

Abstract
GABAA receptor-mediated presynaptic depolarization is believed to induce presynaptic inhibition of excitatory synaptic transmission. We report here the functional roles of presynaptic GABAA receptors in glycinergic transmission of the rat spinal cord. In mechanically dissociated rat sacral dorsal commissural nucleus (SDCN) neurons attached with native glycinergic and GABAergic nerve terminals, glycinergic spontaneous inhibitory postsynaptic currents (sIPSCs) were isolated from a mixture of both glycinergic and GABAergic sIPSCs by perfusing the SDCN nerve cell body with ATP-free internal solution. Under such experimental conditions, exogenously applied muscimol (0.5 μM) depolarized glycinergic presynaptic nerve terminals and significantly increased glycinergic sIPSC frequency to 542.7 ± 47.3 % of the control without affecting the mean current amplitude. The facilitatory effect of muscimol on sIPSC frequency was completely blocked by bicuculline (10 μM) or SR95531 (10 μM), selective GABAA receptor antagonists. This muscimol-induced presynaptic depolarization was due to a higher intraterminal Cl− concentration, which is maintained by a bumetanide-sensitive Na-K-Cl cotransporter. On the contrary, when electrically evoked, this muscimol-induced presynaptic depolarization was found to decrease the action potential-dependent glycine release evoked by focal stimulation of a single terminal. The results suggest that GABAA receptor-mediated presynaptic depolarization has two functional roles: (1) presynaptic inhibition of action potential-driven glycinergic transmission, and (2) presynaptic facilitation of spontaneous glycinergic transmission.
GABA is a primary inhibitory neurotransmitter throughout the mammalian CNS. GABAA receptor-mediated inhibitory actions are generally due to an increase in the Cl− conductance leading to direct postsynaptic hyperpolarization. In sensory afferent terminals, the activation of presynaptic GABAA receptors also induces presynaptic inhibition to reduce the electrically stimulated neurotransmitter release resulting from inactivation of Na+ channels and/or shunt of the presynaptic membrane by depolarizing the presynaptic terminals (Segev, 1990; Stuart & Redman, 1992; Graham & Redman, 1994; Cattaert & El Manira, 1999). Such a GABAA receptor-mediated presynaptic depolarization is based on a higher intracellular Cl− concentration ([Cl−]i) which results from a Na-K-Cl cotransporter (NKCC) (for review, Alvarez-Leefmans, 1990; Kaila, 1994; Russell, 2000). Electron and confocal microscope studies have also provided some evidence for the existence of GABAergic axo-axonic synapses or GABA immunoreactivity on the excitatory nerve terminals or en passant boutons (Atwood et al. 1984; Lamotte d'Incamps et al. 1998). These observations support the idea that GABAA receptors on the excitatory nerve axon or terminal represent morphological substrates for presynaptic inhibition.
Most spinal neurons receive both GABAergic and glycinergic inputs (Todd & Sullivan, 1990; Schneider & Fyffe, 1992; Bohlhalter et al. 1994). In addition, there is much convincing evidence that GABAergic boutons make axo-axonic synapses with other presynaptic nerve terminals in the spinal cord (Todd et al. 1995; Maxwell et al. 1997). These results suggest the possibility that GABA, through GABAA receptors, can modulate the inhibitory GABAergic or glycinergic neurotransmissions at presynaptic sites. To test this hypothesis, we utilized mechanically isolated sacral dorsal commissural nucleus (SDCN) neurons retaining both glycinergic and GABAergic nerve terminals. In such SDCN neurons, we isolated the glycinergic sIPSCs from a mixture of both glycinergic and GABAergic sIPSCs without using bicuculline, a selective GABAA receptor antagonist. This procedure allowed us to selectively focus on the functional action of presynaptic GABAA receptors on glycinergic transmission. In addition, we also attempted to determine whether GABAA receptor-mediated presynaptic depolarization could induce presynaptic inhibition for glycinergic transmission elicited by the selective focal electrical stimulation of a single glycinergic bouton.
METHODS
All experiments conformed to the guiding principles for the care and use of animals approved by the Council of the Physiological Society of Japan and all efforts were made to minimize both the number of animals used and their suffering.
Preparation
Wistar rats (10-12 days old) were decapitated under pentobarbital anaesthesia (50 mg kg−1, i.p.). A segment of the lumbosacral (L6-S2) spinal cord was dissected and transversely sliced at a thickness of 350 μm using a microslicer (VT1000S; Leica, Nussloch, Germany). Slices containing the sacral dorsal commissural nucleus (SDCN) were kept in a control incubation medium (see below) saturated with 95 % O2 and 5 % CO2 at room temperature (21-24 °C) for at least 1 h before mechanical dissociation. For dissociation, slices were transferred into a 35 mm culture dish (Primaria 3801; Becton Dickinson, Rutherford, NJ, USA) containing the standard external solution (see below). The region of the SDCN was identified under a binocular microscope (× 20, SMZ-1; Nikon, Tokyo, Japan). The details of mechanical dissociation have previously been described (Rhee et al. 1999). Briefly, mechanical dissociation was accomplished using a custom-built vibration device and a fire-polished glass pipette oscillating at about 40-60 Hz (0.1-0.2 mm). The tip of the fire-polished glass pipette was lightly placed on the surface of the SDCN region using a micromanipulator. The tip of the glass pipette was then vibrated horizontally for about 2 min. The slices were next removed, and mechanically dissociated neurons (5-10 neurons per dish) were allowed to settle and adhere to the bottom of the dish for 15 min. Such neurons undergoing dissociation retained a short portion of their proximal dendrites (Fig. 1A).
Figure 1. Mechanically dissociated SDCN neurons and the method for detection of sIPSCs.
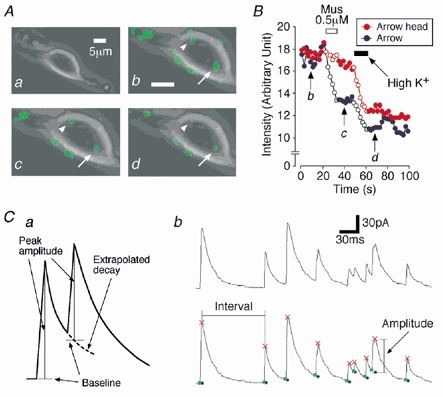
A, representative photographs of a mechanically dissociated SDCN neuron before loading FM 1-43 (a), after loading FM 1-43 (b), after application of 0.5 μM muscimol (c) and after application of 20 mm K+ solution (d). In b, c and d, the fluorescence signals were superimposed on a. B, typical time courses of the fluorescence intensities of two presynaptic nerve terminals during application of muscimol or high K+ solution. Red and blue points were analysed from arrowhead and arrow in A, respectively. Open circles represent periods during the drug application. C, schematic illustration of the sIPSC detection method (a), and a typical trace of sIPSCs at expanded time scale (upper) and its detection result (lower) (b). Red: peak of events; green: onset; blue: calculated baseline.
Electrical measurements
All electrical measurements were performed using the conventional whole-cell patch recording mode at a holding potential of −30 to −40 mV using a patch clamp amplifier (CEZ-2300; Nihon Kohden, Tokyo, Japan), except when examining I-V relationships. Patch pipettes were made from borosilicate capillary glass (1.5 mm o.d., 0.9 mm i.d.; G-1.5; Narishige, Tokyo, Japan) in two stages on a vertical pipette puller (PB-7; Narishige). The resistance of the recording pipettes filled with internal solution was 5-6 MΩ. Electrode capacitance and liquid junction potential were compensated, but the series resistance was not. Neurons were viewed under phase contrast on an inverted microscope (×400, Diaphot; Nikon). Current and voltage were continuously monitored on an oscilloscope (VC-60 23; Hitachi) and a pen recorder (RECTI-HORIT-8K; Sanei, Tokyo, Japan) and recorded on a digital-audio tape recorder (RD-120TE; TEAC). The membrane currents were filtered at 1 kHz (E-3201A Decade Filter; NF Electronic Instruments, Tokyo, Japan), digitized at 4 kHz, and stored on a computer equipped with pCLAMP 8.0 (Axon Instruments). When recording, 10 mV hyperpolarizing step pulses (30 ms in duration) were periodically delivered to monitor the access resistance. All experiments were performed at room temperature (21-24 °C).
Identification and focal stimulation of a single bouton
Identification and focal stimulation of single boutons were performed as described in our previous report (Akaike et al. 2002). Briefly, FM 1-43 (10 μM) in 45 mm high K+ solution, in which 40 mm NaCl in a standard external solution was replaced with equimolar KCl, was perfused across a dissociated single neuron for 30 s, and then washed out with a standard external solution. The fluorescent spots indicating the existence of putative presynaptic nerve terminals were visualized with an inverted microscope equipped with barrier filters (excitation of 390-490 nm, long-pass filter of 590 nm). Fluorescence imaging was recorded by a CCD camera (CoolSNAP, Photometrics, AZ, USA) and the fluorescence intensity was measured by Lumina Vision software (Mitani Corporation, Chiba, Japan) (Fig. 1A).
The stimulation pipette was made by the same method as described for the recording pipette. Its inner diameter was 0.5-0.6 μm. Short negative pulses (5-10 μA, 100 μs in duration) at 0.2 Hz through the glass pipette filled with a standard external solution were applied using a stimulus isolation unit (SS-202J; Nihon Koden). The stimulation pipette was placed close to the surface of a single bouton identified with FM 1-43. The evoked current was elicited in an ‘all-or-none’ fashion when the stimulation pipette was positioned just above a bouton, and was highly sensitive to 0.3 μM tetrodotoxin (TTX) (Akaike et al. (2002).
Data analysis
Spontaneous inhibitory postsynaptic currents (sIPSCs) were detected and analysed in pre-set epochs before, during and after each experimental condition, using the MiniAnalysis Program (Synaptosoft, NJ, USA). Briefly, spontaneous events were automatically screened using an amplitude threshold of 5 pA and were then visually accepted or rejected based upon the rise (0.5-1.5 ms) and decay times (8.0-20.0 ms). In complex waveforms where the event starts to rise before the previous event goes back to the baseline, the baseline for the second event was estimated by extrapolating the decay of the first peak at the location of the double peak. Then the peak amplitude of the second event was determined from this calculated baseline but not from the onset point of the event (Fig. 1C). The average values of sIPSC frequency and amplitude during the control period (10-15 min) were calculated, and the frequency and amplitude of all the events during agonist application (1-2 min) were normalized to these values. The effect of the agonist was quantified as a percentage increase in sIPSC frequency compared to the control value. The amplitude of evoked IPSCs (eIPSCs) was analysed using pCLAMP 8.0. Numerical values were reported as means ± standard error of the mean (s.e.m.) using values normalized to the control levels. Possible differences in the amplitude and frequency distribution were tested by Student's paired two-tailed t test using their absolute values but not normalized ones. Values of P < 0.05 were considered to be significant. On the other hand, the inter-event intervals and amplitudes of a large number of sIPSCs obtained from the same neuron were examined by constructing cumulative probability distributions and compared using the Kolmogorov-Smirnov (K-S) test with StatView software (SAS Institute, Inc., Cary, NC, USA).
Solutions
The incubation medium consisted of (mm): 124 NaCl, 5 KCl, 1.2 KH2PO4, 24 NaHCO3, 2.4 CaCl2, 1.3 MgSO4 and 10 glucose saturated with 95 % O2 and 5 % CO2. The pH was about 7.45. The standard external solution consisted of (mm): 150 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 10 glucose and 10 Hepes. The Ca2+-free external solution consisted of (mm): 150 NaCl, 5 KCl, 5 MgCl2, 2 EGTA, 10 glucose and 10 Hepes. The Na+-free external solution consisted of (mm): 150 N-methyl-d-glucamine-Cl, 5 KCl, 2 CaCl2, 1 MgCl2, 10 glucose and 10 Hepes. These external solutions were adjusted to a pH of 7.4 with Tris-base. For recording sIPSCs, external solutions contained 10 μM 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX) and 20 μM dl-2-amino-5-phosphonovaleric acid (AP5) to block glutamatergic currents. The internal (patch-pipette) solution for the whole-cell patch recording consisted of (mm) 145 caesium methanesulfonate, 5 TEA-Cl, 5 CsCl, 2 EGTA and 10 Hepes, and was adjusted to a pH of 7.2 with Tris-base. For the depression of the postsynaptic GABAA response, most experiments were performed with the above internal solution, which did not contain ATP (Shirasaki et al. 1992; Harata et al. 1997; Jang et al. 2001).
Drugs
The drugs used in the present study included TTX, bicuculline, strychnine, CNQX, AP5, EGTA, muscimol, diazepam, bumetanide, cis-aminocrotonic acid (CACA), baclofen and 2-[3-carboxypropyl-3-amino-6-[4-methoxyphenyl]pyridazinium bromide (SR95531) from Sigma (St Louis, MO, USA), and FM 1-43 (Molecular probes, OR, USA). CNQX, bicuculline and bumetanide were dissolved in dimethyl sulfoxide at 10 mm as a stock solution. All solutions containing drugs were applied by the ‘Y-tube system’ for rapid solution exchange (Akaike & Harata, 1994).
RESULTS
Mechanically dissociated SDCN neurons preserve the functional presynaptic nerve terminals
After brief mechanical dissociation of the SDCN region, we found that neurons were bipolar (Fig. 1Aa) or triangular in shape (10-15 μm in somatic diameter). To identify the functional presynaptic nerve terminals, we applied FM 1-43 to dissociated SDCN neurons (Fig. 1Ab). Note that several fluorescent spots regarded as the putative presynaptic nerve terminals were located on the soma and proximal dendrites of a neuron. When 0.5 μM muscimol was applied to a neuron, one of the fluorescent spots (arrow in Fig. 1Ac) was quickly destained but the other (arrowhead in Fig. 1Ac) was not (Fig. 1B). However, these two fluorescent spots were further destained by subsequent addition of 20 mm K+ solution (Fig. 1Ad and B). The results suggest that mechanically dissociated SDCN neurons preserve the functional presynaptic nerve terminals which are sensitive to the stimulation of muscimol or high K+ solution.
Separation of glycinergic sIPSCs from a mixture of both glycinergic and GABAergic sIPSCs
To investigate whether exogenously applied GABA influences the glycinergic transmission, glycinergic sIPSCs were isolated from GABAergic ones by perfusing the SDCN nerve cell body with ATP-free internal solution, which rapidly induces a selective rundown of the GABAA response (Shirasaki et al. 1992; Harata et al. 1997; Jang et al. 2001). Note that under such experimental conditions the presynaptic nerve terminals maintain the native intraterminal environment. In SDCN neurons well perfused with ATP-free pipette solution, the spontaneous outward currents were recorded at a holding potential of −30 mV in the presence of both 10 μM CNQX and 20 μM AP5. By about 20 min after membrane rupture, these sIPSCs were a mixture of both GABAergic and glycinergic ones, since the cumulative application of 1 μM strychnine and 10 μM bicuculline substantially eliminated all sIPSCs (Fig. 2A and B). However, sIPSCs at about 50 min after membrane rupture were completely eliminated by adding only strychnine (Fig. 2A and B). These results clearly indicate that most of the sIPSCs are glycinergic about 50 min after membrane rupture, and that the ATP-free pipette solution does not affect glycinergic sIPSCs. Under such experimental conditions, the reversal potential for sIPSCs estimated from the current-voltage (I-V) relationship was about −64.8 mV (n = 4, data not shown), which was consistent with the theoretical Cl− equilibrium potential (ECl) of −69.9 mV calculated by the Nernst equation using 161 mm [Cl−]o and 10 mM [Cl−]i.
Figure 2. Separation of glycinergic sIPSCs.
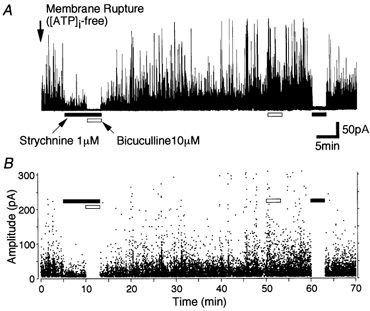
A, typical trace of sIPSCs obtained from a single SDCN neuron at a holding potential, VH, of −30 mV. The pipette solution did not contain ATP. B, all points scatter plots of sIPSCs obtained from A. Each point represents a single IPSC. Note that the cumulative application of strychnine (1 μM) and bicuculline (10 μM) eliminated sIPSCs 10 min after membrane rupture, whereas 50 min after membrane rupture, bicuculline itself did not affect sIPSCs but strychnine completely blocked all sIPSCs.
On the other hand, we also tested the rundown of the GABAA receptor-mediated postsynaptic currents. The postsynaptic responses induced by the exogenous application of 0.5 μM muscimol, a GABAA receptor agonist, were gradually attenuated and substantially occluded within 50 min of the membrane rupture (data not shown). In all subsequent experiments, muscimol and related drugs were applied at least 50 min after the membrane rupture in order to completely abolish the contamination of both GABAergic sIPSCs and postsynaptic GABA currents.
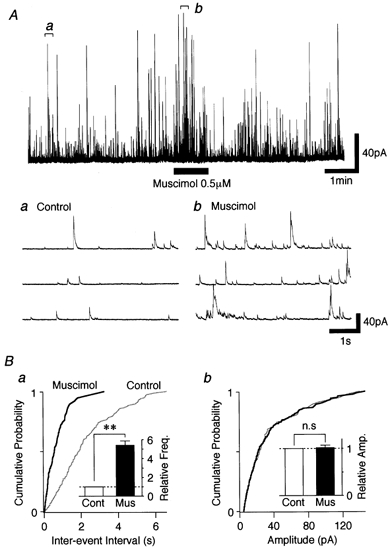
GABAergic modulation of glycinergic sIPSCs
In most of the neurons tested (107 of 131 neurons), 0.5 μM muscimol greatly increased glycinergic sIPSC frequency to 542.7 ± 47.3 % of the control (n = 10, P < 0.01) without affecting the mean current amplitude (101.9 ± 5.5 % of the control, P = 0.901) (Fig. 3A and B). Figure 3Ba and b shows cumulative probability distributions of the inter-event interval and amplitude of glycinergic sIPSCs, respectively. Muscimol significantly shifted the distribution of sIPSC frequency to the left but did not affect the amplitude, thus suggesting that muscimol acts presynaptically to increase the probability of spontaneous glycine release.
Figure 3. Muscimol acts presynaptically on the glycinergic nerve terminals.
A, typical trace of glycinergic sIPSCs observed before, during and after application of 0.5 μM muscimol. Insets (a and b) represent the regions indicated in the upper trace with an expanded time scale. B, cumulative probability plots for the inter-event interval (a; P < 0.01, K-S test) and amplitude (b; P = 0.46, K-S test) of glycinergic sIPSCs shown in A; 603 events for the control and 328 events for muscimol were plotted. Insets, each bar represents the mean ± s.e.m. (n = 12), normalized to the control. Cont: control; Mus: muscimol; ** P < 0.01; n.s: not significant.
To confirm the involvement of presynaptic GABAA receptors in an increase of glycinergic sIPSC frequency, we tested the effect of diazepam, a GABAA receptor modulator, on glycinergic sIPSCs. As predicted, 1 μM diazepam increased the facilitatory action of muscimol on glycinergic sIPSCs (Fig. 4Aa and b, and B). In the presence of diazepam, 0.5 μM muscimol further increased sIPSC frequency to 718.3 ± 107.8 % of the diazepam condition (n = 5, P < 0.01) without affecting the mean current amplitude (Fig. 4B). Muscimol action on glycinergic sIPSC frequency was completely blocked in the presence of 10 μM bicuculline (data not shown) or 10 μM SR95531 (n = 5), specific GABAA receptor antagonists (Fig. 4Ac and B). Baclofen (30 μM), a selective GABAB receptor agonist, reduced the sIPSC frequency to 51.3 ± 3.8 % of the control (n = 9, P < 0.01) without affecting the mean current amplitude (Fig. 4B). However, CACA (10 μM, n = 6), a selective GABAC receptor agonist, had no effect (Fig. 4B). In postnatal 30-day-old SDCN neurons, on the other hand, we also found that 0.5 μM muscimol increased glycinergic sIPSC frequency to 441.3 ± 75.4 % of the control (n = 4, P < 0.05). These results clearly indicate that an increase in sIPSC frequency results from GABAA receptor activation of the glycinergic presynaptic nerve terminals.
Figure 4. GABAA receptors are responsible for glycinergic sIPSC facilitation.
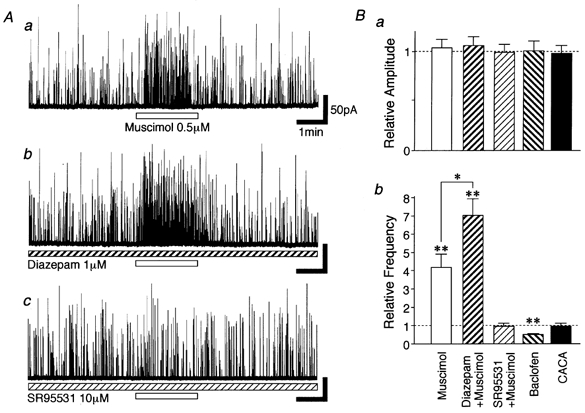
A, typical traces of glycinergic sIPSCs observed before, during and after application of 0.5 μM muscimol in the absence of (a) and in the presence of (b) 1 μM diazepam, and in the presence of 10 μM SR95531 (c). All traces were recorded from the same neuron. B, each bar for amplitude (a) and frequency (b was normalized) to the control. Muscimol (n = 5), diazepam + muscimol (n = 5), SR95531 + muscimol (n = 5), 30 μM baclofen (n = 9), 10 μM CACA (n = 6). * P < 0.05, ** P < 0.01.
Muscimol depolarizes glycinergic presynaptic nerve terminals
We examined which mechanisms are involved in the GABAA receptor-mediated sIPSC frequency facilitation. Firstly, the effect of Cd2+, a general voltage-dependent Ca2+ channel (VDCC) blocker, on muscimol-induced facilitation of sIPSC frequency was tested. The application of 100 μM Cd2+ itself reduced the frequency of sIPSCs to 59.3 ± 8.7 % of the control (n = 5, P < 0.01) without affecting the mean current amplitude (90.9 ± 5.9 %, n = 5, P = 0.13) (Fig. 5B). In the presence of Cd2+, muscimol action on sIPSC frequency (Fig. 5Aa) was completely occluded to 112.6 ± 11.7 % of the Cd2+ condition (n = 5, P = 0.24, Fig. 5Ab and B). Since Cd2+ itself is known to block GABAA receptors (Kumamoto & Murata, 1995; Fisher & Macdonald, 1998), the effect of Ca2+-free external solution on GABAA receptor-mediated sIPSC frequency facilitation was also tested. In a Ca2+-free external solution, sIPSC frequency was greatly reduced to 53.5 ± 6.1 % (n = 6, P < 0.01) of the control without affecting the mean current amplitude (90.4 ± 7.2 %, n = 6, P = 0.22) (Fig. 5C). The facilitatory effect of muscimol on sIPSC frequency was also completely occluded to 106.7 ± 5.8 % of the Ca2+-free condition (n = 6, P = 0.45, Fig. 5Ac and C). These results suggest that muscimol-induced sIPSC frequency facilitation is mediated by Ca2+ influx from the extracellular space via VDCC activation.
Figure 5. Muscimol-induced sIPSC facilitation is mediated by Ca2+ influxes through VDCCs.
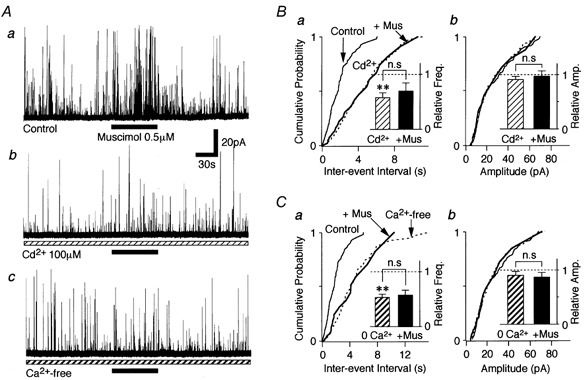
A, typical traces of glycinergic sIPSCs observed before, during and after adding 0.5 μM muscimol in a standard solution (a), in the presence of 100 μM Cd2+ (b) and in a Ca2+-free external solution (c). All traces were obtained from the same neuron. B, cumulative probability plots for inter-event interval (a; P = 0.68) and amplitude (b; P = 0.17) of sIPSCs shown in Aa and b; 402 events for control, 335 events for Cd2+ and 28 events for muscimol were plotted. Insets, all columns represent the mean of 5 neurons and are normalized to the respective control. C, cumulative probability plots for the inter-event interval (a; P = 0.15) and amplitude (b; P = 0.33) of sIPSCs shown in Aa and c; 402 events for the control, 225 events for Ca2+-free and 26 events for muscimol were plotted. Insets, each bar is the mean of 6 neurons and normalized to the control. 0 Ca2+: Ca2+-free; ** P < 0.01; n.s: not significant.
We next tested whether muscimol-induced sIPSC frequency facilitation results from the direct VDCC opening or occurs via the activation of TTX-sensitive Na+ channels. TTX (0.3 μM) also significantly decreased the glycinergic sIPSC frequency to 56.9 ± 7.7 % of the control (P < 0.01, n = 8) without affecting the mean current amplitude (91.5 ± 6.0 % of the control, P = 0.25). This suggests that TTX-sensitive Na+ channels contribute to the generation of glycinergic sIPSCs. In the presence of TTX however, muscimol could still facilitate glycinergic sIPSC frequency to 272.7 ± 20.4 % of the TTX condition (n = 8, P < 0.01, Fig. 6A and B), but this facilitation ratio was significantly lower than the control ratio (P < 0.05, ANOVA test). These results suggest that muscimol-induced depolarization activates TTX-sensitive Na+ channels and, in part, directly activates VDCCs to facilitate glycine release.
Figure 6. Muscimol-induced sIPSC facilitation in the presence of TTX.
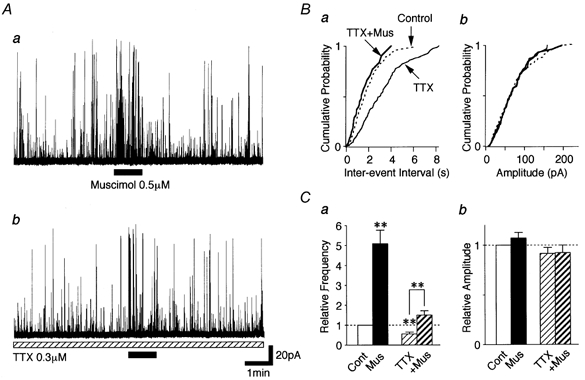
A, typical traces of glycinergic sIPSCs observed before, during and after adding 0.5 μM muscimol to a standard solution (a), and in the presence of 0.3 μM TTX (b). Both traces were obtained from the same neuron. B, cumulative probability plots for the inter-event interval (a; P < 0.01) and amplitude (b; P = 0.41) of sIPSCs shown in A; 458 events for the control, 346 events for TTX and 61 events for TTX plus muscimol were plotted. C, each bar is the mean of 8 neurons and normalized to the control. ** P < 0.01.
The involvement of NKCC in GABAA receptor-mediated presynaptic depolarization
Since muscimol-induced facilitation of sIPSC frequency seems to be mediated by presynaptic depolarization, the activation of presynaptic GABAA receptors may depolarize the glycinergic nerve terminals. This suggests the possibility that the [Cl−]i of presynaptic nerve terminals might be maintained at higher levels than predicted for a passive distribution. In immature CNS neurons or peripheral sensory neurons, the NKCC has been reported to play a pivotal role in the generation of higher [Cl−]i and GABA-induced depolarization (Alvarez-Leefmans, 1990; Plotkin et al. 1997; Clayton et al. 1998; Russell, 2000). We thus investigated the effect of bumetanide, a selective NKCC blocker (Haas, 1989; Xu et al. 1994), on the muscimol-induced facilitation of sIPSC frequency.
Muscimol action on sIPSC frequency was gradually attenuated in the presence of 10 μM bumetanide (Fig. 7A and B), in which the facilitation ratios were reduced from 445.7 ± 89.1 % for the first application to 227.4 ± 68.1 and 179.4 ± 46.4 % for the second and third applications, respectively (P < 0.05, n = 6, Fig. 7B). On the other hand, the facilitatory effect of muscimol also slowly recovered after washout of bumetanide (258.1 ± 84.7 for 10 min and 444.8 ± 135.9 % for 20 min, respectively, Fig. 7A and B). However, these slow effects of bumetanide were probably not due to the direct blockade of presynaptic GABAA receptors, since 10 μM bumetanide itself did not affect GABAergic sIPSC amplitude or frequency (data not shown).
Figure 7. Effects of NKCC blocker and Na+-free external solution on muscimol-induced sIPSC facilitation.
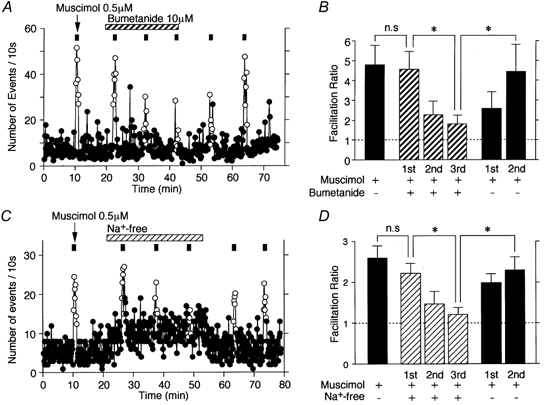
A, typical time course of the event frequency of sIPSCs before, during and after application of 0.5 μM muscimol (○, presence of muscimol; •, absence of muscimol) in the standard external solution with or without 10 μM bumetanide. The number of events for every 10 s time interval is plotted. B, facilitation ratios obtained by application of 0.5 μM muscimol with or without bumetanide. Each bar is the mean of 6 neurons and normalized to the control. The dotted line indicates the respective control. * P < 0.05. C, typical time course of the event frequency of sIPSCs before, during and after the application of 0.5 μM muscimol (○, presence of muscimol; •, absence of muscimol) in Na+-free external solution. The number of events for every 10 s time interval is plotted. D, facilitation ratios obtained by the application of 0.5 μM muscimol in the standard external solution or in Na+-free external solution. Each bar is the mean of 5 neurons and normalized to the control. The dotted line indicates the respective control. * P < 0.05. Note that the muscimol-induced facilitation of sIPSC frequency gradually decreased in the presence of bumetanide or in Na+-free external solution.
Since NKCC carries Na+, K+, and 2Cl− simultaneously and electroneutrally, many studies regarding the function of NKCC have been performed by removing such ions from the external solution (Alvarez-Leefmans, 1990; Rohrbough & Spitzer, 1996; Altamirano et al. 1999). To confirm the involvement of NKCC in the maintenance of higher [Cl−]i, we investigated whether or not the action of muscimol on sIPSC frequency is maintained in a Na+-free external solution. Since muscimol could directly activate VDCCs (Fig. 6), all experiments were performed in the presence of 0.3 μM TTX to exclude the recruitment of Na+ channels. In the presence of TTX, muscimol facilitated sIPSC frequency to 259.1 ± 29.5 % of the TTX condition (Fig. 7C). As predicted, the action of muscimol on sIPSC frequency was also gradually attenuated in the Na+-free external solution (Fig. 7C and D), in which the facilitation ratios were reduced from 221.6 ± 24.8 % for the first application to 146.8 ± 30.8 and 121.7 ± 15.8 % for the second and third applications, respectively (P < 0.05, n = 5, Fig. 7D). On the other hand, the action of muscimol quickly recovered after washout of the Na+-free external solution (198.6 ± 21.6 for 10 min and 229.7 ± 31.4 % for 20 min, respectively, Fig. 7C and D). On the other hand, the replacement of standard external solution with Na+-free external solution gradually increased the sIPSC frequency to 179.4 ± 46.4 % of the control (P < 0.05, n = 5, Fig. 7C). This increase in sIPSC frequency might be due to a Na+-Ca2+ exchanger operating in a reverse mode (Fontana et al. 1995).
Effect of GABAA receptor-mediated presynaptic depolarization on glycinergic eIPSCs evoked by focal electrical stimulation of a single bouton
We tested whether GABAA receptor-mediated presynaptic depolarization induces presynaptic inhibition of electrically evoked glycinergic transmission. When focal stimulating pulses were applied to a single glycinergic nerve terminal (bouton) visualized with FM 1-43 (Fig. 8Aa), eIPSCs resulting from a single synapse were observed and reversibly blocked by adding 1 μM strychnine (n = 6, Fig. 8Ab), thus indicating that the eIPSCs are glycinergic but not GABAergic. Under such experimental conditions, muscimol (5 μM) significantly reduced eIPSC amplitude to 30.1 ± 13.5 % of the control in a concentration-dependent manner (n = 6, P < 0.01, Fig. 8B and Ca). In addition, muscimol (5 μM) greatly increased the failure rate of the evoked glycinergic transmission (33.8 ± 8.6 % for the control and 81.6 ± 8.7 % for 5 μM muscimol, n = 6, P < 0.01, Fig. 8B and Cb). These results clearly indicate that the activation of presynaptic GABAA receptors induces presynaptic inhibition of the evoked glycinergic transmission. In the same recorded neuron, on the other hand, muscimol also facilitated sIPSC frequency without affecting the distribution of sIPSC amplitude (Fig. 8D).
Figure 8. Muscimol inhibits evoked glycinergic transmission.
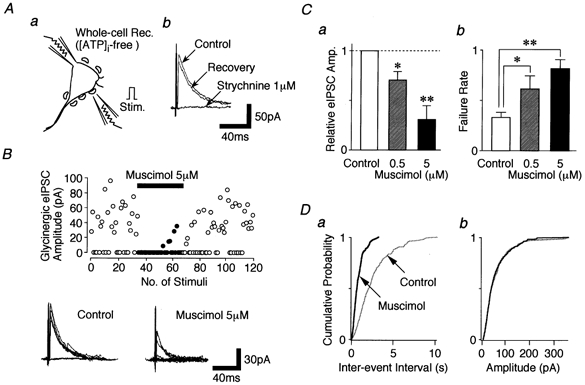
Aa, schematic illustration of the focal stimulation. b, effect of 1 μM strychnine on eIPSCs obtained from a single synapse. B, scatter plot of the glycinergic eIPSC amplitude before, during and after the application of 5 μM muscimol (○, absence of muscimol; •, presence of muscimol). Insets, typical traces of glycinergic eIPSCs in the absence (left) or presence (right) of 5 μM muscimol. Ten traces were superimposed, respectively. C, pooled data for the eIPSC amplitude (a) and its failure rate (b). Each bar is the mean of 6 neurons. Cont: control; * P < 0.05, ** P < 0.01. D, cumulative probability plots for inter-event interval (a; P < 0.01) and amplitude (b; P = 0.74) of sIPSCs recorded from B; 332 events for the control and 165 events for muscimol were plotted.
DISCUSSION
The present study has demonstrated that the activation of presynaptic GABAA receptors increases spontaneous glycine release but inhibits electrically stimulated glycine release, presumably by depolarizing presynaptic nerve terminals. In addition, this GABAA receptor-mediated depolarization is due to a higher [Cl−]i within the glycinergic nerve terminals that is regulated by the inwardly directed Cl− transporter NKCC.
Activation of GABAA receptors depolarizes the glycinergic presynaptic nerve terminals
In mature CNS neurons, GABAA receptor activation generally hyperpolarizes the neuronal membrane by increasing an inwardly directed Cl− conductance based on a lower [Cl−]i. In immature neurons, however, GABA depolarizes and excites the neuronal membrane (Obrietan & van den Pol, 1995; Chen et al. 1996). In addition, GABAA receptor-mediated depolarization is observed in adult dorsal root ganglia and sympathetic neurons as well as hippocampal neurons (Ballanyi & Grafe, 1985; Misgeld, et al. 1986; Alvarez-Leefmans, 1990). In developing neurons, either synaptically released or exogenously applied GABA induces membrane depolarization, which results in an increase of intracellular Ca2+ concentration ([Ca2+]i) (Leinekugel et al. 1995; Obrietan & van den Pol, 1995). Alternatively, GABA-induced depolarization could elicit an increase of [Ca2+]i by the activation of Ca2+ channels via the recruitment of Na+ channels (Hales et al. 1994; Owens et al. 1996). These neurons accumulate Cl− at a higher concentration than that predicted for passive distribution, and GABA equilibrium potential (EGABA) is also more positive than the resting membrane potential (Vm). As a result, the depolarizing action of GABA is believed to result from activation of GABAA receptors, with a consequent efflux of Cl−, which transiently drives Vm toward EGABA. In the present study, the action of muscimol on glycinergic sIPSC frequency may be closely dependent on the Ca2+ influx passing through VDCCs, since exposure to either Cd2+ or Ca2+-free external solution completely occluded the muscimol-induced sIPSC frequency facilitation. On the other hand, muscimol increased sIPSC frequency even in the presence of TTX, although its facilitation ratio was relatively small. In addition, muscimol still increased the sIPSC frequency even in a Na+-free external solution. These results suggest that muscimol-induced depolarization is large enough to surpass the threshold for activation of VDCCs, and that the consequent Ca2+ influx through VDCCs are responsible for muscimol-induced sIPSC frequency facilitation.
Intraterminal [Cl−]i is maintained higher by NKCC
Which mechanisms are involved in the accumulation of [Cl−]i in the glycinergic nerve terminals? In immature neurons, the [Cl−]i remains higher than predicted for passive distribution, and this results from the presence of inwardly directed Cl− transporters, especially the NKCC (Alvarez-Leefmans, 1990; Kaila, 1994; Plotkin et al. 1997; Clayton et al. 1998; Kakazu et al. 1999; Russell, 2000; Jang et al. 2001). In the experiments examining the involvement of the NKCC, muscimol-induced facilitation of sIPSC frequency was gradually reduced either in the presence of bumetanide or in a Na+-free external solution. These results suggest that repeated applications of muscimol gradually reduce the transmembrane Cl− gradient after blockade of the NKCC. However, such slow effects were not mediated by direct GABAA receptor blockade because 10 μM bumetanide had no influence on GABAA receptor-mediated currents (data not shown). Accordingly, a bumetanide-sensitive NKCC might play an important role in the accumulation of a higher [Cl−]i within the glycinergic nerve terminals projecting to SDCN neurons. This conclusion is consistent with our previous report that the NKCC contributes to the higher [Cl−]i and to GABAA receptor-mediated presynaptic depolarization in the glutamatergic presynaptic nerve terminals (Jang et al. 2001).
On the other hand, we found that muscimol still increased glycinergic sIPSC frequency even in postnatal 30-day-old SDCN neurons. It would be interesting to know whether NKCC regulates the [Cl−]i within these glycinergic presynaptic nerve terminals in mature neurons, because the increased expression of the K-Cl cotransporter, which is a major Cl− extrusion mechanism, contributes to the conversion of the GABA-induced postsynaptic response from depolarization to hyperpolarization during postnatal development (Kakazu et al. 1999; Vu et al. 2000).
Physiological implications
The present results indicate that GABAA receptor-mediated presynaptic depolarization has differential effects on spontaneous versus evoked glycine release, that is, presynaptic facilitation of spontaneous glycinergic transmission and presynaptic inhibition of action potential-driven glycinergic transmission. One possible explanation for the reduction of eIPSC amplitude by muscimol is the depletion of the releasable pool due to a large increase in sIPSC frequency. However, this might not be the case because sIPSCs were continuously increased during muscimol application while eIPSCs were inhibited immediately after muscimol application. Alternatively, this presynaptic inhibitory action on evoked glycine release may result from the inactivation of Na+ or Ca2+ channels based on membrane depolarization and/or shunt of the presynaptic membrane (see also Segev, 1990; Graham & Redman, 1994; Cattaert & El Manira, 1999). The present results suggest that the facilitation of spontaneous glycine release by presynaptic GABAA receptor activation leads to tonic inhibition of postsynaptic neurons, while the inhibition of evoked glycine release leads to disinhibition of postsynaptic neurons. Such differential effects on glycinergic transmission suggest that GABAA receptors on the glycinergic presynaptic nerve terminals might be involved in more complex regulation of inhibitory inputs and neuronal excitability.
In the spinal cord, GABAergic boutons onto the primary afferent terminals have been well described either morphologically (Todd et al. 1995; Maxwell et al. 1997) or electrophysiologically (Cattaert & El Manira, 1999). Although the present study has clearly demonstrated that exogenous muscimol depolarizes the glycinergic nerve terminals, it does not reveal the origin of the GABA that activates presynaptic GABAA receptors. One possibility is that GABAergic axo-axonic synapses are responsible for the activation of GABAA receptors on the glycinergic presynaptic nerve terminals. However, little is known about whether or not GABAergic axo-axonic synapses exist on the inhibitory presynaptic nerve terminals. Alternatively, synaptically released GABA might act on neighbouring glycinergic terminals via diffusion (spill-over). In the spinal cord, GABA is often colocalized with glycine (Todd & Spike, 1993) and the corelease of GABA and glycine has been demonstrated (Jonas et al. 1998; Chery & de Koninck, 1999). Chery & de Koninck (1999) suggested that coreleased GABA acts on extrasynaptic GABAA receptors by spill-over. Further study is necessary to resolve the origin of GABA for the presynaptic GABAA receptor activation.
The SDCN is known to be closely involved in nociceptive transmission (Honda, 1985; Ding et al. 1994; Lu et al. 1995; Vizzard et al. 1995). The SDCN receives abundant afferent inputs from both visceral and somatic organs. The convergence of visceral and somatic inputs onto SDCN neurons has been reported both electrophysiologically (Honda, 1985) and anatomically (Lu et al. 1995). The SDCN also integrates the regulatory influences from the brainstem descending pathways (Jones & Light, 1990). Accordingly, GABAA receptor-mediated presynaptic modulation of glycinergic transmission may play an important role in regulation of the sensory process within the spinal cord.
Acknowledgments
This study was supported by Grants-in-Aid for Scientific Research (no. 13307003) from The Ministry of Education, Science and Culture, Japan, The Japan Health Sciences Foundation (no. 21279 and Research on Brain Science), and Kyushu University Interdisciplinary Programs in Education and Projects in Research Development for N. Akaike.
REFERENCES
- Akaike N, Harata N. Nystatin perforated patch recording and its application to analysis of intracellular mechanism. Japanese Journal of Physiology. 1994;44:433–473. doi: 10.2170/jjphysiol.44.433. [DOI] [PubMed] [Google Scholar]
- Akaike N, Murakami N, Katsurabayashi S, Jin Y-H, Imazawa T. Focal stimulation of GABAergic single presynaptic boutons on the rat hippocampal neuron. Neuroscience Research. 2002;42:187–195. doi: 10.1016/s0168-0102(01)00320-0. [DOI] [PubMed] [Google Scholar]
- Altamirano AA, Breitwieser GE, Russell JM. Activation of Na+, K+, Cl− cotransport in squid giant axon by extracellular ions: evidence for ordered binding. Biochimica et Biophysica Acta. 1999;1416:195–207. doi: 10.1016/s0005-2736(98)00222-3. [DOI] [PubMed] [Google Scholar]
- Alvarez-Leefmans FJ. Intracellular Cl− regulation and synaptic inhibition in vertebrate and invertebrate neurons. In: Alvarez-Leefmans FJ, Russell JM, editors. Chloride Channels and Carriers in Nerve, Muscle and Glial cells. New York: Plenum; 1990. pp. 109–158. [Google Scholar]
- Atwood HL, Stevens JK, Marin L. Axoaxonal synapse location and consequences for presynaptic inhibition in crustacean motor axon terminals. Journal of Comparative Neurology. 1984;225:64–74. doi: 10.1002/cne.902250108. [DOI] [PubMed] [Google Scholar]
- Ballanyi K, Grafe P. An intracellular analysis of γ-aminobutyric-acid-associated ion movements in rat sympathetic neurons. Journal of Physiology. 1985;365:41–58. doi: 10.1113/jphysiol.1985.sp015758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohlhalter S, Mohler H, Fritschy JM. Inhibitory neurotransmission in rat spinal cord: co-localization of glycine- and GABAA-receptors at GABAergic synaptic contacts demonstrated by triple immunofluorescence staining. Brain Research. 1994;642:59–69. doi: 10.1016/0006-8993(94)90905-9. [DOI] [PubMed] [Google Scholar]
- Cattaert D, El Manira A. Shunting versus inactivation: Analysis of presynaptic inhibitory mechanisms in primary afferents of t he crayfish. Journal of Neuroscience. 1999;19:6079–6089. doi: 10.1523/JNEUROSCI.19-14-06079.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Trombley PQ, van den Pol AN. Excitatory actions of GABA in developing rat hypothalamic neurons. Journal of Physiology. 1996;494:451–464. doi: 10.1113/jphysiol.1996.sp021505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chery N, de Koninck Y. Junctional versus extrajunctional glycine and GABAA receptor-mediated IPSCs in identified lamina I neurons of the adult rat spinal cord. Journal of Neuroscience. 1999;17:7342–7355. doi: 10.1523/JNEUROSCI.19-17-07342.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton GH, Owens GC, Wolff JS, Smith RL. Ontogeny of cation-Cl− cotransporter expression in rat neocortex. Brain Research. Developmental Brain Research. 1998;109:281–292. doi: 10.1016/s0165-3806(98)00078-9. [DOI] [PubMed] [Google Scholar]
- Ding Y-Q, Qin B-Z, Li J-S, Mizuno N. Induction of c-fos-like protein in spinoparabrachial tract-neurons locating within the sacral parasympathetic nucleus in the rat. Brain Research. 1994;659:283–286. doi: 10.1016/0006-8993(94)90894-x. [DOI] [PubMed] [Google Scholar]
- Fisher JL, Macdonald RL. The role of an α subtype M2-M3 His in regulating inhibition of GABAA receptor current by zinc and other divalent cations. Journal of Neuroscience. 1998;18:2944–2953. doi: 10.1523/JNEUROSCI.18-08-02944.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontana G, Rogowski RS, Blaustein MP. Kinetic properties of the sodium-calcium exchanger in rat brain synaptosomes. Journal of Physiology. 1995;485:349–364. doi: 10.1113/jphysiol.1995.sp020734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham B, Redman SA. Simulation of action potentials in synaptic boutons during presynaptic inhibition. Journal of Neurophysiology. 1994;71:538–549. doi: 10.1152/jn.1994.71.2.538. [DOI] [PubMed] [Google Scholar]
- Haas M. Properties and diversity of (Na-K-Cl). cotransporters. Annual Review of Physiology. 1989;51:443–457. doi: 10.1146/annurev.ph.51.030189.002303. [DOI] [PubMed] [Google Scholar]
- Hales TG, Sanderson MJ, Charles AC. GABA has excitatory actions on GnRH-secreting immortalized hypothalamic (GT1–7) neurons. Neuroendocrinology. 1994;59:297–308. doi: 10.1159/000126671. [DOI] [PubMed] [Google Scholar]
- Harata N, Wu J, Ishibashi H, Ono K, Akaike N. Run-down of the GABAA response under experimental ischemia in acutely dissociated CA1 pyramidal neurons of the rat. Journal of Physiology. 1997;500:673–688. doi: 10.1113/jphysiol.1997.sp022052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda CN. Visceral and somatic afferent convergence onto neurons near the central canal in the sacral spinal cord of the cat. Journal of Neurophysiology. 1985;53:1059–1078. doi: 10.1152/jn.1985.53.4.1059. [DOI] [PubMed] [Google Scholar]
- Jang I-S, Jeong H-J, Akaike N. Contribution of the Na-K-Cl cotransporter on GABAA receptor-mediated presynaptic depolarization in excitatory nerve terminals. Journal of Neuroscience. 2001;21:5962–5972. doi: 10.1523/JNEUROSCI.21-16-05962.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonas P, Bischofberger J, Sandk ühler J. Corelease of two fast neurotransmitters at a central synapse. Science. 1998;281:419–424. doi: 10.1126/science.281.5375.419. [DOI] [PubMed] [Google Scholar]
- Jones SL, Light AR. Termination patterns of serotoninergic medullary raphespinal fibers in the rat lumbar spinal cord: an anterograde immunohistochemical study. Journal of Comparative Neurology. 1990;297:267–282. doi: 10.1002/cne.902970209. [DOI] [PubMed] [Google Scholar]
- Kaila K. Ionic basis of GABAA receptor channel function in the nervous system. Progress in Neurobiology. 1994;42:489–537. doi: 10.1016/0301-0082(94)90049-3. [DOI] [PubMed] [Google Scholar]
- Kakazu Y, Akaike N, Komiyama S, Nabekura J. Regulation of intracellular chloride by cotransporters in developing lateral superior olive neurons. Journal of Neuroscience. 1999;19:2843–2851. doi: 10.1523/JNEUROSCI.19-08-02843.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumamoto E, Murata Y. Characterization of GABA current in rat septal cholinergic neurons in culture and its modulation by metal cations. Journal of Neurophysiology. 1995;74:2012–2027. doi: 10.1152/jn.1995.74.5.2012. [DOI] [PubMed] [Google Scholar]
- Lamotte d'Incamps B, Destombes J, Thiesson D, Hellio R, Lasserre X, Kouchtir-Devanne N, Jami L, Zytnicki D. Indications for GABA-immunoactive axo-axoinc contacts on the intraspinal arborization of a Ib fiber in cat: a confocal microscope study. Journal of Neuroscience. 1998;18:10030–10036. doi: 10.1523/JNEUROSCI.18-23-10030.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leinekugel X, Tseeb V, Ben-Ari Y, Bregestovski P. Synaptic GABAA activation induces Ca2+ rise in pyramidal cells and interneurons from rat hippocampal slices. Journal of Physiology. 1995;487:319–329. doi: 10.1113/jphysiol.1995.sp020882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Jin S-X, Xu T-L, Qin B-Z, Li J-S, Ding Y-Q, Shigemoto R, Mizuno N. Expression of c-fos protein in substance P receptor-like immunoreactive neurons in response to noxious stimuli on the urinary bladder: an observation in the lumbosacral cord segments of the rat. Neuroscience Letters. 1995;198:139–142. doi: 10.1016/0304-3940(95)11991-5. [DOI] [PubMed] [Google Scholar]
- Maxwell DJ, Kerr R, Jankowska E, Riddell JS. Synaptic connections of dorsal horn group II spinal interneurons: synapses formed with the interneurons and by their axon collaterals. Journal of Comparative Neurology. 1997;380:51–69. [PubMed] [Google Scholar]
- Misgeld U, Deisz RA, Dodt HU, Lux HD. The role of chloride transport in postsynaptic inhibition of hippocampal neurons. Science. 1986;232:1413–1415. doi: 10.1126/science.2424084. [DOI] [PubMed] [Google Scholar]
- Obrietan K, van den Pol AN. GABA neurotransmission in the hypothalamus: developmental reversal from Ca2+ elevating to depressing. Journal of Neuroscience. 1995;15:5065–5077. doi: 10.1523/JNEUROSCI.15-07-05065.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owens DF, Boyce LH, Davis BE, Kriegstein AR. Excitatory GABA responses in embryonic and neonatal cortical slices demonstrated by gramicidin perforated-patch recordings and calcium imaging. Journal of Neuroscience. 1996;16:6414–6423. doi: 10.1523/JNEUROSCI.16-20-06414.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plotkin MD, Snyder EY, Hebert SC, Delpire E. Expression of the Na-K-2Cl cotransporter is developmentally regulated in postnatal rat brain: a possible mechanism underlying GABA's excitatory role in immature brain. Journal of Neurobiology. 1997;33:781–795. doi: 10.1002/(sici)1097-4695(19971120)33:6<781::aid-neu6>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- Rhee JS, Ishibashi H, Akaike N. Calcium channels in the GABAergic presynaptic nerve terminals projecting to Meynert neurons of the rat. Journal of Neurochemistry. 1999;72:800–807. doi: 10.1046/j.1471-4159.1999.0720800.x. [DOI] [PubMed] [Google Scholar]
- Rohrbough J, Spitzer NC. Regulation of intracellular Cl− levels by Na+-dependent Cl− cotransport distinguishes depolarizing from hyperpolarizing GABAA receptor-mediated responses in spinal neurons. Journal of Neuroscience. 1996;16:82–91. doi: 10.1523/JNEUROSCI.16-01-00082.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell JM. Sodium-potassium-chloride cotransport. Physiological Reviews. 2000;80:211–276. doi: 10.1152/physrev.2000.80.1.211. [DOI] [PubMed] [Google Scholar]
- Schneider SP, Fyffe RE. Involvement of GABA and glycine in recurrent inhibition of spinal motoneurons. Journal of Neurophysiology. 1992;68:397–406. doi: 10.1152/jn.1992.68.2.397. [DOI] [PubMed] [Google Scholar]
- Segev I. Computer study of presynaptic inhibition controlling the spread of action potentials into axon terminals. Journal of Neurophysiology. 1990;63:987–998. doi: 10.1152/jn.1990.63.5.987. [DOI] [PubMed] [Google Scholar]
- Shirasaki T, Aibara K, Akaike N. Direct modulation of GABAA receptor by ATP in dissociated nucleus tractus solitarii neurons of rat. Journal of Physiology. 1992;449:551–572. doi: 10.1113/jphysiol.1992.sp019101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart GJ, Redman SJ. The role of GABAA and GABAB receptors in presynaptic inhibition of Ia EPSPs in cat spinal motoneurones. Journal of Physiology. 1992;447:675–692. doi: 10.1113/jphysiol.1992.sp019023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd AJ, Spike RC. The localization of classical transmitters and neuropeptides within neurons in laminae I-III of the mammalian spinal dorsal horn. Progress in Neurobiology. 1993;41:609–645. doi: 10.1016/0301-0082(93)90045-t. [DOI] [PubMed] [Google Scholar]
- Todd AJ, Spike RC, Chong D, Neilson M. The relationship between glycine and gephyrin in synapses of the rat spinal cord. European Journal of Neuroscience. 1995;7:1–11. doi: 10.1111/j.1460-9568.1995.tb01014.x. [DOI] [PubMed] [Google Scholar]
- Todd AJ, Sullivan AC. Light microscope study of the coexistence of GABA-like and glycine-like immunoreactivities in the spinal cord of the rat. Journal of Comparative Neurology. 1990;296:496–505. doi: 10.1002/cne.902960312. [DOI] [PubMed] [Google Scholar]
- Vizzard MA, Erdman SL, de Groat WC. Increased expression of neuronal nitric oxide synthase (NOS) in visceral neurons after nerve injury. Journal of Neuroscience. 1995;15:4033–4045. doi: 10.1523/JNEUROSCI.15-05-04033.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vu TQ, Payne JA, Copenhagen DR. Localization and developmental expression patterns of the neuronal K-Cl cotransporter (KCC2) in the rat retina. Journal of Neuroscience. 2000;20:1414–1423. doi: 10.1523/JNEUROSCI.20-04-01414.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J-C, Lytle C, Zhu TT, Payne JA, Benz E., Jr Molecular cloning and functional expression of the bumetanide-sensitive Na-K-Cl cotransporter. Proceedings of the National Academy of Sciences of the USA. 1994;91:2201–2205. doi: 10.1073/pnas.91.6.2201. [DOI] [PMC free article] [PubMed] [Google Scholar]