

Abstract
Presynaptic and postsynaptic membrane activities during experimental metabolic inhibition were analysed in mechanically dissociated rat hippocampal neurons using nystatin-perforated and conventional whole-cell patch clamp recordings. NaCN, an inhibitor of mitochondrial ATP synthesis, induced an outward current across the postsynaptic soma membrane. This current was blocked by tolbutamide, a sulfonylurea, which blocks ATP-sensitive K+ (KATP) channels. The presynaptic effect of metabolic inhibitors such as NaCN, NaN3, or glucose-free solution was to increase the frequency of GABAergic miniature inhibitory postsynaptic currents (mIPSCs). Tolbutamide had no effect on this increase in mIPSC frequency induced by metabolic inhibition. Diazoxide, a KATP channel opener, evoked a similar somatic outward current in a dose-dependent manner. In addition, diazoxide decreased the frequency of mIPSCs in a dose-dependent fashion. Both these pre- and postsynaptic effects of diazoxide were reversed by tolbutamide, suggesting the existence of KATP channels on both pre- and postsynaptic membranes. These results confirm the presence of KATP channels on both the pre- and postsynaptic membranes but indicate that the channels have significantly different sensitivities to metabolic inhibition.
ATP-sensitive K+ (KATP) channels belong to a family of inward rectifying potassium channels whose structure is believed to comprise a tetramer of four inwardly rectifying K+ channel subunits, each coupled to a sulfonylurea binding subunit (Ashcroft & Gribble, 1998; Inagaki & Seino, 1998). While KATP channels were first discovered in cardiac myocytes (Noma, 1983), they are also widespread in the central nervous system (Garcia de Arriba et al. 1999). Within the brain, KATP channels exist both pre- and postsynaptically in many regions, including putative GABAergic hippocampal interneurons (Zawar et al. 1999; Zawar & Neumcke, 2000). Activation of postsynaptic KATP channels causes membrane hyperpolarization, which is thought to be protective against excitotoxicity, while activation of presynaptic KATP channels can directly modulate neurotransmitter release from nerve terminals (Ohno-Shosaku et al. 1993; Watts et al. 1995; Ye et al. 1997). KATP channels are regulated by the intracellular concentration of ATP ([ATP]i), being opened as [ATP]i decreases (for review Aguilar-Bryan et al. 1998), and thus by linking neuronal metabolism and excitability they are thought to play an important role in protecting cells from neuronal death caused by hypoxia, ischaemia or metabolic inhibition.
Although it is generally agreed that postsynaptic KATP channels serve as one of the key mechanisms for neuroprotection during cell-damaging conditions, precise evaluation of presynaptic KATP channel activity during such conditions has been difficult. Direct patch clamp recording from presynaptic terminals is not possible at most central neuronal synapses and hence most studies have analysed spontaneous or evoked synaptic events using various slice preparations. However, the slice preparation suffers from difficulties in accurately and rapidly perfusing the soma and neuronal terminals impinging on the recorded neuron(s) as well as difficulties in distinguishing between direct effects and more indirect effects arising from any altered activity in adjacent cells. In the present study, we have overcome these limitations by using the ‘synaptic bouton preparation’ in which single neurons can be isolated with their presynaptic nerve terminals attached and functional (Koyama et al. 1999b; Rhee et al. 1999). In combination with a rapid perfusion system (the ‘Y-tube method’, Murase et al. 1990), this preparation enabled us to precisely investigate presynaptic effects, by analysing synaptic events, while concurrently observing any postsynaptic membrane currents evoked by the various experimental protocols. In this report we demonstrate the effects of metabolic inhibition on presynaptic and postsynaptic KATP channels and their functional consequences.
METHODS
Preparation
Our institutional Ethics Review Committee for Animal Experimentation approved all the following experimental protocols in accordance with the Guiding Principles for the Care and Use of Animals in the Field of Physiological Sciences of the Physiological Society of Japan.
Twelve- to 15-day-old Wistar rats were decapitated under pentobarbital anaesthesia (50 mg kg−1i.p.). The brain was quickly removed from the skull and sliced in a coronal plane at a thickness of 400 μm with a microslicer (VT1000S; Leica, Nussloch, Germany). The brain slices were incubated for at least 1 h at room temperature (20-25 °C) in a medium saturated with 95 % O2 and 5 % CO2 and of the following composition (mm): 124 NaCl, 5 KCl, 1.2 KH2PO4, 24 NaHCO3, 2.4 CaCl2, 1.3 MgSO4 and 10 glucose. Following incubation, slices were transferred into a 35 mm culture dish (Primaria; Falcon, Lincoln Park, NJ, USA) and the hippocampal CA1 region was identified under a binocular microscope (SMZ-1; Nikon, Tokyo, Japan). The fine tip of a fire-polished glass pipette was placed lightly onto the surface of the hippocampal CA1 region and horizontally vibrated at 50-60 Hz. The slice was removed from the dish after dissociation and the liberated single neurons were left for about 20 min to settle and adhere to the bottom of the dish before electrophysiological experiments were started. Note that the method required no enzyme treatment.
Electrical measurements
Electrical measurements were performed using either the nystatin-perforated patch recording mode (Akaike & Harata, 1994), or the conventional whole-cell recording method. The glass electrodes (patch pipettes) were made from borosilicate capillary glass (1.5 mm o.d., 0.9 mm i.d., G-1.5, Narishige, Tokyo, Japan) using a vertical pipette puller (PB-7, Narishige, Tokyo, Japan). The electrodes for nystatin-perforated patch recordings were filled with the following solution containing nystatin (mm): 80 potassium methanesulfonate, 70 KCl, 5 MgCl2 and 10 Hepes. Nystatin was initially dissolved in acidified methanol at 10 mg ml−1, and the stock solution was diluted with the internal solution just before use to a final concentration of 200 μg ml−1. For conventional whole-cell recordings, electrodes were filled with the following solution (mm): 75 caesium methanesulfonate, 75 CsCl, 5 ethylene glycol-bis N,N,N′,N′-tetraacetic acid (EGTA), 5 tetraethylammonium chloride (TEA-Cl), 2 ATP-Mg, and 10 Hepes. The pH of these internal solutions was adjusted to 7.2 with tris(hydroxymethyl)aminomethane (Tris-OH). The resistance between the recording electrode, filled with these pipette solutions, and the reference electrode was 5-7 MΩ. Neurons were visualized using phase contrast optics on an inverted microscope (Diaphot, Nikon, Tokyo, Japan). Current and voltage were measured under voltage clamp conditions with a patch clamp amplifier (EPC-7; HEKA, Lambrecht, Germany). Membrane currents were filtered at 1 kHz (E-3201A Decade Filter, NF Electronic Instruments, Tokyo, Japan) and digitized at 4 kHz using Digidata 1200 with pCLAMP software (version 8.0, Axon Instruments, Union City, CA, USA). All experiments were performed at room temperature (20-25 °C).
Data analysis
Miniature inhibitory postsynaptic currents (mIPSCs) were collected from digitized records in pre-set epochs before, during, and after each experimental condition. Each epoch included at least 50 events except for conditions in which event frequency was strongly inhibited and very low. Synaptic events were automatically detected using a minimum event amplitude set at 5 pA in MiniAnalysis software (Synaptosoft, Decatur, GA, USA). All detected events were then visually verified before the data were subjected to further analysis. This procedure minimized inclusion of occasional artifacts. Event amplitude was calculated by subtracting baseline current from the peak current value. Baseline current for each event was obtained by averaging a segment of current 2.5 ms just preceding the event. When two or more events overlapped, the baseline current of the latter event was calculated by extrapolating the decay phase of the preceding event to baseline. Note that baseline current was calculated for every detected event which minimized the influence of changes in baseline current on event amplitude. Event frequency and mean amplitude were also analysed using the MiniAnalysis software. Experimental values were normalized to control values obtained in the same cell under the same conditions and are presented as means ± standard error of the mean (s.e.m.). For analysis of the concentration-response relationship for the diazoxide-induced postsynaptic outward current, data were fitted to the following equation using a least squares method:
where IA is the current amplitude normalized to the control response, M is the maximal response normalized to the control response, CA is the concentration of diazoxide, EC50 is the concentration for half maximal response and nH is the Hill coefficient. For analysis of the concentration-response relationship for the diazoxide-induced presynaptic inhibition, data were fitted to the following equation using a least squares method:
where IB is the mIPSC frequency normalized to the control value in the absence of diazoxide, CB is the concentration of diazoxide, IC50 is the concentration for half-maximal inhibition of mIPSC frequency and nH is the Hill coefficient.
External solutions
The ionic composition of the standard external solution was (mm): 150 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 10 glucose and 10 Hepes. The ionic composition of the glucose-free solution was the same as the standard external solution except that glucose was replaced by an equimolar amount of 2-deoxyglucose. The ionic composition of the Ca2+-free external solution was (mm): 146 NaCl, 5 KCl, 5 MgCl2, 2 EGTA, 10 glucose and 10 Hepes. The pH of these solutions was adjusted to 7.4 with Tris-OH. Tetrodotoxin (TTX, 300 nm), 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX, 1 μM) and dl-2-amino-5-phosphonovaleric acid (AP-5, 10 μM) were routinely added to the external solutions throughout the experiments.
Drugs
The drugs used in the present study included sodium azide (NaN3) and sodium cyanide (NaCN) purchased from Ishizu Seiyaku (Osaka, Japan); AP-5, BAPTA-AM, CNQX, 2-deoxy-glucose, diazoxide, EGTA, EGTA-AM, nystatin and ouabain from Sigma (St. Louis, MO, USA); bicuculline methochloride from Tocris Cookson (Bristol, UK); TEA-Cl from Tokyo Kasei (Tokyo, Japan) and glibenclamide, tolbutamide and TTX from Wako Pure Chemicals Industries (Tokyo, Japan). Drugs that are insoluble in water were first dissolved in dimethylsulfoxide (DMSO) and then diluted in the external solution. The final concentration of DMSO was always less than 0.1 %, a solvent concentration which had no effect on membrane potential or electrical activities. Drug solutions were applied using a rapid application system termed the ‘Y-tube method’ (Murase et al. 1990). By this technique the external solution surrounding a neuron could be exchanged within 20 ms.
RESULTS
GABAergic mIPSCs in dissociated hippocampal neurons
Using the nystatin whole-cell recording technique at a holding potential (VH) of −60 mV and in the presence of TTX, CNQX and AP-5, prominent inward synaptic currents were observed that were completely blocked by bicuculline (30 μM), a specific GABAA receptor antagonist (Fig. 1B). mIPSC amplitude was linearly related to the holding potential and reversed polarity at −25 ± 1.51 mV (n = 3, Fig. 1A), quite close to the theoretical Cl− equilibrium potential (ECl; −17.6 mV) based on external and internal Cl− concentrations of 161 and 80 mm, respectively. Thus, the results indicate that these spontaneous events are GABAA receptor-mediated miniature inhibitory postsynaptic currents (mIPSCs).
Figure 1. GABAergic miniature inhibitory postsynaptic currents.

A, current-voltage relationship of spontaneous synaptic events in the presence of TTX, CNQX and AP-5. The data were obtained using nystatin-perforated patch recording mode in voltage clamp conditions. Each point, and the associated vertical bar, represents the mean ± s.e.m of data from 3 neurons. B, representative traces showing bicuculline-sensitive spontaneous synaptic events.
Effect of NaCN on pre- and postsynaptic membrane activities
NaCN is often used as a chemical model of hypoxia or ischaemia. It binds to cytochrome c oxidase in mitochondria and blocks electron transport which is essential for oxidative ATP synthesis (Chen et al. 1999). The intracellular ATP concentration ([ATP]i) is thereby reduced, leading to the opening of KATP channels, which typically induces outward K+ currents in neurons voltage clamped close to their resting membrane potential (Murai et al. 1997; Zawar & Neumcke, 2000). In the present study, when using the nystatin whole-cell recording mode, NaCN (1 mm) evoked an outward current in 13 out of 19 of neurons which ranged from 16.3 to 38.6 pA at a holding potential of −60 mV. This outward current was sensitive to tolbutamide (100 μM), a sulfonylurea known to block KATP channels (Sturgess et al. 1988; Fig. 2A and B). Such a postsynaptic membrane effect is compatible with previous studies on metabolic inhibition (Koyama et al. 1999a; Sun et al. 2000).
Figure 2. Pre- and postsynaptic effects of NaCN.

A, a representative trace showing pre- and postsynaptic responses in the presence of NaCN. B, analysis of postsynaptic (baseline) outward current. Tol: tolbutamide. C, analysis of mIPSCs during NaCN application. In B and C, each column and the associated vertical bar represent the mean + s.e.m. of data from 6 neurons. P value was obtained by using Student's unpaired two-tailed t test.
Mitochondria are known to exist also in presynaptic terminals (Gioio et al. 2001). If KATP channels are present in the presynaptic nerve terminals and are similarly activated by NaCN, one may predict that this might also hyperpolarize the presynaptic membrane, resulting in a decrease in mIPSC frequency. However, NaCN actually significantly increased mIPSC frequency to 185.2 ± 21.3 % of control (Fig. 2C). Mean mIPSC amplitude also increased slightly during this period (to 112 ± 8.0 % of control). The increase in mIPSC frequency caused by NaCN was not affected by co-application of tolbutamide (Fig. 2C) suggesting that this facilitation was not the result of activation of presynaptic KATP channels.
Pre- and postsynaptic KATP channel-mediated events during metabolic inhibition
One possibility for the apparent lack of activation of presynaptic KATP channels is that there may be residual presynaptic [ATP]i during NaCN application. In an attempt to block any remaining oxidative ATP synthesis, we added NaN3, another metabolic poison that blocks electron transport in mitochondria. Using nystatin recordings the application of both NaCN and NaN3 activated two different types of outward current: a more rapid tolbutamide-sensitive current and a slower tolbutamide-insensitive outward current (Fig. 3A and B). The tolbutamide-sensitive current was observed in 10 out of 22 cells, the tolbutamide-insensitive current in 9 out of 22 cells and no outward current was observed in the remaining 3 neurons. A combination of the two types of currents was not observed.
Figure 3. Pre- and postsynaptic effects during NaCN and NaN3 application.

A, representative traces of the tolbutamide(Tol)-sensitive (a) and insensitive (b) postsynaptic outward current evoked by NaCN and NaN3 application. B, time course of postsynaptic current from 5 min before, to 10 min after adding NaCN and NaN3. The average current value during the 5 min control period (before application of metabolic inhibitors) has been subtracted from each point. Each point and the associated vertical error bar represent the mean ± s.e.m. of data from 4 (Tol-sensitive) or 6 (Tol-insensitive) neurons. C, representative trace showing tolbutamide-sensitive outward current and mIPSCs before and during perfusion with Ca2+-free external solution. D, summary of presynaptic effects of NaCN and NaN3 and tolbutamide, in normal and Ca2+-free external solutions. Each column and the associated vertical error bar represent the mean + s.e.m. of data from 17 (in normal solution) or 7 (in Ca2+-free external solution) neurons.
The combined application of NaCN and NaN3 caused a marked increase in mIPSC frequency, somewhat larger than that observed during NaCN alone (Fig. 3D). We next investigated the basis for this increase in mIPSC frequency. In a Ca2+-free external solution, mIPSC frequency was significantly reduced, as previously reported (Koyama et al. 2000). mIPSC frequency, however, was still enhanced by NaCN and NaN3 application in this Ca2+-free condition. A similar poison-induced potentiation was also observed in an external solution containing Cd2+ (data not shown), which non-selectively blocks the voltage-dependent Ca2+ channels (VDCCs). These results show that the increased mIPSC frequency during metabolic inhibition is independent of Ca2+ influxes from the extracellular solution.
Presynaptic KATP channel-mediated events during metabolic inhibition
To more clearly distinguish any direct actions of metabolic inhibition on the presynaptic terminal we used the conventional whole-cell patch clamp recording technique in which our internal solution was modified to markedly reduce any postsynaptic activity during metabolic poisoning. Specifically, the pipette solution contained 2 mm ATP to block postsynaptic KATP channels (and to also prevent GABAA receptor rundown), the 150 mm K+ was replaced by 145 mm Cs+ and 5 mm TEA+ (see Methods) to further block any postsynaptic K+ channels, and EGTA was added to minimize any effects of accumulated Ca2+ during metabolic inhibition. Under these recording conditions, NaCN did not evoke any postsynaptic outward currents. Application of NaCN increased the mIPSC frequency to a degree comparable to that observed with nystatin recordings and again this facilitation was insensitive to tolbutamide (Figs 4Aa and b, and B). Glucose-free solution also gradually increased mIPSC frequency, although the effect was slower than that of NaCN application (Fig. 4Ac), and this increase in mIPSC frequency was again unaffected by additional application of tolbutamide (Fig. 4B). To rule out the possibility that tolbutamide (100 μM) might not be sufficient to block presynaptic KATP channels, we employed a higher concentration of tolbutamide (300 μM) and also investigated the effects of glibenclamide (10 μM), another sulfonylurea, which is 630 times more potent than tolbutamide at eliciting insulin secretion (Sturgess et al. 1988). Neither of these drugs had any effect on the NaCN-induced synaptic facilitation (Fig. 4B). Diazoxide, an opener of KATP channels which binds to the sulfonylurea receptor (Ammala et al. 1996), could still decrease the mIPSC frequency during metabolic inhibition (Fig. 4C). Thus, KATP channels do exist on presynaptic terminals and are still functional during metabolic inhibition. However they do not seem to be activated by acute metabolic inhibition sufficient to activate postsynaptic KATP channels.
Figure 4. Presynaptic KATP channel-mediated events during metabolic inhibition.

Data shown in this figure were obtained using the conventional whole-cell patch clamp recording mode. A, representative traces showing presynaptic facilitation by metabolic inhibitors and their insensitivity to tolbutamide (Tol) or glibenclamide (Glib). B, summary of the effect of sulfonylureas on NaCN-induced presynaptic facilitation. Each column and the associated vertical error bar represent the mean + s.e.m. of data from 22 (NaCN), 9 (+ Glib), 6 (+ Tol 100 μM) and 3 (+ Tol 300 μM) neurons. No statistically significant differences were detected. C, summary of presynaptic KATP channel-mediated effect during metabolic inhibition. Data for NaCN are copied from Fig. 4B for comparison. Each column and the associated vertical error bar represent the mean + s.e.m. of data from 22 (NaCN), 11 (NaCN+DZ), 10 (Glucose-free) and 3 (Glucose-free + Tol) neurons. P value was obtained by Student's unpaired two-tailed t test. Only treatment with NaCN and diazoxide was significantly different from NaCN alone.
Presynaptic facilitation during metabolic inhibition
The possible mechanisms for presynaptic facilitation during metabolic inhibition were also investigated. Since release of neurotransmitter is sensitive to intracellular Ca2+ concentration ([Ca2+]i), we again investigated whether Ca2+ influx from the extracellular solution was required, this time using the conventional whole-cell recording mode. The application of NaCN still increased mIPSC frequency in this Ca2+-free solution (Fig. 5Aa). A similar result was obtained using nystatin-perforated patch recordings and using combined NaCN and NaN3 as the metabolic poisons (Fig. 3D). This clearly indicates that NaCN-induced synaptic facilitation does not require Ca2+ influx from the extracellular solution. We next investigated the effects of the membrane permeant Ca2+ chelators EGTA-AM and BAPTA-AM (Naraghi, 1997). Applying EGTA-AM (10 μM) for more than 15 min had no effect on the NaCN-induced mIPSC facilitation. In contrast, application of BAPTA-AM (10 μM) for more than 15 min markedly reduced the mIPSC frequency facilitation. These results suggest that the presynaptic effects of NaCN, and probably of other metabolic inhibitors, on mIPSC frequency, is mediated via an increase in [Ca2+]i which does not depend on Ca2+ influx. By comparing the effect of ouabain (100 μM) before and during metabolic inhibition we also observed that the activity of Na+-K+ ATPase was altered during metabolic inhibition. Ouabain facilitated mIPSC frequency to 158 ± 22 % but this effect was abolished during NaCN application (97.5 ± 4.3 %; Fig. 5C). This result suggests that the observed synaptic facilitation also involves Na+-K+ ATPase, and that presynaptic [ATP]i is indeed decreased during this experimental metabolic inhibition.
Figure 5. Presynaptic facilitation during metabolic inhibition.

Data shown in this figure were obtained using the conventional whole-cell patch clamp recording mode. A, representative traces showing NaCN-induced effect in Ca2+-free solution (a) and in the BAPTA-AM-pretreated neuron (b). B, summary of effect of Ca2+-free solution and Ca2+ chelators on NaCN-induced synaptic facilitation. Data for NaCN is copied from Fig. 4B for comparison. Each column and the associated vertical error bar represents the mean + s.e.m. of data from 22 (NaCN), 6 (Ca2+-free), 4 (EGTA-AM), and 3 (BAPTA-AM) neurons. C, effect of ouabain (100 μM) before and during NaCN application. Each column and the associated vertical error bar represent the mean + s.e.m. of data from 4 neurons.
Comparative pre- and postsynaptic effects of diazoxide, a KATP channel opener
We next investigated the postsynaptic effects of diazoxide, again utilizing the nystatin-perforated patch recording technique and holding the cells at −60 mV. Similar to the effects of NaCN and/or NaN3, the application of diazoxide rapidly elicited a small outward current which ranged in amplitude from about 9 to 36.3 pA (Fig. 6A). The amplitude of this diazoxide-induced outward current was dose dependent with a half-maximal response (EC50) at 51.1 μM (Fig. 6Ba) and was almost totally blocked by tolbutamide (Fig. 6Bb). Unlike the effect of NaCN however, mIPSCs seemed to be reduced in frequency during diazoxide application, and this action was studied in more detail using conventional whole-cell recordings (below).
Figure 6. Postsynaptic KATP channel-mediated current.

A, representative traces showing postsynaptic baseline current during diazoxide (Dz) application with or without tolbutamide (Tol). Ba, concentration-response relationship for the postsynaptic outward current evoked by diazoxide. Each point, and the associated vertical bar, represents the mean ± s.e.m. of data from 4 neurons. The continuous line is a least squares fit to the Hill equation (see Methods). Dashed line shows EC50 (5.11 ± 0.63 × 10−6m). b, analysis of postsynaptic outward current during diazoxide with or without tolbutamide. Each column and the associated vertical bar represents the mean + s.e.m. of data from 4 neurons. P value was obtained using Student's unpaired two-tailed t test.
In conventional whole-cell recordings, diazoxide clearly decreased mIPSC frequency in a dose-dependent manner (with a half-maximal inhibitory concentration, IC50, of 935 nm) and this inhibition was abolished in the presence of tolbutamide (Fig. 7B). Diazoxide had no significant effect on mIPSC amplitude (Fig. 7A). These results clearly indicate that KATP channels do indeed exist on these GABAergic nerve terminals.
Figure 7. Presynaptic KATP channel-mediated inhibition.
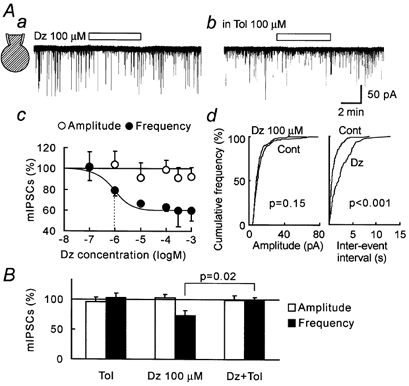
Data shown in this figure were obtained using the conventional whole-cell patch clamp recording mode. Aa and b, representative traces during diazoxide (Dz) application with or without tolbutamide (Tol) co-application. c, concentration-response relationship for the effects of diazoxide on mIPSC frequency and amplitude. Each point and the associated vertical error bar represents the mean ± s.e.m. of data from 3 to 9 neurons. The continuous line is a least squares curve fit (described in the Methods). Dashed line shows IC50 (9.35 ± 1.28 × 10−7m). d, cumulative plots of amplitude and inter-event interval before (Cont) and during diazoxide application. P value was obtained by Kolmogorov-Smirnov test. B, summary of change in mIPSC amplitude and frequency. Each column and the associated vertical error bar represents the mean + s.e.m. of data from 13 (Tol), 10 (Dz), and 12 (Dz + Tol) neurons. P value was obtained with Student's unpaired two-tailed t test.
DISCUSSION
In the present study, we used the ‘synaptic bouton’ preparation in which neurons are isolated with their presynaptic terminals attached and functional. Using nystatin-perforated patch recordings, which preserve the intracellular constituents, we could observe the activity of functional KATP channels on both the presynaptic terminals and the postsynaptic membrane at the same time. Using the more conventional open-patch whole-cell recording mode, in which we could block any postsynaptic currents, we could observe, in isolation, presynaptic activity during metabolic inhibition.
Postsynaptic effects of metabolic inhibition
A prominent outward current was observed as a result of inhibition would produce the following changes in the cell: decreased [ATP]i, a change in intracellular pH and a change in protein phosphorylation states. All of these changes have been reported to affect KATP channel activity (Baukrowitz et al. 1999; Baukrowitz & Fakler, 2000; Loussouarn et al. 2001; Xu et al. 2001). In rat locus coeruleus neurons, the postsynaptic current observed during experimental ischaemia comprised a mixture of KATP channel current and Ca2+-dependent K+ (KCa) current (Murai et al. 1997). KCa current would be expected to become more prominent during prolonged metabolic inhibition, which leads to either increased Ca2+ influx or decreased extrusion, or both, resulting in accumulation of intracellular Ca2+. In the present study we concentrated on the more acute effects of metabolic inhibition. The outward current induced by brief application of NaCN was virtually completely blocked by tolbutamide, indicating that it was due to KATP channel activation.
Diazoxide also evoked a tolbutamide-sensitive outward current. The amplitude of this current peaked at around 20 pA at a maximal dose (300 μM) of diazoxide. This value was close to the amplitude of the tolbutamide-sensitive current evoked by NaCN and/or NaN3. In a previous study in the locus coeruleus (LC), KATP channel-mediated current evoked by experimental ischaemia was much larger in amplitude (Murai et al. 1997). This may be explained by the different amount of KATP channel expressed in hippocampal neurons and LC neurons, differences in the sensitivity to ATP or differences in the extent of metabolic inhibition.
We observed two kinds of outward current during simultaneous application of NaCN and NaN3 (Fig. 5B). The tolbutamide-sensitive current component had a clear onset, a well-defined peak amplitude which was reached by about 5 min after the onset of metabolic inhibition and readily recovered after washout. In contrast, the tolbutamide-insensitive current had a rather gradual onset before more rapidly increasing towards the latter part of the 10 min metabolic inhibition. Furthermore this current was often irreversible after washout. An outward current also seen in Fig. 2A after washout of NaCN and tolbutamide may be a similar tolbutamide-insensitive current. We speculate that these tolbutamide-insensitive currents were KCa currents (Murai et al. 1997), although we made no further investigations as to their precise nature.
Direct effects on GABAA receptors
It is well known that GABAA receptors mediate current ‘run down’ upon depletion of [ATP]i (Harata et al. 1997) with the amplitude of GABAergic mIPSCs thus gradually decreasing and eventually disappearing in the absence of [ATP]i. In the present study, however, mIPSC amplitude was clearly augmented during the first 10 min of metabolic inhibition when recordings were made with the nystatin patch. Furthermore we observed that the current evoked by exogenously applied GABA was also augmented during the initial 5-10 min of metabolic inhibition before decreasing in amplitude (data not shown). In contrast, mean mIPSC amplitude during 10 min of metabolic inhibition was not affected when recorded using conventional whole-cell recordings. This suggests that the increased mIPSC amplitude is due to an increased response of GABAA receptors and not an increase in the size or number of released synaptic vesicles.
Presynaptic effects of metabolic inhibition
If metabolic inhibition activates presynaptic KATP channels in a fashion similar to that we observed for the postsynaptic KATP channels, then we would expect to observe a decrease in neurotransmitter release as a result of KATP channel-induced hyperpolarization and less Ca2+ influx through voltage-dependent Ca2+ channels (VDCCs). Indeed, application of diazoxide decreased the mIPSC frequency in a tolbutamide-sensitive manner. However, metabolic inhibition actually increased mIPSC frequency. This increase in mIPSC frequency during metabolic inhibition was not changed by concurrent application of tolbutamide or glibenclamide. A tolbutamide-insensitive increase in mIPSC frequency was also observed in response to both NaCN and NaN3. In contrast, diazoxide decreased mIPSC frequency even during NaCN-induced synaptic facilitation. This observation suggests that metabolic poisoning, sufficient to activate postsynaptic KATP channels, has little functional effect on presynaptic KATP channels.
Functional KATP channels are clearly present in presynaptic terminals (as judged by the diazoxide effects) yet do not seem to be activated during metabolic inhibition. A number of possibilities exist for the lack of tolbutamide-sensitive presynaptic effect of metabolic poisoning. Firstly, mechanically-dissociated synaptic terminals may not have ‘normal’ energy metabolism and may not use mitochondrial oxidative metabolism to generate ATP. Hence they may be relatively insensitive to NaCN. We consider this unlikely as NaCN did have some presynaptic action which was similar to that of zero glucose and combined NaCN and NaN3. Furthermore, basal presynaptic metabolism seems sufficient to maintain presynaptic ATP levels high enough to keep the presynaptic KATP closed, as application of tolbutamide had no effect on basal mIPSC frequency. Secondly, the possibility exists that presynaptic [ATP]i was not sufficiently decreased under our conditions. While we cannot definitively rule out this possibility in the absence of direct ATP measurements, it seems unlikely considering the similarity of the effects of NaCN, NaCN + NaN3 and zero glucose and the fact that the doses we employed are commonly used to deplete ATP sufficiently to activate KATP channels. The difference in ouabain-induced synaptic facilitation before and during metabolic inhibition also supports a hypothesis that presynaptic [ATP]i was indeed decreased at least enough to alter Na+-K+ ATPase activity. Thirdly, it may be that the presynaptic action of diazoxide was mediated via mitochondrial KATP channels, which have also been reported to be more sensitive to diazoxide than sarcolemmal KATP channels (Garlid et al. 1996). If this is indeed the case one would argue that these channels are not activated during mitochondrial poisoning. However, since diazoxide can still decrease mIPSC frequency during NaCN application one must still explain why they are not activated during metabolic inhibition. Finally, given the presence of presynaptic plasma membrane KATP channels, the data strongly suggest that they have a much reduced ATP sensitivity. Presumably this results from some modification of the channels’ ATP sensitivity rather than from their being composed of novel ATP-insensitive channel subtypes.
It should be recalled that KATP channels lose their activity during prolonged depletion of [ATP]i (Koyama et al. 1999a). While this rundown was not observed in postsynaptic KATP channels during the acute phase of metabolic inhibition (10-20 min after drug perfusion), it is possible that [ATP]i in presynaptic terminals was more rapidly depleted and that presynaptic KATP channels rapidly ran down and ceased their activity. However, diazoxide still could open these presynaptic KATP channels during metabolic inhibition to a degree similar to that observed in control conditions. Since KATP channels are only sensitive to pharmacological openers when they are operable (Kamouchi & Kitamura, 1994), presynaptic KATP channels were unlikely to be in a run down (or inoperable) state in our experimental conditions.
Mechanisms underlying presynaptic facilitation
Presynaptic facilitation of GABA release during metabolic inhibition was also seen in Ca2+-free external solution. In fact, the extent of mIPSC frequency facilitation during metabolic inhibition was not altered by Ca2+-free external solution. Thus, Ca2+ entry into the presynaptic terminal is not required for presynaptic facilitation during metabolic inhibition. Hypoxia directly enhances the release of synaptic vesicles in neocortical neurons (Fleidervish et al. 2001). In snake motor nerve endings, [Ca2+]i was elevated by oxidative metabolic inhibitors due to disruption of energy-dependent Ca2+ buffering (Calupca et al. 2001). In our study, chelating [Ca2+]i by application of BAPTA-AM for more than 15 min significantly inhibited the facilitatory effect of metabolic inhibition (Fig. 5B). In the locus coeruleus, NaCN evoked outward Ca2+-activated K+ currents, which were blocked by thapsigargin but not by ryanodine, indicating the involvement of inositol triphosphate-induced Ca2+ release from the intracellular Ca2+ stores (Murai et al. 1997). Thus, release of Ca2+ from intracellular Ca2+ stores, or a decrease in Ca2+ buffering and/or extrusion, could explain mIPSC frequency facilitation during metabolic inhibition. Since the frequency of spontaneous Ca2+ release from intracellular Ca2+ stores is sensitive to plasma membrane depolarization even when VDCC activity is blocked by Cd2+ (Arima et al. 2001), presynaptic membrane depolarization during metabolic inhibition may directly enhance Ca2+ release from Ca2+ stores and contribute to synaptic facilitation.
Functional relevance
There is general agreement that KATP channels in cardiac muscle and neurons have a protective effect during cell-damaging conditions, such as ischaemia. KATP channels play a significant role in preventing seizures after hypoxia in rats (Yamada et al. 2001). Activation of KATP channels in inhibitory synaptic terminals would be predicted to result in decreased release of inhibitory transmitters with a consequent further depolarization of the postsynaptic membrane. This would be counterproductive in protecting against excitotoxicity. Our present study shows that, in GABAergic nerve terminals during metabolic inhibition, presynaptic KATP channels are not involved in regulating synaptic release, at least at the same time as postsynaptic KATP channels are clearly being activated. In hippocampal slices, GABA release is enhanced by hypoxia, hypoglycaemia and ischaemia and this effect was observed both in Ca2+-containing and Ca2+-free external solutions (Saransaari & Oja, 1997). The enhanced synaptic GABA release seen in this study and in the present study, along with the hyperpolarization of the postsynaptic membrane, would all be expected to reduce excitability and to be neuroprotective. However, in the somatosensory cortex, brief periods of hypoxia not only increased the frequency of spontaneous IPSCs but also increased spontaneous EPSC frequency (Fleidervish et al. 2001). Further investigation of the specific roles of KATP channels in the nervous system should contribute to our understanding of their neuroprotective actions and may also contribute to the development of novel neuroprotective therapies.
Acknowledgments
The authors would like to thank Drs Andrew Moorhouse and Susumu Koyama for critical reading and language assistance for the manuscript. This study was supported by Grants-in-Aid for Scientific Research (No.13307003) from The Ministry of Education, Science and Culture, Japan, The Japan Health Sciences Foundation (No. 21279, Research on Brain Science), and Kyushu University Interdisciplinary Programs in Education and Projects in Research Development for N.A.
REFERENCES
- Aguilar-Bryan L, Clement JPT, Gonzalez G, Kunjilwar K, Babenko A, Bryan J. Toward understanding the assembly and structure of KATP channels. Physiological Reviews. 1998;78:227–245. doi: 10.1152/physrev.1998.78.1.227. [DOI] [PubMed] [Google Scholar]
- Akaike N, Harata N. Nystatin perforated patch recording and its applications to analyses of intracellular mechanisms. Japanese Journal of Physiology. 1994;44:433–473. doi: 10.2170/jjphysiol.44.433. [DOI] [PubMed] [Google Scholar]
- Ammala C, Moorhouse A, Ashcroft FM. The sulphonylurea receptor confers diazoxide sensitivity on the inwardly rectifying K+ channel Kir6. 1 expressed in human embryonic kidney cells. Journal of Physiology. 1996;494:709–714. doi: 10.1113/jphysiol.1996.sp021526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arima J, Matsumoto N, Kishimoto K, Akaike N. Spontaneous miniature outward currents in mechanically dissociated rat Meynert neurons. Journal of Physiology. 2001;534:99–107. doi: 10.1111/j.1469-7793.2001.00099.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashcroft FM, Gribble FM. Correlating structure and function in ATP-sensitive K+ channels. Trends in Neurosciences. 1998;21:288–294. doi: 10.1016/s0166-2236(98)01225-9. [DOI] [PubMed] [Google Scholar]
- Baukrowitz T, Fakler B. KATP channels gated by intracellular nucleotides and phospholipids. European Journal of Biochemistry. 2000;267:5842–5848. doi: 10.1046/j.1432-1327.2000.01672.x. [DOI] [PubMed] [Google Scholar]
- Baukrowitz T, Tucker SJ, Schulte U, Benndorf K, Ruppersberg JP, Fakler B. Inward rectification in KATP channels: a pH switch in the pore. EMBO Journal. 1999;18:847–853. doi: 10.1093/emboj/18.4.847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calupca MA, Prior C, Merriam LA, Hendricks GM, Parsons RL. Presynaptic function is altered in snake K+-depolarized motor nerve terminals containing compromised mitochondria. Journal of Physiology. 2001;532:217–227. doi: 10.1111/j.1469-7793.2001.0217g.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YR, Sturgeon BE, Gunther MR, Mason RP. Electron spin resonance investigation of the cyanyl and azidyl radical formation by cytochrome c oxidase. Journal of Biological Chemistry. 1999;274:24611–24616. doi: 10.1074/jbc.274.35.24611. [DOI] [PubMed] [Google Scholar]
- Fleidervish IA, Gebhardt C, Astman N, Gutnick MJ, Heinemann U. Enhanced spontaneous transmitter release is the earliest consequence of neocortical hypoxia that can explain the disruption of normal circuit function. Journal of Neuroscience. 2001;21:4600–4608. doi: 10.1523/JNEUROSCI.21-13-04600.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia De Arriba S, Franke H, Pissarek M, Nieber K, Illes P. Neuroprotection by ATP-dependent potassium channels in rat neocortical brain slices during hypoxia. Neuroscience Letters. 1999;273:13–16. doi: 10.1016/s0304-3940(99)00603-5. [DOI] [PubMed] [Google Scholar]
- Garlid KD, Paucek P, Yarov-Yarovoy V, Sun X, Schindler PA. The mitochondrial KATP channel as a receptor for potassium channel openers. Journal of Biological Chemistry. 1996;271:8796–8799. doi: 10.1074/jbc.271.15.8796. [DOI] [PubMed] [Google Scholar]
- Gioio AE, Eyman M, Zhang H, Lavina ZS, Giuditta A, Kaplan BB. Local synthesis of nuclear-encoded mitochondrial proteins in the presynaptic nerve terminal. Journal of Neuroscience Research. 2001;64:447–453. doi: 10.1002/jnr.1096. [DOI] [PubMed] [Google Scholar]
- Harata N, Wu J, Ishibashi H, Ono K, Akaike N. Run-down of the GABAA response under experimental ischaemia in acutely dissociated CA1 pyramidal neurones of the rat. Journal of Physiology. 1997;500:673–688. doi: 10.1113/jphysiol.1997.sp022052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inagaki N, Seino S. ATP-sensitive potassium channels: structures, functions, and pathophysiology. Japanese Journal of Physiology. 1998;48:397–412. doi: 10.2170/jjphysiol.48.397. [DOI] [PubMed] [Google Scholar]
- Kamouchi M, Kitamura K. Regulation of ATP-sensitive K+ channels by ATP and nucleotide diphosphate in rabbit portal vein. American Journal of Physiology. 1994;266:H1687–1698. doi: 10.1152/ajpheart.1994.266.5.H1687. [DOI] [PubMed] [Google Scholar]
- Koyama S, Jin YH, Akaike N. ATP-sensitive and Ca2+-activated K+ channel activities in the rat locus coeruleus neurons during metabolic inhibition. Brain Research. 1999a;828:189–192. doi: 10.1016/s0006-8993(99)01303-7. [DOI] [PubMed] [Google Scholar]
- Koyama S, Kubo C, Rhee JS, Akaike N. Presynaptic serotonergic inhibition of GABAergic synaptic transmission in mechanically dissociated rat basolateral amygdala neurons. Journal of Physiology. 1999b;518:525–538. doi: 10.1111/j.1469-7793.1999.0525p.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyama S, Matsumoto N, Kubo C, Akaike N. Presynaptic 5-HT3 receptor-mediated modulation of synaptic GABA release in the mechanically dissociated rat amygdala neurons. Journal of Physiology. 2000;529:373–383. doi: 10.1111/j.1469-7793.2000.00373.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loussouarn G, Pike LJ, Ashcroft FM, Makhina EN, Nichols CG. Dynamic sensitivity of ATP-sensitive K+. channels to ATP. Journal of Biological Chemistry. 2001;276:29098–29103. doi: 10.1074/jbc.M102365200. [DOI] [PubMed] [Google Scholar]
- Murai Y, Ishibashi H, Koyama S, Akaike N. Ca2+-activated K+ currents in rat locus coeruleus neurons induced by experimental ischemia, anoxia, and hypoglycemia. Journal of Neurophysiology. 1997;78:2674–2681. doi: 10.1152/jn.1997.78.5.2674. [DOI] [PubMed] [Google Scholar]
- Murase K, Randic M, Shirasaki T, Nakagawa T, Akaike N. Serotonin suppresses N-methyl-d-aspartate responses in acutely isolated spinal dorsal horn neurons of the rat. Brain Research. 1990;525:84–91. doi: 10.1016/0006-8993(90)91323-9. [DOI] [PubMed] [Google Scholar]
- Naraghi M. T-jump study of calcium binding kinetics of calcium chelators. Cell Calcium. 1997;22:255–268. doi: 10.1016/s0143-4160(97)90064-6. [DOI] [PubMed] [Google Scholar]
- Noma A. ATP-regulated K+ channels in cardiac muscle. Nature. 1983;305:147–148. doi: 10.1038/305147a0. [DOI] [PubMed] [Google Scholar]
- Ohno-Shosaku T, Sawada S, Yamamoto C. ATP-sensitive K+ channel activators suppress the GABAergic inhibitory transmission by acting on both presynaptic and postsynaptic sites in rat cultured hippocampal neurons. Neuroscience Letters. 1993;159:139–142. doi: 10.1016/0304-3940(93)90818-6. [DOI] [PubMed] [Google Scholar]
- Rhee JS, Ishibashi H, Akaike N. Calcium channels in the GABAergic presynaptic nerve terminals projecting to meynert neurons of the rat. Journal of Neurochemistry. 1999;72:800–807. doi: 10.1046/j.1471-4159.1999.0720800.x. [DOI] [PubMed] [Google Scholar]
- Saransaari P, Oja SS. Enhanced GABA release in cell-damaging conditions in the adult and developing mouse hippocampus. International Journal of Developmental Neuroscience. 1997;15:163–174. doi: 10.1016/s0736-5748(97)80001-9. [DOI] [PubMed] [Google Scholar]
- Sturgess NC, Kozlowski RZ, Carrington CA, Hales CN, Ashford ML. Effects of sulphonylureas and diazoxide on insulin secretion and nucleotide-sensitive channels in an insulin-secreting cell line. British Journal of Pharmacology. 1988;95:83–94. doi: 10.1111/j.1476-5381.1988.tb16551.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun XD, Lee EW, Wong EH, Lee KS. ATP-sensitive potassium channels in freshly dissociated adult rat striatal neurons: activation by metabolic inhibitors and the dopaminergic receptor agonist quinpirole. Pflügers Archiv. 2000;440:530–547. doi: 10.1007/s004240000322. [DOI] [PubMed] [Google Scholar]
- Watts AE, Hicks GA, Henderson G. Putative pre- and postsynaptic ATP-sensitive potassium channels in the rat substantia nigra in vitro. Journal of Neuroscience. 1995;15:3065–3074. doi: 10.1523/JNEUROSCI.15-04-03065.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Cui N, Yang Z, Wu J, Giwa LR, Abdulkadir L, Sharma P, Jiang C. Direct activation of cloned KATP channels by intracellular acidosis. Journal of Biological Chemistry. 2001;276:12898–12902. doi: 10.1074/jbc.M009631200. [DOI] [PubMed] [Google Scholar]
- Yamada K, Ji JJ, Yuan H, Miki T, Sato S, Horimoto N, Shimizu T, Seino S, Inagaki N. Protective role of ATP-sensitive potassium channels in hypoxia-induced generalized seizure. Science. 2001;292:1543–1546. doi: 10.1126/science.1059829. [DOI] [PubMed] [Google Scholar]
- Ye GL, Leung CK, Yung WH. Pre-synaptic effect of the ATP-sensitive potassium channel opener diazoxide on rat substantia nigra pars reticulata neurons. Brain Research. 1997;753:1–7. doi: 10.1016/s0006-8993(96)01473-4. [DOI] [PubMed] [Google Scholar]
- Zawar C, Neumcke B. Differential activation of ATP-sensitive potassium channels during energy depletion in CA1 pyramidal cells and interneurones of rat hippocampus. Pflügers Archiv. 2000;439:256–262. doi: 10.1007/s004249900184. [DOI] [PubMed] [Google Scholar]
- Zawar C, Plant TD, Schirra C, Konnerth A, Neumcke B. Cell-type specific expression of ATP-sensitive potassium channels in the rat hippocampus. Journal of Physiology. 1999;514:327–341. doi: 10.1111/j.1469-7793.1999.315ae.x. [DOI] [PMC free article] [PubMed] [Google Scholar]