, Shimoyama M, Shibasaki F, Nagai R, Yazaki Y, Komuro I. Calcineurin plays a critical role in the development of pressure overload-induced cardiac hypertrophy. Circulation. 2001a;104:97–101. doi: 10.1161/01.cir.104.1.97. [DOI] [PubMed] [Google Scholar]
- Zou Y, Yao A, Zhu W, Kudoh S, Hiroi Y, Shimoyama M, Uozumi H, Kohmoto O, Takahashi T, Shibasaki F, Nagai R, Yazaki Y, Komuro I. Isoproterenol activates extracellular signal-regulated protein kinases in cardiomyocytes through calcineurin. Circulation. 2001b;104:102–108. doi: 10.1161/hc2601.090987. [DOI] [PubMed] [Google Scholar]