

Abstract
Nitric oxide (NO) regulates the release of catecholamines from the adrenal medulla but the molecular targets of its action are not yet well identified. Here we show that the NO donor sodium nitroprusside (SNP, 200 μM) causes a marked depression of the single CaV1 L-channel activity in cell-attached patches of bovine chromaffin cells. SNP action was complete within 3-5 min of cell superfusion. In multichannel patches the open probability (NPo) decreased by ∼60 % between 0 and +20 mV. Averaged currents over a number of traces were proportionally reduced and showed no drastic changes to their time course. In single-channel patches the open probability (Po) at +10 mV decreased by the same amount as that of multichannel patches (∼61 %). Such a reduction was mainly associated with an increased probability of null sweeps and a prolongation of mean shut times, while first latency, mean open time and single-channel conductance were not significantly affected. Addition of the NO scavenger carboxy-PTIO or cell treatment with the guanylate cyclase inhibitor ODQ prevented the SNP-induced inhibition. 8-Bromo-cyclicGMP (8-Br-cGMP; 400 μM) mimicked the action of the NO donor and the protein kinase G blocker KT-5823 prevented this effect. The depressive action of SNP was preserved after blocking the cAMP-dependent up-regulatory pathway with the protein kinase A inhibitor H89. Similarly, the inhibitory action of 8-Br-cGMP proceeded regardless of the elevation of cAMP levels, suggesting that cGMP/PKG and cAMP/PKA act independently on L-channel gating. The inhibitory action of 8-Br-cGMP was also independent of the G protein-induced inhibition of L-channels mediated by purinergic and opiodergic autoreceptors. Since Ca2+ channels contribute critically to both the local production of NO and catecholamine release, the NO/PKG-mediated inhibition of neuroendocrine L-channels described here may represent an important autocrine signalling mechanism for controlling the rate of neurotransmitter release from adrenal glands.
Nitric oxide (NO) is a highly diffusible and reactive free radical (Ignarro et al. 1987; Palmer et al. 1987), recognized as a key intercellular messenger in central and peripheral neurons. NO is involved in synaptic plasticity phenomena, such as long-term potentiation and long-term depression (Shuman & Madison, 1994), and in the modulation of sensory transmission (Haley et al. 1992), including modulation of acoustic and proprioceptive signals (Grassi et al. 1995; Azzena et al. 2000).
NO effectively also modulates the activity of neuroendocrine cells. In bovine chromaffin cells, NO production can be induced autocrinally (Oset-Gasque et al. 1994; Schwarz et al. 1998) or paracrinally by both the afferent nerves (Dun et al. 1993) and surrounding endothelial cells (Torres et al. 1994). When either applied directly or produced by NO donors, NO affects the release of catecholamines in a distinct manner depending on cell stimulation. NO increases the basal secretion of catecholamines (O'Sullivan & Burgoygne, 1990; Oset-Gasque et al. 1994), while inhibiting the exocytosis evoked by high doses of ACh (Oset-Gasque et al. 1994; Rodriguez-Pascual et al. 1996; Nagayama et al. 1998), sustained KCl depolarizations (Rodriguez-Pascual et al. 1996) or application of Ba2+ ions (Machado et al. 2000). The origins of the reduced release during strong stimuli are still unclear, although there is evidence for a cGMP-mediated inhibition of P/Q-type Ca2+ currents (Rodriguez-Pascual et al. 1994) and a drastic slow-down of the emptying of granules (Machado et al. 2000). Since Ca2+ is crucial for NO synthase activation and consequent NO production (Bredt & Snyder, 1990), the negative control of NO on voltage-gated Ca2+ channels could represent an effective autocrine mechanism to limit the rate of Ca2+ entry and catecholamine release during sustained adrenal gland stimulation (Schwarz et al. 1998).
The inhibitory action of NO on voltage-gated Ca2+ channels is well documented, although the mechanism of action is not yet well identified. This is due to the complexity of the system and to a number of unresolved controversial results. In rat pinealocytes, NO inhibits the whole-cell L-type currents via a cGMP-dependent mechanism (Chik et al. 1995), while in glomus cells of rabbit carotid body the specific action of NO on L-channels is direct and cGMP independent (Summers et al. 1999). In rat insulinoma RINm5F cells NO and 8-bromo-cyclicGMP (8-Br-cGMP) are very effective in inhibiting both L- and non-L-type channels (Grassi et al. 1999). NO and 8-Br-cGMP are also effective in inhibiting cardiac and smooth muscle L-type channels, but the action seems to proceed through three different mechanisms in a rather contradictory manner (Tohse & Sperelakis, 1991; Han et al. 1994; Hu et al. 1997; Tewari & Simard, 1997; Gallo et al. 1998; Jiang et al. 2000). Early studies on cardiac L-channels suggest that the inhibitory effect of NO/cGMP derives from the activation of a cGMP-dependent phosphodiesterase (PDE), which lowers the level of cAMP/protein kinase A (PKA) and the corresponding L-channel activity (Méry et al. 1993; Han et al. 1994). In contrast, other reports suggest that 8-Br-cGMP inhibits cardiac L-channel activity via a protein kinase G (PKG)-mediated phosphorylation regardless of the cAMP/PKA pathway (Tohse & Sperelakis, 1991; Jiang et al. 2000), or that NO directly inhibits the cardiac L-channels expressed in heterologous systems independently of cGMP and cAMP (Hu et al. 1997).
Since the neuroendocrine L-channel plays a critical role in the control of catecholamine release (García et al. 1984) and NO preferentially acts on this channel type, we considered it of interest to study the molecular mechanisms that form the basis of neuroendocrine L-channel gating modulation by NO. Given the existence of multiple modulatory pathways, we also examined the possible cross-talk between the NO/PKG-mediated signalling and both the autocrine G-protein-induced inhibition and the cAMP/PKA-mediated potentiation, which all markedly affect neuroendocrine L-channel gating (Carabelli et al. 2001). As before, we followed the single-channel approach with the dual purpose of studying the NO/PKG signalling pathway in an intact intracellular environment and to gain further information about the effects of NO at the unitary L-current level. Data on the action of NO on single L-channels are quite limited and incomplete (Tohse & Sperelakis, 1991; Tewari & Simard, 1997), although essential for clarifying a number of controversial issues about the molecular mechanisms controlling the NO-induced inhibition of L-channels in various tissues (Han et al. 1994; Hu et al. 1997; Gallo et al. 1998; Jiang et al. 2000).
Here, we show for the first time that the NO/PKG signalling pathway inhibits the single L-channel activity in bovine chromaffin cells by driving the channel into a gating mode of low probability of opening regardless of the level of available cAMP and activated Gi/Go proteins. This action widens the possibility of modulating neuroendocrine L-channels, which also experience up- and down-regulation by locally activated Gi/Go proteins or remotely stimulated cAMP/PKA signalling (Carbone et al. 2001), and may furnish a rationale for an autoregulatory role of NO in controlling Ca2+ channel activity and catecholamine secretion in adrenal glands.
METHODS
Cell cultures
Bovine chromaffin cells were obtained by digestion with collagenase from adrenal glands of 6- to 18-month-old cows and successively purified by density gradient centrifugation as previously described (Carabelli et al. 1998). The cells were plated at a density of 105 ml−1 in plastic dishes pretreated with poly-l-ornithine (1 mg ml−1) and laminin (5 μg ml−1 in L-15 carbonate) and maintained in an incubator at 37 °C in a water-saturated 5 % CO2 atmosphere. The culture medium contained: DMEM, supplemented with 10 % fetal calf serum (GIBCO, Grand Island, NY, USA), 50 I.U. ml−1 penicillin, 50 μg ml−1 streptomycin (GIBCO), 2.5 μg ml−1 gentamicin (Sigma Chemical Co., St Louis, MO, USA), 10 μM cytosine arabinoside and 10 μM fluorodeoxyuridine (Sigma).
Cell-attached recordings
The activity of single L-type channels was recorded in the cell-attached configuration of the patch-clamp technique (Hamill et al. 1981) using an EPC-9 amplifier (HEKA Elektronik, Lambrecht, Germany). Electrodes of 4-8 MΩ resistance were made from thick borosilicate glass (Hilgenberg, Mansfield, Germany) as previously described (Carabelli et al. 1996). The pipette-filling control solution contained (mm): 100 BaCl2, 10 TEA-Cl, 1 MgCl2, 10 Na-Hepes, 10 μM ω-conotoxin-MVIIC (ω-CTx-MVIIC) and 300 nm TTX (pH adjusted to 7.3 with TEAOH). (-)-Bay K 8644 (5 μM) was always present in the pipette solution to better resolve L-channel openings, otherwise hardly detected. In most experiments, opioidergic and purinergic receptor antagonists (10 μM naloxone and 100 μM suramin) were added to prevent autocrine L-channel inhibition (Carabelli et al. 2001). In a different set of experiments, in which the receptor-coupled G proteins were activated, the antagonists were replaced with a mixture of opioidergic and purinergic agonists (10 μM DAMGO, 1 μM DPDPE and 100 μM ATP) in the pipette solution. The cell-attached condition was achieved with the cell bathed in a Tyrode's solution containing (mm): 140 NaCl, 4 KCl, 2 MgCl2, 2 CaCl2 and 10 Hepes (pH adjusted to 7.3 with NaOH). Membrane potential was zeroed by perfusing the cell with a control solution containing (mm): 135 KAsp, 1 MgCl2, 10 Hepes, 5 EGTA and 300 nm TTX (pH adjusted to 7.3 with KOH). Voltages were not corrected for the liquid junction potential, which was −16 mV between KAsp and the pipette solution (Barry & Lynch, 1991; Neher, 1992). In this way we could compare our results with previously published data (Carabelli et al. 1998, 2001).
To induce NO production we employed sodium nitroprusside (SNP), which is one of the NO donors most widely used in the literature. To reach maximal effects, SNP was added to the external solution at near saturating concentration (200 μM; Grassi et al. 1999) and illuminated with an optic fibre beam of dim light directed onto the cells (Bates et al. 1991). The culture dish was replaced after each trial with SNP. This allowed each set of recordings to be performed on cells that were not previously challenged with the NO donor.
Current traces were acquired at 5-10 kHz and filtered at 1 kHz with an 8-pole low-pass Bessel filter. Membrane stimulation and data acquisition were performed using PULSE programs (HEKA Elektronik). L-channel activity was recorded by applying 200 ms (or 600 ms) depolarizing pulses to 0, +10 or +20 mV from −40 mV holding potential (Vh). Consecutive depolarizations were applied every 6 s for 7-10 min. Except for a series of experiments in which channel activity was monitored for 20 min to check channel rundown in control conditions, in most experiments the data shown refer to the first 7 min of recordings. The first minute was in control conditions and the following six during drug application. All the experiments were performed at room temperature (22-24 °C).
Data analysis
Data analysis was performed using TAC and TACFIT software (version 3.04; Bruxton Corporation, Seattle, WA, USA). Fast capacitative transients were minimized on-line by the patch-clamp analogue compensation. Uncompensated capacitative currents were corrected by averaging sweeps with no channel activity (nulls) and subtracting them from each active sweep. Event detection was performed with the 50 % threshold detection method, with each transition visually inspected before being accepted.
Most of the present data derive from patches containing two or, less frequently, three channels. They refer to the experiments in which the channel activity in control conditions was compared to that during the application of specific compounds on the same patch (SNP, carboxy-PTIO, 8-Br-cGMP). Under these conditions, there was no strict requirement to limit our analysis to patches containing only one channel, so the NPo was calculated by adding the time duration of single, double and even triple openings and dividing the sum by the duration of the analysed time interval (Lambert & Feltz, 1995). NPo was evaluated sweep by sweep, excluding the first and the last closure. Null traces were included in the calculation of mean NPo for both controls and drug application. This was suggested by the fact that the percentage of nulls significantly increased during cell perfusion with NO donors and PKG activators and thus the NPo reduction with time could be better evaluated (see Fig. 2B).
Figure 2. Mean NPo and null sweeps probability versus time before and during SNP application.
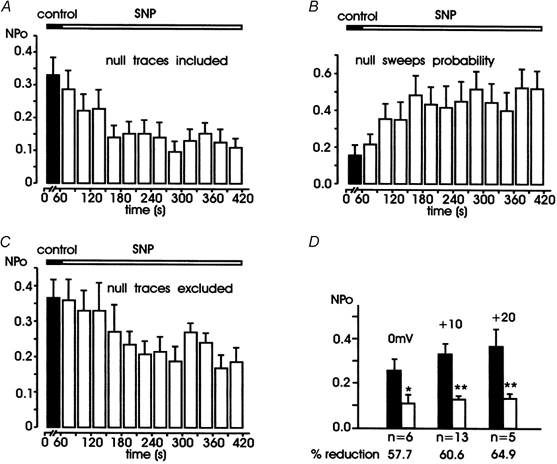
Filled bars are data in control conditions obtained by averaging data collected during 1 min of recording from 13 patches. Open bars are values during SNP application obtained at intervals of 30 s (A-C) or by grouping all the values from the third to the sixth minute (D). In A-C, depolarizations were at +10 mV. A shows the mean NPo calculated including the null sweeps (see Methods) and B shows the probability of null sweeps versus time. Notice the marked decrease of NPo and the almost threefold increase of null sweeps probability with time. C shows the values of NPo calculated by excluding the null sweeps from the analysis. NPo decreased from 0.37 ± 0.05 to 0.21 ± 0.01 with a 43.2 % reduction with respect to controls, P < 0.05. D shows the mean values of NPo at 0, +10 and +20 mV in control conditions and during SNP application, with the percentage of reduction indicated below (* P < 0.05, ** P < 0.01). The difference between minimal and maximal reduction (57.7 vs. 64.9 %) was not statistically significant (P > 0.05).
Patches containing unitary openings (n = 9) were identified following the criteria previously described (Carabelli et al. 1996, 2001) and were used to calculate the mean open time, the mean closed time and the single-channel open probability (Po) at a fixed potential (+10 mV; Fig. 3). As for NPo, Po was evaluated by excluding the first and last closure and mean open probability was calculated including null sweeps. This furnished lower values of mean Po than those previously reported, in which null sweeps were not included (Carabelli et al. 2001). The inclusion of nulls was required for studying time-dependent reductions of mean Po and did not alter the final interpretation of the data. To better resolve the longest closed time component (Fig. 3C), in five patches the depolarizing pulse was prolonged to 600 ms.
Figure 3. Single L-channel parameters in control conditions and during application of SNP.
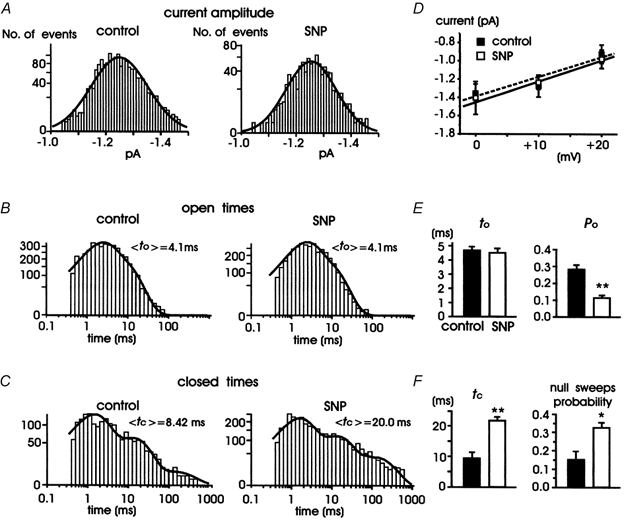
A, histograms showing distribution of single L-channel amplitudes measured at +10 mV before (left) and during exposure to 200 μM SNP (right) collected from 13 patches. The curves are best-fitted Gaussian functions with a mean of −1.24 ± 0.07 pA in controls and −1.27 ± 0.18 pA with SNP. B, open time distribution at +10 mV in control conditions (left) and during SNP application (right). The data were collected from 13 patches: 4 patches with two channel openings and 9 with single channel openings (of these latter, 4 were depolarized with pulses of 200 ms and 5 with pulses of 600 ms). The distributions were fitted with a two-exponential function with the following time constants: tO1 = 1.9 ms (57 % of channel openings) and tO2 = 7.1 ms (43 %) in controls, and tO1 = 1.9 ms (60 %) and tO2 = 7.5 ms (40 %) with SNP. Mean open times (<tO>) derived from the fit are given to the top right of each distribution. C, closed time distribution at +10 mV in control conditions (left) and during SNP application (right). The data were collected from 5 patches displaying single channel openings and depolarized with pulses of 600 ms to +10 mV. The distributions were fitted with a three-exponential function with the following time constants: tC1 = 1.3 ms (65 %), tC2 = 12.7 ms (32 %) and tC3 = 127 ms (3 %) in controls, and tC1 = 1.4 ms (56 %), tC2 = 12.5 ms (32 %) and tC3 = 127 ms (12 %) with SNP. Mean closed times (<tC>) derived from the fit are given to the top right of each distribution. D, mean unitary current amplitudes plotted versus voltage. The linear regressions through data points have mean slope conductances of 21.3 ± 2.6 pS (control) and 21.5 ± 6.8 pS (SNP) (n = 5-13). E and F, average tO, tC, Po and null sweeps probability in controls and with SNP obtained from the arithmetic mean of the values calculated from patches containing a single Bay K-modified L-channel (* P < 0.05, ** P < 0.01).
Open time and closed time histograms were plotted on square root-log coordinates and constructed as previously described (Carabelli et al. 2001). Data were not corrected for missed events and distributions of open and closed times were fitted by the sum of decaying exponentials. To increase the number of events of the open time distributions, unitary data events were also pooled from patches containing two channels. In these patches, single-channel openings were frequent and usually occurred at the end of the depolarizing pulse, where the degree of channel inactivation was sufficiently high to favour the occurrence of single events. The mean amplitude of the unitary current was determined by fitting the amplitude histograms with a Gaussian distribution. The unitary conductance was evaluated by linear regression of the mean unitary currents at 0, +10 and +20 mV.
Data are presented as means ± s.e.m. for number of patches (n). Statistical significance was calculated using Student's paired t test and P values less than 0.05 were considered significant. The statistical significance of open probability (NPo) changes during drug application was assessed by ANOVA for repeated measurements.
Drugs and solutions
(-)-Bay K 8644, ATP, naloxone, suramin, SNP, 8-bromoguanosine 3′,5′-cyclic monophosphate sodium salt (8-Br-cGMP), 8-(4-chlorophenylthio)-cAMP (8-CPT-cAMP), [d-Pen2-Pen5]-enkephalin (DPDPE) and [d-Ala2,N-Me-Phe4,Gly5-ol]-enkephalin (DAMGO) were purchased from Sigma. 2-(4-Carboxyphenyl)-4,4,5,5-tetramethyl-imidazoline-1-oxyl-3-oxide potassium salt (carboxy-PTIO) was obtained from Affiniti Research Products Ltd (Mamhead, UK) and 1H-[1,2,4]oxadiazole[4,3-a]quinoxalin-1-one (ODQ) from Alexis Corporation (Läufelfingen, Switzerland). H89 and KT-5823 were purchased from CN Biosciences Inc. (Darmstadt, Germany). ω-CTx-MVIIC was obtained from Tocris Cookson (Bristol, UK).
RESULTS
SNP inhibits L-channel activity in multichannel recordings
Single L-channel activity can be resolved in cell-attached patches of bovine chromaffin cells using step depolarizations to +10 mV from −40 mV (Vh) and pipette solutions containing 100 mm BaCl2, 5 μM Bay K 8644 and 10 μM ω-CTx-MVIIC (Carabelli et al. 1998, 2001). Under these conditions, single L-channel currents have a mean amplitude of −1.24 pA and mean open time of ∼4 ms, which make them easily distinguishable from the brief openings of N- and P/Q-channels observed occasionally in some patches (mean open time ∼0.6 ms). Despite these advantages, the activity of L-channels in chromaffin cells is usually inhibited in control conditions, due to the presence of endogenous neurotransmitters (ATP and opioids) directly released inside the recording pipette (Carabelli et al. 2001). This autocrine inhibition is mediated by PTX-sensitive G proteins and switches the channel into a low-Po mode in which the open probability is reduced by about a factor of two. To prevent this, except where otherwise indicated, all the experiments were performed in the presence of purinergic and opioidergic receptor antagonists (100 μM suramin and 10 μM naloxone) in the patch pipette, to keep the autoreceptor-coupled G proteins inactive. This caused a scaling-up of reconstituted averaged currents without changing channel activation and inactivation. Under these control conditions the L-channel activity could persist unaltered for 10-20 min (n = 5), without showing signs of channel rundown. The time course of the open probability recorded from a representative control patch is shown in Fig. 1D.
Figure 1. The NO donor SNP markedly inhibits the single L-channel activity in bovine chromaffin cells.
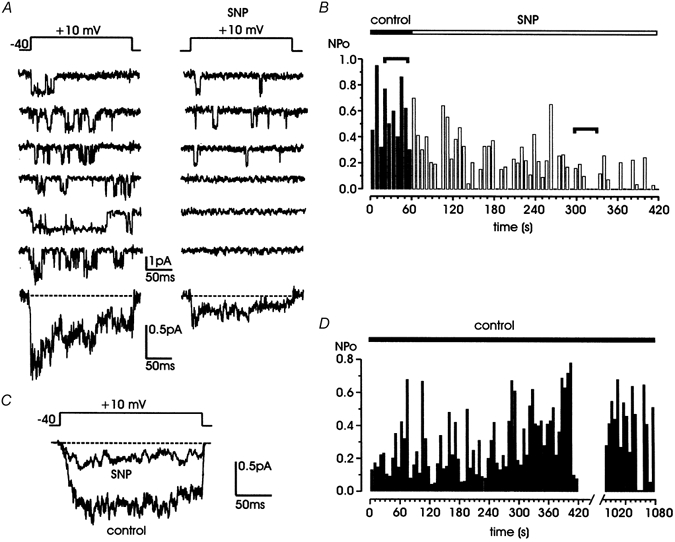
A, representative traces of L-channel activity, recorded in a cell-attached patch containing more than one channel under control conditions (left) and during exposure to 200 μM SNP (right). ω-CTx-MVIIC (10 μM), Bay K 8644 (5 μM) and a mixture of purinergic and opioidergic receptor antagonists (100 μM suramin, 10 μM naloxone) were present in the pipette solution. Bottom traces are averaged currents calculated over 10 (control) and 40 sweeps (SNP) from the same patch. B, NPoversus time before (▪) and during SNP exposure (□). Horizontal segments indicate the selected traces shown in A. C, averaged currents obtained from 13 patches, taking 10 traces in control and 40 traces from the third to the sixth minute of SNP application from each individual patch. D, NPoversus time for a representative control cell. Channel activity was tested for rundown for 18 min.
The action of SNP was tested using the protocol illustrated in Fig. 1. L-channel activity was evoked every 6 s with pulses of 200 ms to +10 mV. After 1 min in control conditions (10 traces) the cell was continuously exposed to 200 μM SNP and the activity of available channels was tested with the same frequency for a period of 6 min. In most of the experiments we focused on two parameters: the open channel probability in multichannel patches (indicated as NPo) and the time course of the averaged currents calculated after the second minute of SNP application, when the activity of L-channels was visibly lowered. In multichannel recordings, NPo ranged from 0.30 to 0.95 but significantly decreased after the second minute of SNP application (ranging from 0 to 0.4 in most traces; Fig. 1A and B). The SNP-induced decrease of NPo appeared with a delay of 1-2 min and reached maximal effects usually between the third and the fifth minute of SNP application. Compared to control traces, the averaged current in the presence of SNP was scaled down, i.e. of smaller size but similar time course. The same thing occurred if the mean currents were obtained by averaging the traces of 13 patches in control conditions and from the third to the sixth minute of SNP addition (Fig. 1C). The half-time-to-peak (t1/2) was 11 ms in control conditions and 9.5 ms with SNP and the percentage of inactivation calculated over the last 30 ms of the recordings was 26 % in control conditions and 34 % with SNP. Similar mean values were obtained from the averaged currents of each individual patch; the mean t1/2 was 10.7 ± 0.9 (control) and 9.1 ± 2.8 ms (SNP) and mean percentage of inactivation was 28.5 ± 5 (control) and 35.5 ± 5 % (SNP).
The analysis of NPo performed over 13 patches showed that mean NPo was 0.33 ± 0.05 in controls and its reduction by SNP was statistically significant starting after the second minute of SNP perfusion (F(4,12) = 3.49; P < 0.05; Fig. 2). The depression of NPo was then estimated by averaging the data collected from the third to the sixth minute of SNP exposure. In this time interval NPo decreased to 0.13 ± 0.01, with a 60.6 % reduction with respect to controls (P < 0.01). After SNP removal, wash out with control solutions did not usually produce a significant recovery of channel activity in the following 2-3 min. This is because, with the high SNP concentration used (200 μM), the recovery required long periods of washing (∼20 min) (Rauch et al. 1997; Tewari & Simard, 1997; Grassi et al. 1999; Lang et al. 2000).
As shown in Fig. 2, the decrease of NPo with SNP addition was associated with a threefold increase in the number of null sweeps (Fig. 2B), which contributed to NPo reduction, although it was clearly not the only cause. In fact, NPo decreased by 43.2 % even when null sweeps were excluded from the determination of NPo (Fig. 2C). Notice that the reduction of NPo was not limited to L-channel activity at +10 mV. NPo was also comparably depressed at 0 and +20 mV (by 57.7 and 64.9 %, respectively; Fig. 2D). The percentage of reduction at various potentials was not significantly different, suggesting that the inhibitory action of SNP was insensitive to voltage in the range between 0 and +20 mV.
SNP increases the shut times and probability of null traces in single-channel recordings
Given that SNP causes a marked diminution in NPo, the next step was to quantify the action of the NO donor on those parameters characterizing single L-channel activity. As shown in Fig. 3, SNP did not cause significant changes to the amplitude distribution at +10 mV. The mean amplitude was −1.24 ± 0.07 pA in control conditions and −1.27 ± 0.18 pA with SNP (Fig. 3A). Similarly, small changes occurred at 0 and +20 mV, giving nearly unaltered single-channel conductance of 21.3 ± 2.6 and 21.5 ± 6.8 pS with and without the NO donor, respectively (Fig. 3D). Almost no changes were also observed in the open time distributions at +10 mV, which were fitted with two exponentials both in control conditions and in the presence of SNP. The fit gave the same mean tO (<tO> = 4.1 ms) in control conditions and with SNP (Fig. 3B). Also, no significant difference was found when the mean open times were calculated by the arithmetic means of all the data in control conditions and with SNP (4.7 vs. 4.5 ms, Fig. 3E).
In nine patches containing single L-channels, SNP markedly inhibited Po (61.2 % reduction, Fig. 3E). Such a reduction is nearly identical to the 60.6 % decrease of NPo at +10 mV reported above and suggests that the predominant action of the NO donor is on L-channel gating (Po) rather than on the number of available channels (N). Most of the effects on Po are due to both a prolongation of mean closed times (Fig. 3C and D) and an increase of null sweep probability (Fig. 3F). To better resolve long closures during application of SNP, in five patches the shut time distribution was constructed with data collected using step depolarizations of 600 instead of 200 ms. A fit with a three-exponential function showed that the main effect of SNP was on the contribution of the longest component, tC3, which increased from 3 to 12 %, while the contribution of the other two components, tC1 and tC2, was little affected (see Fig. 3C, legend). Notice that the mean tC derived from the fit, <tC>, compared well with the arithmetic means of the shut times calculated from five patches in control conditions (8.42 vs. 9.45 ms) and in the presence of SNP (20.0 vs. 21.9 ms; Fig. 3F), confirming that the prolongation of mean tC is one of the primary causes of Po reduction by SNP.
Finally, we found that SNP did not alter the latency of first openings. In the nine patches containing only single channels, the mean first latency was 37.3 ± 6.9 ms in control conditions and 37.4 ± 2.7 ms with SNP. Considering that these values are partly derived from step depolarizations of 600 ms (n = 5), they appear in good agreement with previous estimates obtained using threefold shorter pulses (33.8 ± 1.6 ms; Carabelli et al. 2001).
NO action on neuroendocrine L-channels is mediated by cGMP and PKG
To prove the involvement of NO production we first tested the effects of SNP in the presence of the NO scavenger carboxy-PTIO (300 μM), which traps the NO produced by SNP breakdown but induces no significant changes in NPo when applied alone (n = 5, Fig. 4D). As shown in Fig. 4A and B, in the presence of carboxy-PTIO, SNP failed to notably change L-channel activity in multichannel patches. On average, in seven patches there was no significant decrease in NPo at +10 mV from the third to the sixth minute of drug addition (Fig. 4C and D). Mean currents with and without the drugs were of comparable size (bottom traces in Fig. 4A). Carboxy-PTIO alone also had no significant effects on mean NPo ( in Fig. 4D).
in Fig. 4D).
Figure 4. The NO scavenger carboxy-PTIO prevents the inhibitory effects of SNP.
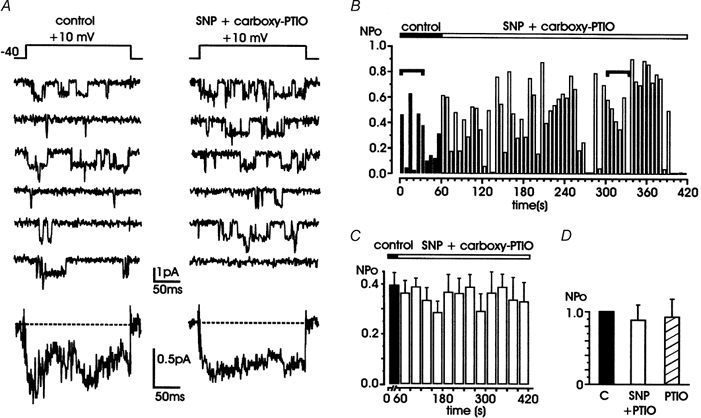
A, representative traces of L-channel activity recorded in control conditions (left) and during the simultaneous application of 200 μM SNP and 300 μM carboxy-PTIO (right). Bottom traces are averaged currents calculated from the same patch over 10 and 40 sweeps, respectively. B, time course of NPo during cell exposure to both solutions. Filled bars are NPo values in control conditions and open bars are NPo values during drug application. C, mean NPo values obtained by averaging data collected from 7 patches over 1 min (control, ▪) and 30 s periods (drug application, □). D, normalized mean NPo with respect to control, for cells exposed to SNP + carboxy PTIO (n = 7; □) or carboxy PTIO alone (n = 4; ). All the patches contained multichannel openings.
The next step was to check whether NO effects on L-channels were mediated by an increase in the intracellular levels of cGMP. To verify this hypothesis, SNP was applied after blocking guanylate cyclase activity with the selective inhibitor ODQ (Garthwaite et al. 1995). To ensure an effective action of the inhibitor, cells were pretreated for 15 min with 10 μM ODQ dissolved in Tyrode solution and then checked for the effects of the NO donor. As shown in Fig. 5, ODQ alone and ODQ plus SNP failed to significantly change NPo. The averaged currents had comparable amplitude and similar time course to controls. On average, NPo was 0.34 ± 0.14 in control conditions and 0.33 ± 0.04 with SNP (n = 6). Thus, blockade of guanylate cyclase activity prevented the inhibitory effects of SNP, suggesting that the action of NO is probably mediated by an increase in the intracellular levels of cGMP. To prove the existence of a cGMP-mediated mechanism, we also tested whether the membrane-permeable analogue, 8-Br-cGMP, was able to depress L-channel gating as well as SNP. As reported in various cell preparations, 8-Br-cGMP starts having effects on Ca2+ currents above 300 μM (Grassi et al. 1999; Tohse & Sperelakis, 1991). We therefore checked whether 400 μM 8-Br-cGMP mimicked the SNP effects and reduced NPo. We found that the cGMP analogue reduced NPo from 0.52 ± 0.11 to 0.20 ± 0.03 (61.5 % reduction, n = 5, P < 0.05), in good agreement with the SNP-induced inhibition. The reduction of NPo was statistically significant starting from the third minute of drug application (F(4,4) = 6.5, P < 0.05; Fig. 6C). Recovery from 8-Br-cGMP-induced inhibition was not evident on a short time scale (2-3 min), as in different experimental models (Rauch et al. 1997).
Figure 5. The guanylate cyclase inhibitor ODQ prevents SNP action.
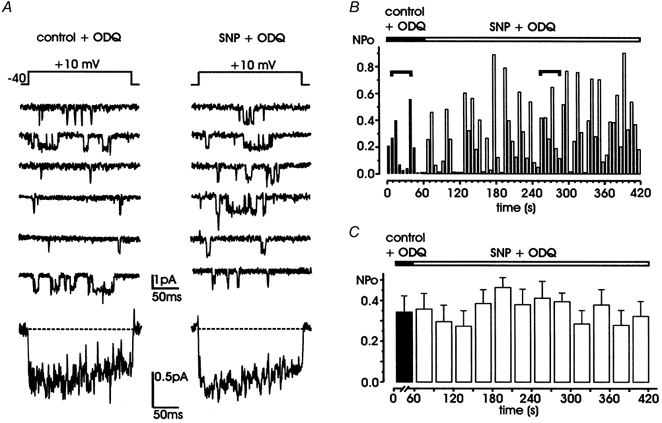
A, during simultaneous application of SNP (200 μM) and ODQ (10 μM; right), L-channel activity is not significantly different from that in the presence of ODQ alone (left). Bottom traces are averaged currents from the same patch over 10 and 40 sweeps, respectively. Notice the close similarities between the two traces. B, NPo values versus time calculated from the same patch shown in A. Horizontal segments indicate the representative traces shown in A. C, mean NPo obtained by averaging data from 5 patches over 1 min (control + ODQ, ▪) and 30 s periods (SNP + ODQ, □), respectively. All the patches contained multichannel openings.
Figure 6. 8-Br-cGMP mimics the effects of SNP by reducing L-channel activity through a PKG-mediated mechanism.
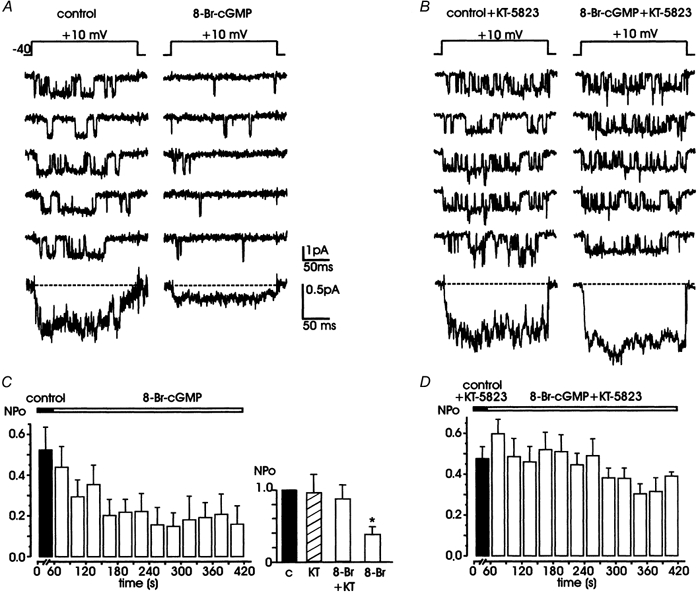
A, cell exposure to 400 μM 8-Br-cGMP (right) reduces the probability of channel opening with respect to controls (left). B, after incubation with the specific PKG inhibitor, KT-5823 (1 μM), 8-Br-cGMP fails to evoke the marked reduction in NPo shown in A. Bottom traces in A and B are averaged currents obtained from 10 (control) and 40 traces (drug) in both cases. C and D show the mean NPo values versus time obtained from 5 patches. Filled bars in C and D are mean control values collected over 1 min; open bars are mean values obtained by averaging data over 30 s periods during application of 8-Br-cGMP and 8-Br-cGMP + KT-5823, respectively. The inset in C shows normalized mean NPo with respect to controls exposed to KT-5823 for about 10 min (n = 4), pretreated with KT-5823 and then exposed to 8-Br-cGMP (n = 5), or exposed to 8-Br-cGMP alone (n = 5). All the patches contained multichannel openings.
Given that the action of NO was probably mediated by cGMP, we next tested whether PKG was involved in this inhibition of NPo. To verify this hypothesis, we tested the action of 8-Br-cGMP (400 μM) in cells pretreated with the membrane-permeable PKG inhibitor, KT-5823 (1 μM for 20 min; Grider, 1993). KT-5823 is widely recognized as a selective PKG inhibitor, although its specificity in platelets and rat mesangial cells has recently been questioned (Burkhardt et al. 2000). We found that pretreatment with the PKG inhibitor was effective in preventing the potent action of 8-Br-cGMP. In fact, in five cells pretreated with KT-5823, we observed only a slight decrease in NPo, which was not statistically significant (0.41 ± 0.03 vs. 0.48 ± 0.10 in controls, P > 0.05). We also tested the action of KT-5823 alone for about 10 min (n = 4) and found no relevant difference in NPo with respect to untreated control cells (Fig. 6C, inset). Thus, KT-5823 per se does not alter L-channel gating but prevents cGMP/PKG-mediated inhibition.
NO/cGMP action is unrelated to the intracellular levels of PKA
Increased levels of cAMP up-regulate the activity of normally available L-channels in bovine chromaffin cells (Carabelli et al. 2001). In cardiac cells, NO is known to decrease L-channel activity by lowering the levels of cAMP through the up-regulation of a cGMP-activated phosphodiesterase (Méry et al. 1993). Thus, any interference of NO with the cAMP/PKA pathway would in principle produce a reduction of L-channel activity independently of the effect of PKG. We checked whether this could also occur in our cell preparation by testing the effects of SNP after having blocked the cAMP/PKA pathway by the PKA inhibitor H89. We found that 20 min cell pretreatment with 1 μM H89 did not alter the inhibitory action of SNP. Indeed, the activity of available L-channels was strongly depressed by SNP (Fig. 7A and B). NPo was markedly reduced and so was the mean current obtained by averaging the traces collected from the third to the sixth minute of drug application (Fig. 7C). In five patches, the mean NPo decrease was 59.0 % (F(4,4) = 3.6, P < 0.05). Thus, in bovine chromaffin cells the inhibitory action of NO on L-channels is unrelated to the cAMP/PKA pathway and can therefore be entirely attributed to a PKG-mediated action on channel gating.
Figure 7. SNP preserves its action even when the PKA inhibitor H89 prevents cAMP-mediated up-regulation of L-channel.
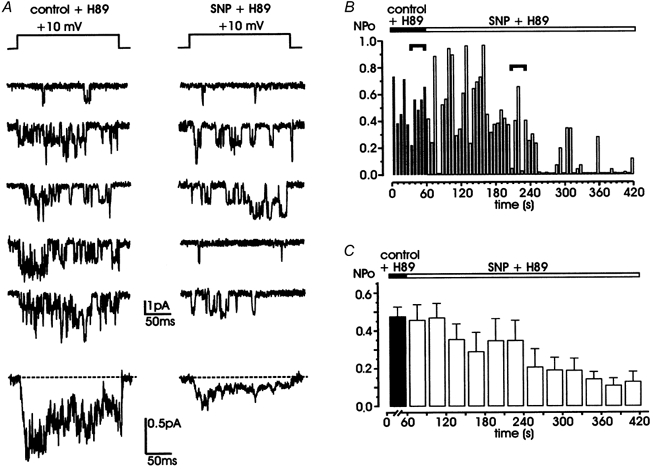
A, L-channel activity in a chromaffin cell incubated for 20 min with 1 μM H89 (left) is effectively inhibited by exposure to SNP (200 μM; right). Bottom traces are averaged currents obtained from 10 (control + H89) and 40 traces (SNP + H89) of the same patch. Notice the strong depression induced by SNP, which is comparable to that of Fig. 1. B and C show the time course of NPo and mean NPo derived from 6 patches following the same protocols as in Fig. 5. Horizontal segments in B indicate the representative traces shown in A. All the patches contained multichannel openings.
cGMP-mediated inhibition is independent of other L-channel modulations
The L-channel of bovine chromaffin cells possesses two distinct modulatory pathways of channel gating: a direct PTX-sensitive G protein down-modulation and a remote PKA-mediated up-regulation (Carabelli et al. 2001; Carbone et al. 2001). We therefore checked whether these modulatory pathways could interfere with the presently described NO-mediated inhibition, by testing whether PKG activation by 8-Br-cGMP was still effective when either one of the two modulations was active. Figure 8 shows that in both cases the PKG-mediated inhibition proceeded independently of the activity of the other pathway. In the case of G protein activation, with the recording pipette containing ATP (100 μM) and μ/δ-opioid receptor agonists (10 μM DAMGO and 1 μM DPDPE), the addition of 400 μM 8-Br-cGMP markedly inhibited L-channel activity after about 3 min of application. There was a clear prolongation of closed times and a robust depression of averaged currents obtained from one patch (Fig. 8A) or six patches (Fig. 8B) in the presence of 8-Br-cGMP. On average, NPo was low in patches containing only the agonists (0.25 ± 0.04) and decreased to 0.1 ± 0.03 after addition of 8-Br-cGMP (Fig. 8B). The same thing occurred in patches pretreated with the membrane-permeable cAMP analogue, 8-CPT-cAMP (1 mm), which significantly increases the activity of L-channels (Fig. 8C). Addition of 8-Br-cGMP produced a marked increase of channel closed times and a net decrease of the averaged currents derived from one patch (Fig. 8C) or four patches (Fig. 8D). NPo was high in the presence of 1 mm 8-CPT-cAMP (0.45 ± 0.08) and was nearly halved after 3 min of exposure to 8-Br-cGMP (0.19 ± 0.04).
Figure 8. 8-Br-cGMP inhibits L-channel activity regardless of the down- and up-modulation induced by Gi/Go proteins and cAMP.
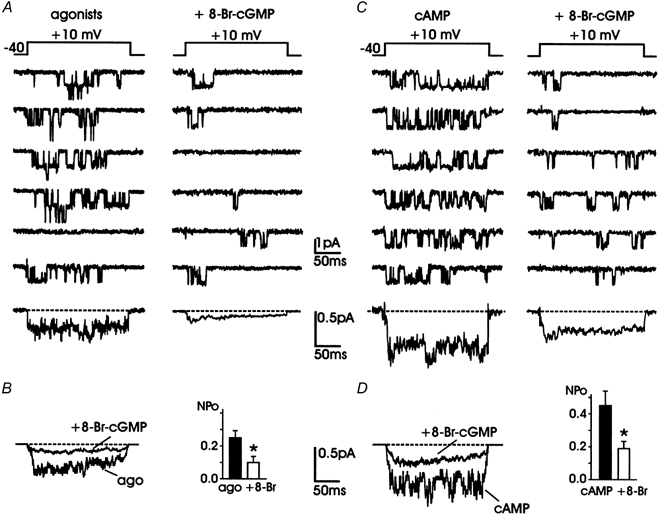
A, L-channel activity recorded in the presence of purinergic and opioidergic receptor agonists (100 μM ATP, 10 μM DAMGO and 1 μM DPDPE) in the patch pipette (left) is inhibited by cell exposure to 400 μM 8-Br-cGMP (right). Averaged currents from 10 traces in the presence of agonists alone and 40 traces in the presence of agonists + 8-Br-cGMP are shown at the bottom. B shows the averaged currents and the NPo values derived from the data of 6 patches. NPo with the agonists was 0.25 ± 0.04 (▪) and decreased to 0.10 ± 0.03 (* P < 0.02) with 8-Br-cGMP (□). C, L-channel activity recorded from a chromaffin cell incubated for 30 min with 8-CPT-cAMP (1 mm) (left) in which a single L-channel displayed high-Po activity. Addition of 400 μM 8-Br-cGMP (right) produced a marked inhibition, which started to become significant from the third minute of application. The selected traces to the right were recorded between the third and sixth minute of drug application. Bottom traces are averaged currents obtained from 10 (cAMP) and 40 traces (cAMP + 8-Br-cGMP; right). Notice their larger amplitude with respect to those in A. D shows the averaged currents and the NPo values derived from 4 patches. NPo with 8-CPT-cAMP was 0.45 ± 0.08 (▪) and decreased to 0.19 ± 0.04 (* P < 0.05) with 8-Br-cGMP (□). All the patches contained multichannel openings.
DISCUSSION
We have provided evidence for the existence of an inhibitory action of NO donors on single L-channel gating of bovine chromaffin cells, through the activation of a cGMP/PKG pathway. The action requires 3-5 min to complete and produces a marked lowering of L-channel activity; the channel opens less frequently and stays more closed than normal. There are no appreciable changes in the lifetime of channel openings, the single-channel conductance and the latency of first openings, suggesting that PKG modifies the gating machinery without altering channel activation and ion permeation. The inhibition of L-channel gating by NO proceeds regardless of other modulatory pathways (cAMP/PKA and G proteins), thus uncovering a possible novel site for channel phosphorylation distinct from others, which may help the understanding of the structure and function of different L-channel isoforms, but whose existence still needs to be proved (see Dolphin, 1998; Jiang et al. 2000; Carbone et al. 2001).
The molecular components of NO action on L-channel gating
Qualitatively, our results compare well with those reported in different neuronal and neuroendocrine cells, in which the NO donors induce a scaling down of the whole-cell Ca2+ currents without altering the activation-inactivation time course (Chik et al. 1995; Grassi et al. 1999; Summers et al. 1999). Except for the case of sympathetic neurons (Chen & Schofield, 1995), retinal ganglion cells (Hirooka et al. 2000) and rod photoreceptors (Kurenny et al. 1994), in which NO is shown to produce a modest scaling-up of N-type Ca2+ currents, there is a general agreement that NO donors produce a robust reduction of Ca2+ currents.
There seem to be three distinct pathways by which NO can reduce Ca2+ channel activity. First, NO may interact directly with neuronal and cardiac L-channels by S-nitrosylation of the channel protein (Hu et al. 1997; Summers et al. 1999), in a way similar to that reported for Na+, K+ and NMDA channels (Bolotina et al. 1994; Aizenman et al. 1998; Li et al. 1998; Choi et al. 2000). This action usually occurs more quickly than in the case of indirect effects and proceeds regardless of the levels of cGMP.
Second, NO interferes with Ca2+ channel activity through the activation of a cytosolic guanylate cyclase and subsequent elevation of cGMP/PKG levels. A PKG-mediated inhibition of L-channel activity has been reported in chick cardiac cells (Tohse & Sperelakis, 1991), guinea-pig smooth muscle cells (Tewari & Simard, 1997) and rat pinealocytes (Chik et al. 1995). A cGMP/PKG-mediated inhibition also occurs on the gating of cloned rabbit L-channels expressed in oocytes (Jiang et al. 2000) and it might be also implicated in the cGMP-mediated reduction of high-threshold Ca2+ currents of rat dorsal root ganglion cells (Kim et al. 2000). Our data fit this mechanism nicely. The inhibitory effects of SNP are mimicked by 8-Br-cGMP and prevented by both the NO scavenger (carboxy-PTIO) and the selective inhibitors of guanylate cyclase (ODQ) and protein kinase G (KT-5823).
Third, NO inhibits L-channels through the activation of cGMP-dependent cAMP-phosphodiesterases (PDE), which lowers the levels of cAMP and reverses the cAMP-mediated up-regulation of L-channel activity (Méry et al. 1993). This action is typical of cardiac L-type channels (Wahler & Dollinger, 1995) and is clearly distinct from the PKG-mediated inhibition of the same channel proposed by other authors (Tohse & Sperelakis, 1991; Jiang et al. 2000).
Given this, we thought it of primary interest to verify whether the two above-mentioned mechanisms (i.e. PKG- and PDE-mediated channel inhibitions) could coexist and interact in neuroendocrine L-channels of chromaffin cells. We found that, at variance with the cardiac L-channel, the cAMP/PKA signalling does not interfere with the NO-induced inhibition of L-channels. In our experimental model, the action of SNP does not require high levels of cAMP to proceed, as in cardiac cells (Méry et al. 1993). In fact, the NO-induced inhibition occurs in control cells in which the basal level of cAMP is usually low (Carabelli et al. 2001) and is fully preserved in cells in which the cAMP-PKA pathway is blocked by the PKA inhibitor H89 (Fig. 7). Thus, at variance with cardiac L-channels, all the data point to a PKG-mediated inhibition of the neuroendocrine L-channel of bovine chromaffin cells. Along this line, it is also interesting to notice that, while in cardiac myocytes the endogenous levels of cGMP are sufficient to down-regulate the amplitude of L-type Ca2+ currents through the activation of a cAMP-PDE (Gallo et al. 1998), in bovine chromaffin cells the basal levels of endogenous NO are insufficient to affect L-channel activity through the cGMP/PKG pathway. In fact, in the presence of the NO scavenger (carboxy-PTIO) and the guanylate cyclase inhibitor (ODQ) the L-channel activity is not significantly different from control values (Fig. 4 and Fig. 5). This implies that, in our experimental conditions, the levels of endogenous NO are low, probably because of the absence of physiological stimuli (e.g. an increase in Ca2+ influx) required to activate local NO synthase.
PKG action on single L-channel parameters
Given that the neuroendocrine L-channels of bovine chromaffin cells are down-modulated by a cGMP/PKG-mediated mechanism, the next issue was to identify the single-channel parameters modified by this action. We found that the depressive effect of PKG-phosphorylation is voltage independent between 0 and +20 mV. The action is due to both an increased contribution of the slow component of closed times and increased probability of null sweeps, while mean open times, single-channel conductance and first latencies are practically unchanged. In other words, PKG-phosphorylation switches the channel into a low-Po gating mode with a reduced number of openings but of otherwise similar lifetime and activation kinetics. Notice that the increased contribution of the slowest closed time component with no change of the time constant (tC3 = 127 ms) by NO is able to produce increased mean closed times (decreased Po) with no changes to the mean first latency, whose probability distribution function depends only on the values of the time constants of closed times distribution (Colquhoun & Hawkes, 1981). The increased number of nulls, however, implies that the PKG-modified channel is likely to enter a closed state which may last for several seconds, as shown by the repeated nulls often observed in SNP-treated patches.
The above effects are more or less the opposite of PKA-phosphorylation, which switches cardiac (Hess et al. 1984), neuronal (Kavalali et al. 1997) and neuroendocrine L-channels (Carabelli et al. 2001) into a gating mode of high Po characterized by an increased frequency of channel openings and greater open channel lifetimes. The PKG effects reported here are in qualitative agreement with the only two studies available on single L-channels of cardiac and smooth muscle cells, in which SNP and 8-Br-cGMP are shown to prolong the slow component of closed times (Tohse & Sperelakis, 1991) and reduce the availability of functioning channels (Tewari & Simard, 1997). At variance with these reports, our data point to a voltage-independent inhibitory action of PKG on L-channel gating (Po) due to an increased contribution (not a prolongation) of the slowest closed times and an increased number of nulls. Our data exclude the possibility of a reduced L-channel availability (N). We observed practically the same degree of NO-induced depression in multichannel and single-channel patches (60.6 vs. 61.2 %; Fig. 2A and Fig. 3E). Assuming a homogeneous population of L-channels, a dual effect of NO on both Po and N would be expected to produce a much greater inhibition in multichannel patches.
Our findings represent the only data available on the effects of cGMP/PKG on single neuroendocrine L-channels. So far, there are no comparable reports about the effects of NO on single neuronal and neuroendocrine L-channels. In our experience, however, SNP exerts qualitatively similar effects on the single L-channel of insulin-secreting RINm5F cells (C. Grassi, M. D'Ascenzo & G. B. Azzena, unpublished results), suggesting that the action reported here may be valid in general for different types of neuroendocrine L-channels. Future studies on neuronal cell preparations are thus required to confirm the present findings, which may be helpful for clarifying the role of NO in the control of neuronal excitability.
Site of action of PKG and cross-talk with other L-channel modulations
The voltage-independent inhibition of neuroendocrine L-channel gating induced by PKG is indicative of a modulatory effect, which is independent of the probability of the channel to be open. Very likely, channel inhibition by PKG occurs while the channel is closed at rest and persists at various membrane potentials, introducing no obvious delay of channel openings and changes to the open channel lifetime. This action is impressively similar to the voltage-independent inhibition induced by PTX-sensitive G proteins recently observed on the same L-channel (Carabelli et al. 2001). Activation of Gi/Go protein subunits by either exogenous application or endogenous release of opioids and ATP causes a marked decrease of Po, mainly due to increased closed times and null sweeps. We found this similarity very interesting for understanding the functioning of L-channel gating and thought it worthwhile to test whether the two signalling pathways acted at distinct or closely related binding sites. As shown in Fig. 8A, the effects of the two inhibitors were additive. 8-Br-cGMP was able to halve the NPo in patches in which the G proteins were activated by the presence of purinergic and opiodergic receptor agonists in the pipette. A reason for this is that the Gi/Go protein subunits (most likely βγ) bind at a channel region well separated from the site of action of PKG, which for the cardiac α1C subunit is located at position Ser533 in the I-II cytoplasmic linker (Jiang et al. 2000). This is in agreement with the idea that there are no apparent consensus sequences for Gβγ subunits in the I-II cytoplasmic linker on both the α1C and α1D L-channel isoforms (Bell et al. 2001). In this case, the alternative is that Gβγ binds at the C-terminal of L-channels, where the site of cAMP phosphorylation is likely to coexist (at Ser1928 in rabbit α1C subunit; Gao et al. 1997). This would be in agreement with two main observations: (1) PKG and PKA act in parallel on L-channel gatings (Tewari & Simard, 1997; see also Fig. 8); and (2) cAMP prevents the action of G protein subunits (Carabelli et al. 2001). However, since there are not yet clear indications of which G protein subunits are involved in neuroendocrine L-channel inhibition, we cannot exclude the possibility that PKG and G protein subunits act on the same channel region (I-II cytoplasmic linker) which, at the moment, appears to be a critical site for controlling Ca2+ channel activation (Dolphin, 1998).
Relevance of L-channel modulation by NO to catecholamine release
The effect of L-channel block by NO-mediated activation of PKG is expected to be relevant in those cells in which neuroendocrine L-channels play a critical role in the regulation of cell activity. L-channels contribute to a variable fraction of the total current (20-50 %) and are shown to be determinant in the control of catecholamine release during nicotinic receptor activation in feline (Lopez et al. 1994), bovine (Lomax et al. 1997) and rat chromaffin cells (Kim et al. 1995). In particular, L-channel activation dominates the nicotinic induced release of catecholamines from rat adrenal glands and the role played by these channels is greater in the secretion of noradrenaline than in that of adrenaline (Nagayama et al. 1999). Thus, the presently described inhibitory action of NO on neuroendocrine L-channel activity may be crucial to the control of catecholamine release in chromaffin cells.
Our findings are in agreement with a number of reports showing that NO and NO donors inhibit Ca2+ entry and the corresponding exocytosis of catecholamines evoked by strong stimuli. In bovine chromaffin cells, NO, SNP and 8-Br-cGMP produce a marked inhibition of ACh- and KCl-stimulated catecholamine secretion and opposite effects on basal release (Oset-Gasque et al. 1994). The action on ACh- and KCl-induced secretion appears linked to a decrease of Ca2+ fluxes associated with P/Q- rather than L-type channels (Rodriguez-Pascual et al. 1994). Notice, however, that these data were derived from fluorescence measurements of intracellular Ca2+ concentrations and not from whole-cell or single-channel current recordings, as in our case. Thus, specific inhibitory effects of NO on L-type channels could be partly overlooked. Our data do not exclude a NO-mediated action on P/Q-type channels, as already reported in RINm5F cells (Grassi et al. 1999). On the contrary, they highlight the existence of a PKG-mediated inhibition of L-channels that might work in concert with the cAMP-dependent up-regulation and G protein-mediated down-modulation of the same channel in the autocrine/paracrine control of catecholamine release (Fig. 7 and Fig. 8).
Since NO can be made available from different sources, including the endothelial cells of closely packed capillary vessels (Torres et al. 1994), the preganglionic sympathetic fibres (Dun et al. 1993) and the chromaffin cells themselves (Dun et al. 1993; Moro et al. 1993; Oset-Gasque et al. 1994), the inhibitory action of neuroendocrine L-channels mediated by the NO/PKG signalling pathway may represent an effective feedback system to regulate Ca2+ entry and catecholamine secretion during massively stimulated adrenal gland activity. However, these effects do not account for the NO-mediated increase of catecholamine release under basal conditions (O'Sullivan & Burgoygne, 1990; Oset-Gasque et al. 1994). A possibility, which needs to be tested, is that the positive effects of NO on basal release derive from an increased Ca2+ mobilization induced via either a PKG-mediated elevation of Ca2+-mobilizing agents (Willmott et al. 1995; Clementi et al. 1996) or direct nitrosylation of ryanodine receptors (Stoyanovsky et al. 1997).
Acknowledgments
We thank Dr Pietro Baldelli and Tiziana Cesetti for helpful discussions and Dr Paola Borgiani for assistance with part of the statistical analysis. This work was supported by the Italian MIUR, the CNR, and by the Turin and Catholic Universities’ local funds.
REFERENCES
- Aizenman E, Brimecombe JC, Potthoff WK, Rosenberg PA. Why is the role of nitric oxide in NMDA receptor function and dysfunction so controversial? Progress in Brain Research. 1998;118:53–71. doi: 10.1016/s0079-6123(08)63200-8. [DOI] [PubMed] [Google Scholar]
- Barry PH, Lynch JW. Liquid junction potentials and small cell effects in patch-clamp analysis. Journal of Membrane Biology. 1991;121:101–117. doi: 10.1007/BF01870526. [DOI] [PubMed] [Google Scholar]
- Bates JN, Baker MT, Guerra R, Harrison DG. Nitric oxide generation from nitroprusside by vascular tissue. Evidence that reduction of the nitroprusside anion and cyanide loss are required. Biochemical Pharmacology. 1991;42:S157–165. doi: 10.1016/0006-2952(91)90406-u. [DOI] [PubMed] [Google Scholar]
- Bell DC, Butcher AJ, Berrow NS, Page KM, Brust PF, Nesterova A, Stauderman KA, Seabrook GR, Nürnberg B, Dolphin AC. Biophysical properties, pharmacology, and modulation of human, neuronal L-Type (α1D, CaV1. 3) voltage-dependent calcium currents. Journal of Neurophysiology. 2001;85:816–827. doi: 10.1152/jn.2001.85.2.816. [DOI] [PubMed] [Google Scholar]
- Bolotina VM, Najibi S, Palacino JJ, Pagano PJ, Cohen RA. Nitric oxide directly activates calcium-dependent potassium channels in vascular smooth muscle. Nature. 1994;368:850–853. doi: 10.1038/368850a0. [DOI] [PubMed] [Google Scholar]
- Bredt DS, Snyder SH. Isolation of nitric oxide synthetase, a calmodulin-requiring enzyme. Proceedings of the National Academy of Sciences of the USA. 1990;87:682–685. doi: 10.1073/pnas.87.2.682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkhardt M, Glazova M, Gambaryan S, Vollkommer T, Butt E, Bader B, Heermeir K, Lincoln TM, Walter U, Palmetshofer A. KT5823 inhibits cGPM-dependent protein kinase activity in vitro but not in intact human platelets and rat mesangial cells. Journal of Biological Chemistry. 2000;275:33 536–33 541. doi: 10.1074/jbc.M005670200. [DOI] [PubMed] [Google Scholar]
- Carabelli V, Carra I, Carbone E. Localised secretion of ATP and opioids revealed through single Ca2+ channel modulation in bovine chromaffin cells. Neuron. 1998;20:1255–1268. doi: 10.1016/s0896-6273(00)80505-x. [DOI] [PubMed] [Google Scholar]
- Carabelli V, Hernández-Guijo JM, Baldelli P, Carbone E. Direct autocrine inhibition and cAMP-dependent potentiation of single L-type Ca2+ channels in bovine chromaffin cells. Journal of Physiology. 2001;532:73–90. doi: 10.1111/j.1469-7793.2001.0073g.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carabelli V, Lovallo M, Magnelli V, Zucker H, Carbone E. Voltage-dependent modulation of single N-type Ca2+ channel kinetics by receptor agonists in IMR32 cells. Biophysical Journal. 1996;70:2144–2154. doi: 10.1016/S0006-3495(96)79780-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbone E, Carabelli V, Baldelli P, Cesetti T, Hernández-Guijo JM, Giusta L. G-protein- and cAMP-dependent L-channel gating modulation: a manyfold system to control calcium entry in neurosecretory cells. Pflügers Archiv. 2001;442:801–813. doi: 10.1007/s004240100607. [DOI] [PubMed] [Google Scholar]
- Chen C, Schofield GG. Nitric oxide donors enhanced Ca2+ currents and blocked noradrenaline-induced Ca2+ current inhibition in rat sympathetic neurons. Journal of Physiology. 1995;482:521–531. doi: 10.1113/jphysiol.1995.sp020537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chik CL, Liu Q-Y, Li B, Karspinski E, Ho AK. cGMP inhibits L-type Ca2+ channel currents through protein phosphorylation in rat pinealocytes. Journal of Neuroscience. 1995;15:3104–3109. doi: 10.1523/JNEUROSCI.15-04-03104.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi YB, Tenneti L, Le DA, Ortiz J, Bai G, Chen HS, Lipton SA. Molecular basis of NMDA receptor-coupled ion channel modulation by S-nitrosylation. Nature Neuroscience. 2000;3:15–21. doi: 10.1038/71090. [DOI] [PubMed] [Google Scholar]
- Clementi E, Riccio M, Sciorati C, Nisticò G, Meldolesi J. Molecular basis of NMDA receptor-coupled ion channel modulation by S-nitrosylation. Journal of Biological Chemistry. 1996;271:17 739–17 745. [Google Scholar]
- Colquhoun D, Hawkes AG. On the stochastic properties of single ion channels. Proceedings of the Royal Society B. 1981;211:205–235. doi: 10.1098/rspb.1981.0003. [DOI] [PubMed] [Google Scholar]
- Dolphin AC. Mechanisms of modulation of voltage-dependent calcium channels by G proteins. Journal of Physiology. 1998;506:3–11. doi: 10.1111/j.1469-7793.1998.003bx.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dun NJ, Dun SL, Wu SY, Forstermann U. Nitric oxide synthase immunoreactivity in rat superior cervical ganglia and adrenal glands. Neuroscience Letters. 1993;158:51–54. doi: 10.1016/0304-3940(93)90610-w. [DOI] [PubMed] [Google Scholar]
- Gallo MP, Ghigo D, Bosia A, Alloatti G, Costamagna C, Penna C, Levi RC. Modulation of guinea-pig cardiac L-type calcium current by nitric oxide synthase inhibitors. Journal of Physiology. 1998;506:639–654. doi: 10.1111/j.1469-7793.1998.639bv.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao TY, Yatani A, Dell'Acqua ML, Sako H, Green SA, Dascal N, Scott J, Hosey MM. cAMP-dependent regulation of cardiac L-type Ca2+ channels requires membrane targeting of PKA and phosphorylation of channel subunits. Neuron. 1997;19:185–196. doi: 10.1016/s0896-6273(00)80358-x. [DOI] [PubMed] [Google Scholar]
- García AG, Sala F, Reig JA, Viniegra S, Frías J, Fonteriz RI, Gandía L. Dihydropyridine BAY-K-8644 activates chromaffin cell calcium channels. Nature. 1984;309:69–71. doi: 10.1038/309069a0. [DOI] [PubMed] [Google Scholar]
- Garthwaite J, Southam E, Boulton CL, Nielsen EB, Schmidt K, Mayer B. Potent and selective inhibition of nitric oxide-sensitive guanylyl cyclase by 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one. Molecular Pharmacology. 1995;48:148–188. [PubMed] [Google Scholar]
- Grassi C, D'Ascenzo M, Valente A, Azzena GB. Ca2+ channel inhibition induced by nitric oxide in rat insulinoma RINm5F cells. Pflügers Archiv. 1999;437:241–247. doi: 10.1007/s004240050775. [DOI] [PubMed] [Google Scholar]
- Grassi C, Santarelli R, Nisticò S, Bagetta G, Azzena GB. Possible modulation of auditory middle latency responses by nitric oxide in the inferior colliculus of anaesthetized rats. Neuroscience Letters. 1995;196:213–217. doi: 10.1016/0304-3940(95)11872-t. [DOI] [PubMed] [Google Scholar]
- Grider JR. Interplay of VIP and nitric oxide in regulation of the descending relaxation phase of peristalsis. American Journal of Physiology. 1993;264:G334–340. doi: 10.1152/ajpgi.1993.264.2.G334. [DOI] [PubMed] [Google Scholar]
- Haley JE, Dickenson AH, Schachter M. Electrophysiological evidence for a role of nitric oxide in prolonged chemical nociception in the rat. Neuropharmacology. 1992;31:251–258. doi: 10.1016/0028-3908(92)90175-o. [DOI] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Archiv. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Han X, Shimoni Y, Giles WR. An obligatory role for nitric oxide in autonomic control of mammalian heart rate. Journal of Physiology. 1994;476:309–314. doi: 10.1113/jphysiol.1994.sp020132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess P, Lansman JB, Tsien RW. Different modes of Ca channel gating behaviour favoured by dihydropyridine Ca agonists and antagonists. Nature. 1984;311:538–544. doi: 10.1038/311538a0. [DOI] [PubMed] [Google Scholar]
- Hirooka K, Kourennyi DE, Barnes S. Calcium channel activation facilitated by nitric oxide in retinal ganglion cells. Journal of Neurophysiology. 2000;83:198–206. doi: 10.1152/jn.2000.83.1.198. [DOI] [PubMed] [Google Scholar]
- Hu H, Chiamvimonvat N, Yamagishi T, Marban E. Direct inhibition of expressed cardiac L-type Ca2+ channels by S-nitrosothiol nitric oxide donors. Circulation Research. 1997;81:742–752. doi: 10.1161/01.res.81.5.742. [DOI] [PubMed] [Google Scholar]
- Ignarro LJ, Buga GM, Wood KS, Byrns RE, Chaudhuri G. Endothelium-derived relaxing factor produced and released from artery and vein is nitric oxide. Proceedings of the National Academy of Sciences of the USA. 1987;84:9265–9269. doi: 10.1073/pnas.84.24.9265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang LH, Gawler DJ, Hodson N, Milligan CJ, Pearson HA, Porter V, Wray D. Regulation of cloned cardiac L-type calcium channels by cGMP-dependent protein kinase. Journal of Biological Chemistry. 2000;275:6135–6143. doi: 10.1074/jbc.275.9.6135. [DOI] [PubMed] [Google Scholar]
- Kavalali ET, Hwang KS, Plummer MR. cAMP-dependent enhancement of dihydropyridine-sensitive calcium channel availability in hippocampal neurons. Journal of Neuroscience. 1997;17:5334–5348. doi: 10.1523/JNEUROSCI.17-14-05334.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SJ, Lim W, Kim J. Contribution of L- and N-type calcium currents to exocytosis in rat adrenal medullary chromaffin cells. Brain Research. 1995;675:289–296. doi: 10.1016/0006-8993(95)00085-5. [DOI] [PubMed] [Google Scholar]
- Kim SJ, Song SK, Kim J. Inhibitory effect of nitric oxide on voltage-dependent calcium currents in rat dorsal root ganglion cells. Biochemical and Biophysical Research Communications. 2000;271:509–514. doi: 10.1006/bbrc.2000.2658. [DOI] [PubMed] [Google Scholar]
- Kurenny DE, Moroz LL, Turner RW, Sharkey KA, Barnes S. Modulation of ion channels in rod photoreceptors by nitric oxide. Neuron. 1994;13:315–324. doi: 10.1016/0896-6273(94)90349-2. [DOI] [PubMed] [Google Scholar]
- Lambert RC, Feltz A. Maintained L-type Ca2+ channel activity in excised patches of PTX-treated granule cells of the cerebellum. Journal of Neuroscience. 1995;15:6014–6022. doi: 10.1523/JNEUROSCI.15-09-06014.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang RJ, Harvey JR, McPhee GJ, Klemm MF. Nitric oxide and thiol reagent modulation of Ca2+-activated K+ (BKCa) channels in myocytes of the guinea-pig taenia caeci. Journal of Physiology. 2000;525:363–376. doi: 10.1111/j.1469-7793.2000.00363.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Chapleau MW, Bates JN, Bielefeldt K, Lee H-C, Abboud M. Nitric oxide as an autocrine regulator of sodium currents in baroreceptor neurons. Neuron. 1998;20:1039–1049. doi: 10.1016/s0896-6273(00)80484-5. [DOI] [PubMed] [Google Scholar]
- Lomax RB, Michelena P, Nunez L, García-Sancho J, García AG, Montiel C. Different contributions of L- and Q-type Ca2+ channels to Ca2+ signals and secretion in chromaffin cell subtypes. American Journal of Physiology. 1997;272:C476–484. doi: 10.1152/ajpcell.1997.272.2.C476. [DOI] [PubMed] [Google Scholar]
- Lopez MG, Villaroya M, Lara B, Sierra RM, Albillos A, García AG, Gandía L. Q- and L-type Ca2+ channels dominate the control of secretion in bovine chromaffin cells. FEBS Letters. 1994;349:331–337. doi: 10.1016/0014-5793(94)00696-2. [DOI] [PubMed] [Google Scholar]
- Machado JD, Segura F, Brioso MA, Borges R. Nitric oxide modulates a late step of exocytosis. Journal of Biological Chemistry. 2000;275:20 274–20 279. doi: 10.1074/jbc.M000930200. [DOI] [PubMed] [Google Scholar]
- Méry PF, Pavoine C, Belhassen L, Pecker F, Fishmeister R. Nitric oxide regulates cardiac Ca2+ current. Involvement of cGMP-inhibited and cGMP-stimulated phosphodiesterases through guanylyl cyclase activation. Journal of Biological Chemistry. 1993;268:26 286–26 295. [PubMed] [Google Scholar]
- Moro MA, Michelena P, Sanchez-García P, Palmer R, Moncada S, García AG. Activation of adrenal medullary l-arginine: nitric oxide pathway by stimuli which induce the release of catecholamines. European Journal of Pharmacology. 1993;246:213–218. doi: 10.1016/0922-4106(93)90033-6. [DOI] [PubMed] [Google Scholar]
- Nagayama T, Hosokawa A, Yoshida M, Suzuki-Kusaba M, Hisa H, Kimura T, Satoh S. Role of nitric oxide in adrenal catecholamine secretion in anesthetized dogs. American Journal of Physiology. 1998;275:R1075–1081. doi: 10.1152/ajpregu.1998.275.4.R1075. [DOI] [PubMed] [Google Scholar]
- Nagayama T, Matsumoto T, Kuwakubo F, Fukushima Y, Yoshida M, Suzuki-Kusaba M, Hisa H, Kimura T, Satoh S. Role of calcium channels in catecholamine secretion in the rat adrenal gland. Journal of Physiology. 1999;520:503–512. doi: 10.1111/j.1469-7793.1999.00503.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neher E. Correction for liquid junction potentials in patch clamp experiments. Methods in Enzymology. 1992;207:123–131. doi: 10.1016/0076-6879(92)07008-c. [DOI] [PubMed] [Google Scholar]
- Oset-Gasque MJ, Parramon M, Hortelano S, Bosca L, Gonzales MP. Nitric oxide implication in the control of neurosecretion by chromaffin cells. Journal of Neurochemistry. 1994;63:1693–1700. doi: 10.1046/j.1471-4159.1994.63051693.x. [DOI] [PubMed] [Google Scholar]
- O'Sullivan AJ, Burgoyne RD. Cyclic GMP regulates nicotine-induced secretion from cultured bovine adrenal chromaffin cells: effects of 8-bromo-cyclic GMP, atrial natriuretic peptide, and nitroprusside (nitric oxide) Journal of Neurochemistry. 1990;54:1805–1808. doi: 10.1111/j.1471-4159.1990.tb01238.x. [DOI] [PubMed] [Google Scholar]
- Palmer RMJ, Ferrige AG, Moncada S. Nitric oxide release accounts for the biological activity of endothelium-derived relaxing factor. Nature. 1987;327:524–526. doi: 10.1038/327524a0. [DOI] [PubMed] [Google Scholar]
- Rauch M, Schmid HA, de Vente J, Simon E. Electrophysiological and immunocytochemical evidence for a cGMP-mediated inhibition of subfornical organ neurons by nitric oxide. Journal of Neuroscience. 1997;17:363–371. doi: 10.1523/JNEUROSCI.17-01-00363.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Pascual F, Miras-Portugal MT, Torres M. Modulation of the dihydropyridine-insensitive Ca2+ influx by 8-bromo-guanosine-3′:5′-monophosphate, cyclic (8-Br-cGMP) in bovine adrenal chromaffin cells. Neuroscience Letters. 1994;180:269–272. doi: 10.1016/0304-3940(94)90536-3. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Pascual F, Miras-Portugal MT, Torres M. Effect of cyclic GMP-increasing agents nitric oxide and C-type natriuretic peptide on bovine chromaffin cell function: inhibitory role mediated by cyclic GMP-dependent protein kinase. Molecular Pharmacology. 1996;49:1058–1070. [PubMed] [Google Scholar]
- Shuman EM, Madison DV. Nitric oxide and synaptic function. Annual Review of Neuroscience. 1994;17:153–183. doi: 10.1146/annurev.ne.17.030194.001101. [DOI] [PubMed] [Google Scholar]
- Schwarz PM, Rodriguez-Pascual F, Koesling D, Torres M, Förstermann U. Functional coupling of nitric oxide synthase and soluble guanylyl cyclase in controlling catecholamine secretion from bovine chromaffin cells. Neuroscience. 1998;82:255–265. doi: 10.1016/s0306-4522(97)00274-1. [DOI] [PubMed] [Google Scholar]
- Stoyanovsky D, Murphy T, Anno PR, Kim Y-M, Salama G. Nitric oxide activates skeletal and cardiac ryanodine receptors. Cell Calcium. 1997;21:19–29. doi: 10.1016/s0143-4160(97)90093-2. [DOI] [PubMed] [Google Scholar]
- Summers BA, Overholt JL, Prabhakar NR. Nitric oxide inhibits L-type Ca2+ current in glomus cells of the rabbit carotid body via a cGMP-independent mechanism. Journal of Neurophysiology. 1999;81:1449–1457. doi: 10.1152/jn.1999.81.4.1449. [DOI] [PubMed] [Google Scholar]
- Tewari K, Simard JM. Sodium nitroprusside and cGMP decrease Ca2+ availability in basilar artery smooth muscle cells. Pflügers Archiv. 1997;433:304–311. doi: 10.1007/s004240050281. [DOI] [PubMed] [Google Scholar]
- Tohse N, Sperelakis N. cGMP inhibits the activity of single calcium channels in embryonic chick heart cells. Circulation Research. 1991;69:325–331. doi: 10.1161/01.res.69.2.325. [DOI] [PubMed] [Google Scholar]
- Torres M, Ceballos G, Rubio R. Possible role of nitric oxide in catecholamine secretion by chromaffin cells in the presence and absence of cultured endothelial cells. Journal of Neurochemistry. 1994;63:988–996. doi: 10.1046/j.1471-4159.1994.63030988.x. [DOI] [PubMed] [Google Scholar]
- Wahler GM, Dollinger SJ. Nitric oxide donor SIN-1 inhibits mammalian cardiac calcium current through cGMP-dependent protein kinase. American Journal of Physiology. 1995;426:C45–54. doi: 10.1152/ajpcell.1995.268.1.C45. [DOI] [PubMed] [Google Scholar]
- Willmott NJ, Galione A, Smith PA. Nitric oxide induces Ca2+ mobilization and increases secretion of incorporated 5-hydroxytryptamine in rat pancreatic β-cells. FEBS Letters. 1995;371:1981–1992. doi: 10.1016/0014-5793(95)00848-4. [DOI] [PubMed] [Google Scholar]