

Abstract
The durations of transmembrane action potentials recorded from single myocytes isolated from the endocardial surface of hypertrophied left ventricles of rats were increased, compared to the durations recorded from normal left ventricular cells at 36-37 °C. Exposure to phalloidin (1-20 μM, < 20 min), a specific stabilizer of the non-myofibrillar actin microfilament component of the cardiac cytoskeleton, had no effect on action potential duration of normal cells, but significantly shortened the prolonged action potentials of hypertrophied cells. Cytochalasin D (5-50 μM), a disrupter of the actin microfilaments, also had little effect on action potential duration of normal cells. However, cytochalasin D further increased the action potential duration of hypertrophied cells at 10 min exposure. The addition of phalloidin to solutions containing cytochalasin D, reduced the latter's increase of action potential duration in hypertrophied cells. Whole-cell transient outward K+ current (Ito1) density was significantly decreased in hypertrophied cells. At a test potential of +60 mV, the mean Ito1 density recorded from normal cells was 13.5 ± 1.1 pA pF−1 (n = 18) compared to 4.17 ± 1.2 pA pF−1 for LVH cells (n = 22; P < 0.05). Phalloidin (20 μM) increased and cytochalasin D (50 μM) decreased whole-cell Ito1 in hypertrophied cells but had no effect on Ito1, in normal cells. When equimolar concentrations were used, phalloidin, 10 μM, reversed the decrease in Ito1 brought about by cytochalasin D, 10 μM, in hypertrophied cells. The L-type calcium current density was reduced in LVH compared to normal cells. Phalloidin (20 μM) and cytochalasin D (50 μM) had no effect on ICa,L in normal or LVH myocytes. The decrease in Ito1 in hypertrophied cells and the altered Ito1 responsiveness to phalloidin and cytochalasin D reflect modification of Ito1 channel function mediated, in part, through hypertrophy-altered cytoskeletal actin microfilament regulation of Ito1.
Ionic currents across membranes of myocytes enlarged in response to global chronic pressure overload of the mammalian heart are altered (Boyden & Jeck, 1995; Tomaselli & Marban, 1999). The underlying mechanisms, however, remain incompletely defined (Nattel et al. 1999). Our laboratory and others (Tomita et al. 1994; reviewed by Tomaselli & Marban, 1999) have reported action potential duration (APD) lengthening in hypertrophied cells of the pressure-overloaded rat left ventricle which we ascribed, at least in part, to a diminution of the Ca2+-independent voltage-gated transient outward K+ current (Ito1) (Tomita et al. 1994).
Abnormalities of cellular structure have been proposed as a contributor to abnormalities of cellular electrophysiology and arrhythmias associated with acute ischaemic or haemodynamic factors in the globally hypertrophied heart (Hart, 1994; Myerburg & Bassett, 1997). The mechanical stability of the cardiac cell, including the anchoring of transducer structures, depends on the integrity of the cytoskeleton (Bennett, 1990, 2000; Janmey, 1998), and geometric changes in the cell membrane may alter membrane electrophysiology (Glogauer et al. 1997). It is therefore reasonable to hypothesize that cardiac transmembrane electrical properties are influenced by interactions between the components of membrane ion channels and elements of the cytoskeleton which stabilize the components in position. Prominent increases in cytoskeletal microtubule density occur during pressure overload-induced cardiac hypertrophy in humans (Zile et al. 2001) and animals (Tagawa et al. 1997); the contractile deficit associated with such pressure overloading may be corrected by exposure to the microtubular depolymerizer, colchicine (Zile et al. 1999). In contrast, the relations between channel components, ionic currents and cortical cytoskeletal elements during the electrophysiological remodelling characteristic of global left ventricular hypertrophy (LVH) are unexplored (Myerburg & Bassett, 1997; Tomaselli & Marban, 1999). Thus, in the present study we test the hypothesis that agents that either disrupt (cytochalasin D) or stabilize (phalloidin) the subsarcolemmal non-myofibrillar actin microfilament component alter action potential configuration. Further, we studied the potential role of two important currents for the maintenance of the action potential, Ito1 and ICa,L, (L-type calcium current) in response to these substances in both normal and LVH cells.
METHODS
Preparation of animals and surgical procedure
Male Sprague-Dawley rats (200-250 g) were used. Normal and sham-operated rats were used as controls and were age- and weight-matched with LVH rats. LVH rats were produced via aseptic surgical banding of the abdominal aorta as described previously (Tomita et al. 1994). Animals were monitored twice daily for the first 3 days post-operatively; acetaminophen was administered when appropriate. Rats were maintained in a colony for 6-12 weeks after the surgery until the day of study. This investigation conformed with the ACUC guidelines of the University of Miami Shcool of Medicine.
Isolation of single cardiac myocytes
The rats were anaesthetized with sodium pentobarbital (40- 45 mg kg−1, i.p.), anticoagulated with heparin sodium (400 i.u. kg−1, i.p.), and ventilated through an endotracheal tube and a Harvard positive-pressure pump. Appropriate blood pressures were measured and the animals were checked for signs of congestive heart failure. The animals were killed by excision of the hearts; each heart was then weighed quickly. The heart weight/body weight ratios were calculated and used to quantify hypertrophy. Single ventricular myocytes were isolated via the Langendorf perfusion method using 300 i.u. ml−1 collagenase; 0.03 % hyaluronidase, 0.02 % trypsin inhibitor and 0.1 % albumin, dissolved in Tyrode solution containing 50 μM Ca2+ (as previously described by Tomita et al. 1994). Small pieces of the subendocardial side of the LV free wall (depth of ∼30 %) (midway between the apex and the base) were dissected from enzyme digested heart using fine scissors. We restricted our experiments to this population of endocardial LV myocytes to avoid potential confounding problems of regional variations in expression of voltage-dependent K+ (Kv) channel subunits encoding Ito (transient outward current) channels (Wickenden et al. 1999; Nerbonne, 2000). Isolated cells were stored in Tyrode solution containing 2 mm Ca2+ at 4 °C. Cells were studied on the day of isolation.
Solutions
The composition of the modified Tyrode solution was (mm): NaCl 144, KCl 4, CaCl2 1.8, MgCl2 0.5, NaHPO4 0.33, glucose 5.5 and Hepes 5.5 (pH 7.4 with NaOH). Nominally Ca2+-free Tyrode solution was prepared by omitting CaCl2 from the modified Tyrode solution. For recording action potentials (AP), cells were perfused with Tyrode solution at 37 ± 0.5 °C. The pipette solution contained the following (mm): KCl 140, MgCl2 5, ATP 5, EGTA 5, and Hepes 10 (pH 7.3 with KOH). For measurements of whole-cell Ito1 (Ca2+-independent component of Ito1), Na+-free Tyrode solution (where NaCl was replaced with equimolar choline chloride) was used externally. Tetrodotoxin (TTX, 0.02 mm) was added to the bath solution to eliminate Na+ current contamination and to exclude the possible contribution of Na+-activated K+ currents or transient currents generated through the Na+-K+ pump. In addition, 0.2-0.3 mm CoCl2 or 0.2 mm CdCl2 was added to the external solution to block Ca2+ currents and 5 mm atropine was added to remove the potential cholinergic effect of choline chloride. For recording ICa,L, 12.5 μM TTX was added to the Na+-free solution, K+-free bath solution containing (mm): 120 CsCl, 1 MgCl2, 10 EGTA and 10 Hepes (pH 7.3 with CsOH); while the pipette solution contained (mm): potassium aspartate 120, KCl 20, MgCl2 1, K2ATP 4, EGTA 10, and Hepes 5 and pH was adjusted to 7.4 with KOH.
Electrophysiologic studies
Experiments were conducted using standard whole-cell patch-clamp techniques and an Axopatch-1D amplifier (Axon Instruments). APs were recorded under current-clamp conditions, while Ito1 and ICa, L were recorded under the whole-cell membrane voltage-clamp mode. Glass pipette electrodes were forged by a micropipette puller (model P-87, Sutter Instrument Co.) and have electrode resistance of 2-4 MΩ (when filled with internal pipette solution).
Bath temperature was maintained at 37 ± 0.5 °C for AP recordings and at room temperature (22-25 °C) for Ito1 and ICa,L recordings. The current signals were filtered with a low pass filter with a cut-off frequency of 1-2 kHz at −3 dB, digitized at a conversion rate of 25-50 kHz with a 12-bit resolution Labmaster A-D converter (Tecmar) under the control of a computer, and stored on computer for off-line analysis. Data were analysed by using the software program pCLAMP (Axon Instruments).
Cell capacitance was determined in the appropriate internal and external solution for the ionic current being recorded by integrating the area under the capacitive transient evoked by a 10 mV hyperpolarizing step. The amplitudes of whole-cell membrane currents were normalized to cell capacitance and reported as pA pF−1. The series resistance was compensated (up to 80 %) in order to minimize the duration of the capacitive surge on the current tracing.
Ito1 and ICa,L current-voltage relationships were obtained by using a single 300 ms for Ito1 or 500 ms for ICa,L depolarizing pulse to various test potentials (-40 to +60 mV, in 10 mV increments) from a holding potential Vh = −80 mV at 5 s intervals. The amplitude of Ito1 and ICa,L was measured as the difference between the peak current and the minimum plateau current level at the end of the depolarizing pulse.
Statistical analysis
Data are reported as mean ± s.e.m. Statistical significance was evaluated by analysis of variance or Student's unpaired t test where appropriate. Two way ANOVA was used to test for a main effect for factor cell type with two levels, normal and hypertrophy, or for an effect of interventions. When a significant F test score was found, the Newman-Keuls test was applied to determine differences in individual means. P < 0.05 was considered significant.
RESULTS
Body weights were similar in control and LVH rats. However, heart weight/body weight ratio (mg g−1) was significantly increased in the LVH rats 6-8 weeks after partial ligation of their abdominal aortas (normal 3.3, n = 18; LVH 4.8, n = 26; P < 0.05). In line with the increase in ratio, subendocardial myocytes from the inner 50 % of the LV free wall (midway between the base and the apex) of rats with ventricular hypertrophy demonstrated an increased cell size, estimated from measurements of capacitance (normal 152 ± 4 pF, n = 40; LVH 198 ± 5 pF, n = 42; P < 0.05).
In pilot experiments, it was determined that phalloidin (> 20 μM) and cytochalasin D (> 50 μM) induced severe loss in resting potential, inexcitability or rapid physical degeneration of the normal and LVH cells. Therefore 20 μM phalloidin and 50 μM cytochalasin D were chosen as the maximal concentrations tested for all experiments.
Effects of phalloidin on APD
Action potentials were recorded from normal and LVH subendocardial muscle cells (stimulation rate = 1 Hz; 37 °C, Tyrode solution), prior to and after exposure to phalloidin (Fig. 1) and cytochalasin D (Fig. 2). APD to 25, 50, 75 and 90 % repolarization (APD25-90) was quantitated and reported in Tables 1 and 2. Consistent with previous reports (Keung & Aronson, 1981; Tomita et al. 1994; Meszaros et al. 2001), LVH cells had longer APDs than normal cells under control condition (Fig. 1 and Fig. 2, Table 1). Phalloidin (10 μM; 10 min exposure) had no effect on AP configuration or APD recorded from LV myocytes isolated from sham-operated or normal rats (Table 1). In contrast, phalloidin (10 μM; 10 min exposure) significantly shortened APD in LVH cells (Table 1). Representative action potential recordings in Fig. 1 demonstrate that phalloidin even at a higher concentration (20 μM) had no effect on AP configuration in normal cells while it shortened AP durations in LVH cells. In addition, the effects of phalloidin in LVH cells were time dependent (Table 1). Exposure to 10 μM phalloidin for 10 and 20 min has no effect on APD of untreated normal cells; whereas, additional shortening of LVH APD occurred and approached steady-state APD shortening at 20 min (Table 1).
Figure 1. Representative action potentials recorded from normal (top) and LVH (bottom) endocardial cells.

Stimulation rate is 1 Hz; action potentials induced via 2 ms suprathreshold stimuli applied through the pipette. Superfusion with 37 °C normal Tyrode solution maintained throughout. Note the longer APD of the LVH cell. Exposure to 20 μM phalloidin (10 min) had no effect on resting or action potential of the normal cell. In contrast, this concentration of phalloidin markedly decreased APD of the LVH cell at 10 min. Identical results were obtained in seven other paired experiments.
Figure 2. APD response to 50 μM cytochalasin D (10 min) for a normal cell (top) and LVH cell (bottom).

Conditions identical to those shown in Fig. 1. Exposure of the normal cell to cytochalasin D induced a slight prolongation in APD. Note the increased control APD of the LVH cell; exposure to cytochalasin D (10 min) markedly lengthened APD of the LVH cell; these results were observed in four other paired experiments. Washout with drug-free solution (10 min) reversed much of the APD prolongation induced by cytochalasin D.
Table 1.
Effects of phalloidin on APD
| APD25 (ms) | APD50 (ms) | APD75 (ms) | APD90 (ms) | |
|---|---|---|---|---|
| Normal (n =3) | ||||
| Control | 9.1 ± 0.7 | 24.9 ± 1.2 | 35.6 ± 0.7 | 52.2 ± 0.7 |
| PHA (10 μM); 10 min | 8.5 ± 0.9 | 20.7 ± 0.8 | 34.5 ± 0.6 | 49.7 ± 0.8 |
| PHA (10 μM); 20 min | 8.5 ± 0.4 | 19.1 ± 0.9 | 33.4 ± 0.9 | 47.2 ± 0.7 |
| LVH (n = 3) | ||||
| Control | 16.4 ± 0.7 | 67.2 ± 0.2 | 85.1 ± 0.8 | 114.8 ± 0.7 |
| PHA (10 μM); 10 min | 17.1 ± 0.4 | 34.1 ± 0.9* | 53.5 ± 0.8* | 79.2 ± 0.7* |
| PHA (10 μM); 20 min | 17.6 ± 0.5 | 16.9 ± 0.5* | 45.5 ± 0.3* | 67.3 ± 0.7* |
P < 0.05 compared to LVH control. The summed responses for the series of experiments at two different exposure times to phalloidin (10 μM) are shown. Note that control APD25–90 is longer in the LVH cells than the normal cells. At 10 and 20 min exposure to 10 μM phalloidin there is little change in APD25–90 of the normal cells. In contrast, APD50, 75, 90 of the LVH cells are significantly shortened at 10 min exposure to 10 μM phalloidin; additional shortening of LVH–APD is observed at 20 min.
Table 2.
Effects of cytochalasin D and phalloidin on APD
| APD25 (ms) | APD50 (ms) | APD75 (ms) | APD90 (ms) | |
|---|---|---|---|---|
| Normal (n = 3) | ||||
| Control | 8.2 ± 0.7 | 15.2 ± 2.4 | 26.7 ± 4.1 | 44.2 ± 4.4 |
| Cyto D (50 μM) | 8.9 ± 0.9 | 15.3 ± 3.2 | 31.7 ± 4.1 | 46.5 ± 5.8 |
| Cyto D + phalloidin (20 μM) | 8.1 ± 0.6 | 15.8 ± 2.2 | 30.2 ± 3.7 | 45.4 ± 1.1 |
| LVH (n = 5) | ||||
| Control | 18.1 ± 2.8 | 39.6 ± 4.8 | 76.4 ± 18.4 | 105.7 ± 18.8 |
| Cyto D (50 μM) | 30.9 ± 1.9* | 74.6 ± 17.9* | 136.7 ± 22.3* | 181.1 ± 15.5* |
| Cyto D + phalloidin (20 μM) | 24.0 ± 1.6** | 46.5 ± 5.9** | 88.3 ± 12.3** | 130.1 ± 6.8** |
P < 0.05 compared to LVH control
P < 0.05 compared to LVH Cyto D. The results of a series of experiments using 50 μM cytochalasin D followed by 20 μM phalloidin are summarized. Note the prolonged APD of LVH cells. Exposure to 50 μM cytochalasin D has no effect on APD25–90 of normal cells (10 min). Subsequent exposure to phalloidin (20 μM) (maintaining cytochalasin D superfusion) has no effect on APD25–90 of the normal cell. In contrast, 50 μMcytochalasin D produces significant lengthening of APD25–90 in LVH cells at 10 min exposure. While maintaining 50 μM cytochalasin D, exposure to 20 μM phalloidin (10 min) significantly but only partially reverses the APD25–90 prolongation noted during exposure to cytochalasin D. Similar results were obtained in four other paired experiments.
Effects of cytochalasin D on APD
Ten minutes exposure to 50 μM cytochalasin D had an effect in LVH cells opposite to that of phalloidin (Fig. 2). At this concentration, cytochalasin D markedly lengthened APD globally (APD25-90) in LVH cells; while having no effect on APD in the normal cells (Table 2). The AP prolonging effects of 50 μM cytochalasin D in LVH cells were partially reversed by phalloidin (20 μM) (Table 2). To determine whether phalloidin and cytochalasin D altered ventricular repolarization via modulation of ionic currents, the effects of phalloidin and cytochalasin D on Ito1 and ICa,L density were examined.
Responsiveness of Ito1 to phalloidin and cytochalasin D
Figure 3 shows representative Ito1 current recordings from a normal and a LVH cell. For both types of cells, the threshold for activation of the current was ∼-20 mV; the peak current amplitude increased as the test potentials were made more positive. The current density (amplitude normalized to cell membrane capacitance) was significantly smaller in LVH cells than in normal cells at all test potentials. At a test potential to +60 mV, Ito1 density was 13.5 ± 1.1 vs. 4.17 ± 1.2 pA pF−1 for normal cells (n = 18) and LVH cells (n = 22), respectively; P < 0.05.
Figure 3. Representative outward current recordings in the absence (control) and presence of 20 μM phalloidin from a normal cell (top traces) and an LVH cell (bottom traces).

Note that the control peak outward current is reduced in the LVH cell (compared to the normal cell). Exposure to 20 μM phalloidin for 10 min, had no effect on peak and plateau currents generated by the normal cell. In contrast, note the marked increase in peak current (plateau current is unaffected) during exposure of the LVH cell to 20 μM phalloidin, for 10 min.
Exposure to phalloidin (20 μM; 10 min) had no effect on Ito1 recorded from normal cells (Fig. 3, top panel); however, phalloidin (20 μM; 10 min) consistently increased the depressed peak Ito1 observed in LVH cells (Fig. 3, bottom panel), but never restored Ito1 to control levels in normal cells. This concentration of phalloidin had little or no effect on the sustained (plateau) outward current in both normal and LVH cells (Fig. 3). At a test potential to +60 mV, 10 min exposure to 20 μM phalloidin increased Ito1 density in LVH cells: 3.1 ± 1.1 vs. 7.2 ± 0.9 pA pF−1 in the absence and present of phalloidin respectively; n = 6; P < 0.05.
Cytochalasin D (50 μM; 10 min exposure) had no effect on Ito1 or plateau current in normal cells (Fig. 4, top panel). Conversely, exposure to cytochalasin D (50 μM; 10 min) decreased the already depressed Ito1 in LVH cells (Fig. 4, bottom panel) with no effect on the plateau currents. Cytochalasin D (50 μM) reduced LVH Ito1 at a test potential of +60 mV: 3.2 ± 0.8 (before) vs. 1.8 ± 0.4 pA pF−1 after exposure to the destabilizer (n = 4); P < 0.05.
Figure 4. Representative current recordings in the absence (control) and presence of 50 μM cytochalasin D from a normal cell (top traces) and an LVH cell (bottom traces).

Note the reduced control peak outward current for the LVH cell compared to the normal cell. Exposure to 50 μM cytochalasin D for 10 min had no effect on outward currents in the normal cell. In contrast, the ‘depressed’ peak outward current in the LVH cell was further diminished during 10 min exposure to 50 μM cytochalasin D (representative of three paired experiments).
In a separate series of experiments, a complete reversal of the depressant effects of 10 μM cytochalasin D on Ito1 in LVH cells over the range of test potentials (+10 to +60 mV) was observed with the application of equimolar (10 μM) phalloidin (Fig. 5).
Figure 5. Summarized data on the effects of cytochalasin D pre-treatment followed by phalloidin treatment on the voltage dependence of Ito1 current density from five paired experiments.

Peak outward current density (pA pF−1) was plotted against test potentials. Note that for the normal cells (filled symbols) exposure to equimolar concentrations of destabilizer/stabilizer, i.e. 10 μM cytochalasin D (10 min), and then 10 μM phalloidin for an additional 10 min in the continued presence of cytochalasin D, had no significant effect on peak outward current. Note the diminished control peak outward current for the LVH cells (□); exposed to 10 μM cytochalasin D (10 min) significantly decreased LVH peak outward current over the range +10 mV to +60 mV (▵). In the continued presence of 10 μM cytochalasin D, exposure to 10 μM phalloidin (10 min), over the same test potential range completely reversed the depression in peak outward induced by the destabilizer (○). * P < 0.05 cf. control (LVH).
Effects of phalloidin and cytochalasin on ICa,L
ICa,L is an important determinant of cardiac repolarization (Swynghedauw, 1999). Consistent with earlier study in feline ventricular myocytes (Nuss & Houser, 1991), un-normalized peak inward ICa,L was smaller in normal cells compared to LVH cells (Fig. 6); yet, when ICa,L was normalized to cell capacitance, it became apparent that ICa,L density in LVH cells was less than normal cells (Fig. 7). Peak ICa,L density for the normal cells was 12.2 ± 0.8 (n = 6) vs. 7.4 ± 1.1 pA pF−1 for LVH cells, n = 6, P < 0.05. There was also a modest change in the current-voltage relation shape for the LVH cells at −30 to −10 mV range hinting at the presence of the calcium T-type current, ICa,T (Fig. 7). This result is consistent with a study by Martinez et al. (1999) that demonstrated the existence of ICa,T in hypertrophied rat ventricle; whereas, ICa,T was not present in normal rat ventricle.
Figure 6. Representative ICa,L recordings from normal (top) and LVH (bottom) cells in the absence (control) and presence of cytochalasin D.

Inward currents were elicited using Vh and test potential protocols described in methods. Note that peak control ICa,L (uncorrected for cell size) for the LVH cell is greater than that elicited in the normal cell; inactivation is lightly prolonged in the LVH cell. Exposure to cytochalasin D (50 μM, 10 min) does not alter the peak current amplitude or inactivation for either the normal or the LVH cell.
Figure 7. Summary data for six paired experiments characterizing the effect of cytochalasin D on ICa,L as shown in Fig. 6.

Protocols described in Methods. Note decreased LVH ICa,L density over the test voltage range. Exposure to cytochalasin D (50 μM, 10 min) had no effect on ICa,L-voltage relations for both types of cells.
Representative current recording traces showing the effects cytochalasin D on ICa,L in normal and LVH cells are shown in Fig. 6. Exposure to cytochalasin D (50 μM, 10 min) does not alter the peak inward Ca2+ current for either the normal or LVH cell. Data from six such paired experiments are summarized in Fig. 7. Similarly, phalloidin had no effect on the evoked inward ICa,L currents in both the normal and LVH cell (Fig. 8).
Figure 8. ICa,L of normal and LVH cells is unresponsive to phalloidin.

ICa,L traces for normal cell (top) and LVH cell (bottom) in the absence and presence of the actin microfilament stabilizer, protocols identical to that shown in Figs 6 and 7. Note that peak ICa,L (uncorrected for cell size) is slightly increased in the LVH cell; inactivation is prolonged in the LVH cell. Exposure to phalloidin (20 μM; 10 min) had no effect on the ICa,L-voltage relations or inactivation of both cells. Representative of three paired experiments.
DISCUSSION
Sudden cardiac death remains a major health problem (Pye & Cobb, 1992; Myerburg & Bassett, 1997). The majority of such events are due to fatal cardiac arrhythmias resulting, at least in part, from ion channel dysfunction. While coronary artery disease is the most common aetiology of sudden cardiac death, the potential for a heart to generate a fatal arrhythmia during acute ischaemia is enhanced when LVH is present (Myerburg & Bassett, 1997; Pinto & Boyden, 1999) and LVH is a strong risk factor for fatal arrhythmias, alone or in conjunction with diseased coronary arteries (Pye & Cobb, 1992; Haider et al. 1998).
Global LVH, produced by a variety of techniques, is characterized by cellular electrical remodelling, most prominently APD prolongation (Tomaselli & Marban, 1999). APD prolongation in LVH is associated with decrease in important macroscopic K+ repolarizing currents: Ito1 (Ca2+-independent transient outward K+ current) and IK (delayed rectifier K+ current) (Boyden & Jeck, 1995; Nattel et al. 1999; Tomaselli & Marban, 1999). Besides electrical modification, structural remodelling may also occur in disease states such as global LVH (Anversa et al. 1986; Janmey, 1998; ter Keurs, 1998).
It is clear that microtubular components of the cytoskeleton, which transduce pressure stress in concert with actin microfilaments and intermediate filaments, are increased in cardiac hypertrophy (reviewed by ter Keurs, 1998). Yet, it is unclear whether the structure/function of the cortical cytoskeletal actin microfilaments and accessory proteins are perturbed during chronic pressure overload (Anversa et al. 1986; Janmey, 1998). In preliminary studies, there is evidence that the expression of cortical actin microfilament is increased in pressure-overload hypertrophied rat ventricular myocytes compared to normal cells (Fig. 9). However, more detailed studies are required to determine the structural and functional ion channel changes that result from the cytoskeletal actin microfilament changes in the hypertrophied myocytes. Nevertheless, we provide evidence that the cytoskeleton may modulate myocardial ionic currents, either directly or indirectly.
Figure 9. Actin microfilaments are increased in LVH compared to normal cells (preliminary results).
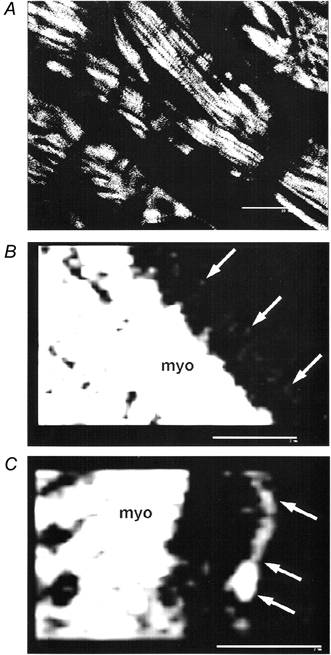
FITC-phalloidin confocal fluorescent microscopic images of frozen sections from normal (A, B) and LVH (C) cardiac muscle. Arrows denote non-myofibrillar actin microfilament labeled with FITC-phalloidin. Photographs are representative of six cells in two normal preparations; five cells in two LVH preparations. B and C are single optical sections from deconvoluted stacks. Scale bars: A, 20 μm; B, C, 2 μm. Procedures for preparing tissues, labelling with FITC-phalloidin, and laser confocal imaging have been described previously (Salas et al. 1997; Salas, 1999).
The cytochalasins are agents which disrupt actin from polymerization (Brenner & Korn, 1979); their specificity for cytoskeletal actin may be concentration-dependent (Calaghan et al. 2000). In contrast, the phalloidins are substances which stabilize the microfilaments against depolymerization. Actin microfilament disrupters, cytochalasin B and DNase I, enhance KATP (ATP-sensitive K+ channel) opening in patches from guinea-pig ventricular myocytes (Furukawa et al. 1996); anti-microtubule agents have no such effect. Cytochalasin D reduces whole-cell peak Na+ current in rat and rabbit ventricular cells (Undrovinas et al. 1995), probably by altering coupling between availability and activation (Maltsev & Undrovinas, 1997). Cytochalasin D also modulates rectification of the inward K+ rectifier current in guinea-pig ventricular cells (Mazzanti et al. 1996). In general, phalloidin opposes the cardiac action of cytochalasins. For example, while cytochalasin D accelerates KATP channel rundown (Furukawa et al. 1996), phalloidin inhibits such rundown (Furukawa et al. 1996). The effects of cytoskeletal-active agents on normal cardiac cellular electrophysiology appear to be channel and species specific.
Thus, LVH-altered cardiac ionic currents may also result (indirectly) from the structural remodelling of transduced cytoskeletal actin microfilaments (Bretscher, 1991) and/or adapter proteins (Bennett & Gilligan, 1993) during cardiac cell enlargement (Collins et al. 1996). In the present study, phalloidin and cytochalasin D were employed to test the hypothesis that LVH electrical remodelling in cardiac cells is, in part, a result of an indirect/direct interaction between the cytoskeleton and cardiac ionic currents (Ito1 and ICa,L).
Effects of phalloidin and cytochalasin D on cardiac electrophysiology
The results of this study demonstrate modulation of APD by phalloidin (20 μM) and cytochalasin D (50 μM) in LVH but not in normal control cells. Similarly, concentrations of cytochalasin D equivalent to those we used have been shown to have no effect on monophasic APD in normal dog ventricle (Wu et al. 1998). Cytochalasin D shortens APD while phalloidin prolongs repolarization in LVH cells. In addition, phalloidin can reverse the AP prolonging effects of cytochalasin D.
Although phalloidin and cytochalasin D had no effect on ICa,L in normal or LVH cells (consistent with earlier studies in rat (Undrovinas & Maltsev, 1998) and guinea-pig ventricular cardiomyocytes (Pascarel et al. 1999)), in this study phalloidin increased and cytochalasin D decreased the already depressed Ito1 density in LVH cells only. These effects on Ito1 correspond to changes in APD induced by phalloidin (i.e. shortening of APD and increasing Ito1) and cytochalasin D (i.e. prolongation of APD and decreasing Ito1) in hypertrophied cells. This suggests that the effects of phalloidin and cytochalasin D are both channel specific and pathological state-specific (i.e. LVH). Furthermore, the fact that phalloidin can reverse the effects of cytochalasin on the action potential and Ito1 in LVH cells on a mole-to-mole basis suggests the actions of these agents are via a similar pathway, presumably through depolymerizing or polymerizing the actin microfilament. Moreover, these data suggest that electrical remodelling in the hypertrophied hearts may, in part, be modulated by pressure overload-induced adaptation of the cortical actin microfilaments and associated components.
Potential cytoskeletal basis for electrical remodelling in LVH cardiac myocytes
That neither cytoskeletal active agents (phalloidin and cytochalasin D) had effect on APD or Ito1 in normal cells suggests that under normal conditions there are no (quantitatively or qualitatively) apparent interactions between Ito1 channels or subunits and the actin microfilaments or associated components of the cytoskeleton. Yet, the condition of hypertrophy-induced adaptation of the cardiomyocytes appears to alter interactions between the cortical cytoskeleton and the components of Ito1 channels and/or the endoexocytic transport of Ito1 channel components (Hille, 1992; Qualman et al. 2000), leading to downregulation and silencing of Ito1 channels (Tomita et al. 1994).
Another possible explanation is that hypertrophy-induced structural remodelling may give rise to a generalized increase in cytoskeletal components (microtubules, intermediate filaments, actin microfilament, etc.) thereby increasing the substrate for these cytoskeletal active agents. Therefore, the law of mass action may explain the greater sensitivity to cytochalasin and phalloidin in hypertrophied myocytes.
Finally, one also cannot rule out altered binding affinities of drugs to the actin microfilaments in LVH. Nonetheless, further experiments are required to elucidate the mechanism(s) whereby cytoskeletal actin microfilament may directly/indirectly modulate ionic currents in LVH.
At first glance, it would appear that generation of an excess of actin microfilaments (preliminary experiments, Fig. 9), or other cytoskeletal proteins that have an association with altered ion channel function, would be a maladaptive response. However, viewed in another context, it might be considered adaptative. Changes in cell volume and geometry associated with hypertrophy process might, in itself, create distortions of transmembrane spanning proteins that form ion channels and other receptors and transporters. Enhancement of the subsarcolemmal supporting structure may thus serve a function of stabilizing cell membrane and transmembrane geometry, and therefore protect the function of transmembrane structures. In this conceptual model, the fact that cytochalasin D, a disrupter of actin microfilaments, impaired current flow across a transmembrane channel (Ito) in a diseased cell suggests that it interferes with a protective mechanism, rather than correcting a maladaptive mechanism. This notion has been tested experimentally (Glogauer et al. 1997) in another model. In another study, alterations of the cytoskeleton in an ankyrin B deficient knockout mouse, adversely affected cardiac sodium current (INa) (Chauhan et al. 2000), suggesting a maladaptive consequence of loss of cytoskeletal elements. Conversely, in the present study, phalloidin, a stabilizer of the structural protein, actually enhances Ito1, supporting a concept that protection of actin microfilaments is beneficially adaptive.
Clinical significance
The rat heart displays a constellation of currents that differ in many respects from those of larger mammals and extrapolation of our results to the clinical situation of human hearts must be made with caution. General electrophysiological-structure principles extracted from the present study may be applicable to the human heart, in view of the demonstrations that Ito1 which is critical in the repolarization process is decreased in both hypertrophied human myocytes (Tomaselli & Marban, 1999; Swynghedauw, 1999) and the aged rat heart (Walker et al. 1993; Cerbai et al. 1994). Nerbonne (2000) has noted that cardiac arrhythmias are frequently treated with pore-blocking drugs. However, the observations that transient outward K+ currents and certain underlying subunits may be down-regulated in cardiac abnormalities such as LVH, have led to the suggestion that novel therapies should be contemplated (Nerbonne, 2000). We propose that consideration of cytoskeletal elements in pharmacotherapies intended to decrease or restore ion channel activity altered by disease states or ageing offers a new therapeutic target for exploration.
Acknowledgments
This work was supported by grants from the National Institutes of Health: HL19044 (A.L.B.), HL21735 (R.J.M.) and the Florida Biomedical Research Program (A.L.B.). R.J.M. is supported in part by the AHA Chair in Cardiovascular Research at the University of Miami and the Louis Lemberg Chair in Cardiology.
REFERENCES
- Anversa P, Ricci R, Olivetti G. Quantitative structural analysis of the myocardium during physiologic growth and induced cardiac hypertrophy: A review. Journal of the American College of Cardiology. 1986;7:1140–1149. doi: 10.1016/s0735-1097(86)80236-4. [DOI] [PubMed] [Google Scholar]
- Bennett PB. Ion channels, cytoskeletal proteins, and cellular excitability. Circulation Research. 2000;86:367–368. doi: 10.1161/01.res.86.4.367. [DOI] [PubMed] [Google Scholar]
- Bennett V. Spectrin-based membrane skeleton: a multipotential adapter between plasma membrane and cytoskeleton. Physiological Reviews. 1990;70:1029–1065. doi: 10.1152/physrev.1990.70.4.1029. [DOI] [PubMed] [Google Scholar]
- Bennett V, Gilligan DM. The spectrin-based membrane skeleton and micron-scale organization of the plasma membrane. Annual Review of Cell Biology. 1993;9:27–66. doi: 10.1146/annurev.cb.09.110193.000331. [DOI] [PubMed] [Google Scholar]
- Boyden PA, Jeck CJ. Ion channel function in disease. Cardiovascular Research. 1995;29:312–318. [PubMed] [Google Scholar]
- Brenner SL, Korn ED. Substoichiometric concentrations of cytochalasin D inhibit actin polymerization. Additional evidence for an F-actin treadmill. Journal of Biological Chemistry. 1979;254:9982–9985. [PubMed] [Google Scholar]
- Bretscher A. Microfilament structure and function in the cortical cytoskeleton. Annual Review of Cell Biology. 1991;7:337–374. doi: 10.1146/annurev.cb.07.110191.002005. [DOI] [PubMed] [Google Scholar]
- Calaghan SC, White E, Bedut S, Le Guennec J-Y. Cytochalasin D reduces Ca2+ sensitivity and maximum tension via interactions with myofilaments in skinned rat cardiac myocytes. Journal of Physiology. 2000;529:405–411. doi: 10.1111/j.1469-7793.2000.00405.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerbai E, Barbieri M, Li Q, Mugelli A. Ionic basis of action potential prolongation of hypertrophied myocytes isolated from the heart of hypertensive rats of different ages. Cardiovascular Research. 1994;28:1180–1187. doi: 10.1093/cvr/28.8.1180. [DOI] [PubMed] [Google Scholar]
- Chauhan VJ, Tuvia S, Buhusi M, Bennett V, Grant AO. Abnormal cardiac Na+ channel properties and QT heart rate adaptation in neonatal ankyrinB knockout mice. Circulation Research. 2000;86:441–447. doi: 10.1161/01.res.86.4.441. [DOI] [PubMed] [Google Scholar]
- Collins JF, Pawloski-Dahm C, Davis MG, Ball N, Dorn GW, Walsh RA. The role of the cytoskeleton in left ventricular pressure overload hypertrophy and failure. Journal of Molecular and Cellular Cardiology. 1996;28:1435–1443. doi: 10.1006/jmcc.1996.0134. [DOI] [PubMed] [Google Scholar]
- Furukawa T, Yamane T-I, Terai T, Katayama Y, Hiroaka M. Functional linkage of the cardiac ATP-sensitive K+ channel to actin cytoskeleton. Pflügers Archiv. 1996;431:504–512. doi: 10.1007/BF02191896. [DOI] [PubMed] [Google Scholar]
- Glogauer M, Arora P, Yao G, Sokholov I, Ferrier J, McCulloch CAG. Calcium ions and tyrosine phosphorylation interact coordinately with actin to regulate cytoprotective responses to stretching. Journal of Cell Science. 1997;110:11–21. doi: 10.1242/jcs.110.1.11. [DOI] [PubMed] [Google Scholar]
- Haider AW, Larson MG, Benjamin EJ, Levy D. Increased left ventricular mass and hypertrophy are associated with increased risk for sudden death. Journal of the American College of Cardiology. 1998;32:1454–1459. doi: 10.1016/s0735-1097(98)00407-0. [DOI] [PubMed] [Google Scholar]
- Hart G. Cellular electrophysiology in cardiac hypertrophy and failure. Cardiovascular Research. 1994;28:933–946. doi: 10.1093/cvr/28.7.933. [DOI] [PubMed] [Google Scholar]
- Hille B. Ionic Channels of Excitable Membranes. Sunderland, MA, USA: Sinauer Associates; 1992. pp. 510–511. [Google Scholar]
- Janmey PA. The cytoskeleton and cell signaling: Component localization and mechanical coupling. Physiological Reviews. 1998;78:763–781. doi: 10.1152/physrev.1998.78.3.763. [DOI] [PubMed] [Google Scholar]
- Keung CH, Aronson RS. Non-uniform electrophysiological properties and electrotonic interactions in hypertrophied rat myocardium. Circulation Research. 1981;49:150–158. doi: 10.1161/01.res.49.1.150. [DOI] [PubMed] [Google Scholar]
- Maltsev VA, Undrovinas AI. Cytoskeleton modulates coupling between availability and activation of cardiac sodium channel. American Journal of Physiology. 1997;273:H1832–1840. doi: 10.1152/ajpheart.1997.273.4.H1832. [DOI] [PubMed] [Google Scholar]
- Martinez ML, Heredia MP, Delgado C. Expression of T-type Ca2+ channels in ventricular cells from hypertrophied rat hearts. Journal of Molecular and Cellular Cardiology. 1999;31:1617–1625. doi: 10.1006/jmcc.1999.0998. [DOI] [PubMed] [Google Scholar]
- Mazzanti M, Assandri R, Ferroni A, DiFrancesco D. Cytoskeletal control of rectification and expression of four substrates in cardiac inward rectifier K+ channels. FASEB Journal. 1996;10:357–361. doi: 10.1096/fasebj.10.2.8641571. [DOI] [PubMed] [Google Scholar]
- Meszaros J, Khananshvili D, Hart G. Mechanism underlying delayed afterdepolarizations in hypertrophied left ventricular myocytes of rats. American Journal Physiology – Heart and Circulatory Physiology. 2001;281:H903–914. doi: 10.1152/ajpheart.2001.281.2.H903. [DOI] [PubMed] [Google Scholar]
- Myerburg RJ, Bassett AL. Cellular electrophysiology, heterogeneity and arrhythmias. Journal of Cardiovascular Electrophysiology. 1997;8:884–886. doi: 10.1111/j.1540-8167.1997.tb00849.x. [DOI] [PubMed] [Google Scholar]
- Nattel S, Roden DM, Escande D. A spotlight on electrophysiological remodeling and the molecular biology of ion channels. Cardiovascular Research. 1999;42:267–269. doi: 10.1016/s0008-6363(99)00072-3. [DOI] [PubMed] [Google Scholar]
- Nerbonne J. Molecular basis of functional voltage-gated K+ channel diversity in the mammalian myocardium. Journal of Physiology. 2000;525:285–298. doi: 10.1111/j.1469-7793.2000.t01-1-00285.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuss HB, Houser SR. Voltage dependence of contraction and calcium current in severely hypertrophied feline ventricular myocytes. Journal of Molecular and Cellular Cardiology. 1991;23:717–726. doi: 10.1016/0022-2828(91)90981-q. [DOI] [PubMed] [Google Scholar]
- Pascarel C, Brette F, Cazorla O, Le Guennec JY. Effects on L-type calcium current of agents interfering with the cytoskeleton of isolated guinea-pig ventricular myocytes. Experimental Physiology. 1999;84:1043–1050. [PubMed] [Google Scholar]
- Pinto JMB, Boyden PA. Electrical remodeling in ischemia and infarction. Cardiovascular Research. 1999;42:294–297. doi: 10.1016/s0008-6363(99)00013-9. [DOI] [PubMed] [Google Scholar]
- Pye MP, Cobbe SM. Mechanisms of ventricular arrhythmias in cardiac failure and hypertrophy. Cardiovascular Research. 1992;26:740–750. doi: 10.1093/cvr/26.8.740. [DOI] [PubMed] [Google Scholar]
- Qualmann B, Kessels MM, Kelly RB. Molecular links between endocytosis and the actin cytoskeleton. Journal of Cell Biology. 2000;150:F111–116. doi: 10.1083/jcb.150.5.f111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salas PJI. Insoluble γ-tubulin containing-structures are anchored to the apical network of intermediate filaments in polarized CACO-2 epithelial cells. Journal of Cell Biology. 1999;146:645–657. doi: 10.1083/jcb.146.3.645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salas PJI, Rodriguez ML, Viciana A, Vega-Salas DE, Hauri H-P. The apical sub-membrane cytoskeleton participates in the organization of the apical pole in epithelial cells. Journal of Cell Biology. 1997;137:359–375. doi: 10.1083/jcb.137.2.359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swynghedauw B. Molecular mechanisms of myocardial remodeling. Physiological Reviews. 1999;79:215–262. doi: 10.1152/physrev.1999.79.1.215. [DOI] [PubMed] [Google Scholar]
- Tagawa H, Wang N, Narishige T, Ingber DE, Zile MR, Cooper G., IV Cytoskeletal mechanics in pressure-overload hypertrophy. Circulation Research. 1997;80:281–289. doi: 10.1161/01.res.80.2.281. [DOI] [PubMed] [Google Scholar]
- ter Keurs HENK. Microtubules in cardiac hypertrophy. A mechanical role in decompensation? Circulation Research. 1998;82:828–831. doi: 10.1161/01.res.82.7.828. [DOI] [PubMed] [Google Scholar]
- Tomaselli GF, Marban E. Electrophysiological remodeling in hypertrophy and heart failure. Cardiovascular Research. 1999;42:270–283. doi: 10.1016/s0008-6363(99)00017-6. [DOI] [PubMed] [Google Scholar]
- Tomita F, Bassett AL, Myerburg RJ, Kimura S. Diminished transient outward currents in rat hypertrophied ventricular myocytes. Circulation Research. 1994;75:296–303. doi: 10.1161/01.res.75.2.296. [DOI] [PubMed] [Google Scholar]
- Undrovinas AL, Maltsev VA. Cytochalasin D alters kinetics of Ca2+ transient in rat ventricular cardiomyocytes. Journal of Molecular and Cellular Cardiology. 1998;30:1655–1670. doi: 10.1006/jmcc.1998.0715. [DOI] [PubMed] [Google Scholar]
- Undrovinas AL, Shander GS, Makielski JC. Cytoskeleton modulates gating of voltage-dependent sodium channel in heart. American Journal of Physiology. 1995;269:H203–214. doi: 10.1152/ajpheart.1995.269.1.H203. [DOI] [PubMed] [Google Scholar]
- Walker KE, Lakatta EG, Houser SR. Age associated changes in membrane currents in rat ventricular myocytes. Cardiovascular Research. 1993;27:1968–1977. doi: 10.1093/cvr/27.11.1968. [DOI] [PubMed] [Google Scholar]
- Wickenden AD, Jegla TJ, Kaprielian R, Backx PH. Regional contributions of Kv1. 4, Kv4.2 and Kv4.3 to transient outward K+ current in rat ventricle. American Journal of Physiology. 1999;276:H1599–1607. doi: 10.1152/ajpheart.1999.276.5.H1599. [DOI] [PubMed] [Google Scholar]
- Wu J, Biermann M, Rubart M, Zipes DP. Cytochalasin D as excitation-contraction uncoupler for optically mapping action potentials in wedges of ventricular myocardium. Journal of Cardiovascular Electrophysiology. 1998;9:1336–1347. doi: 10.1111/j.1540-8167.1998.tb00109.x. [DOI] [PubMed] [Google Scholar]
- Zile MR, Green GR, Schuyler GT, Aurigemma GP, Miller DC, Cooper G., IV Cardiocyte cytoskeleton in patients with left ventricular pressure overload hypertrophy. Journal of the American College of Cardiology. 2001;37:1080–1084. doi: 10.1016/s0735-1097(00)01207-9. [DOI] [PubMed] [Google Scholar]
- Zile MR, Koide M, Sato S, Ishigwo Y, Conrad CH, Buckley JM, Morgan JP, Cooper G., IV Role of microtubules in the contractile dysfunction of hypertrophied myocardium. Journal of the American College of Cardiology. 1999;33:250–260. doi: 10.1016/s0735-1097(98)00550-6. [DOI] [PubMed] [Google Scholar]