

Abstract
To compare the anorectic effectiveness of leptin and the amylin analogue salmon calcitonin (sCT), rodents were treated on 1 day with subcutaneous injections. In chow-fed C57Bl/6J mice, leptin and sCT reduced energy intake and acted additively. After C57Bl/6J mice had become leptin-resistant on being fed chocolate as a palatable high-caloric supplement to chow, their sCT-induced decrease in energy intake was more pronounced than in chow-fed mice with differential changes in the intake of chocolate (strong reduction) and chow (slight increase). Dose-response relationships for sCT-induced reductions in energy intake were analysed in chow-fed C57Bl/6J mice and two obese strains, ob/ob mice and melanocortin-4 receptor knockout (MC4-r-KO) mice, as well as in wild-type and fatty (fa/fa) rats. Compared to C57Bl/6J mice, reduction in food intake induced by sCT was attenuated in MC4-r-KO mice, and nearly absent in ob/ob mice, over the dose range investigated. Compared to C57Bl/6J mice, wild-type rats responded more sensitively to sCT and its efficiency was only slightly reduced in fatty (fa/fa) rats. Thus, while genetically induced failures of leptin signalling reduce the action of sCT, it effectively inhibits the intake of a palatable, high fat-high sugar diet even in states of diet-induced obesity with functional leptin resistance.
When lean and certain strains of obese rodents, chow-fed ad libitum, are acutely treated with recombinant leptin, the leptin immediately suppresses food intake by acting on arcuate nucleus (ARC) neurones as its brain target, and long-term continuation of the treatment results in dose-dependent reductions in body fat content (Pelleymounter et al. 1998). Models of genetic obesity in which leptin itself is absent or its receptors are non-functional highlight the physiological contribution of leptin to the maintenance of energy balance (Zhang et al. 1994; Chua et al. 1996). On the other hand, in humans with access to the type of food that is typical of industrialised societies, most forms of acquired obesity are not caused by deficient leptin production or primary receptor defects but rather by functional leptin resistance (Sinha & Caro, 1998). In obese humans, leptin treatment, in combination with dietary restriction, exerts little or no enhancing action on body weight loss, similar to its weak effect in lean subjects, and it does not enhance the compliance with caloric dietary restriction in obese subjects (Heynsfield et al. 1998; Hukshorn et al. 2000). As a laboratory analogue, it was shown that primarily leptin-responsive rodents maintained on high-caloric diets became resistant within 8-15 weeks to leptin applied systemically, i.e. reaching the brain via its normal route (Widdowson et al. 1997; Lu et al. 1998; Lin et al. 2000).
However, despite leptin resistance in humans and laboratory animals acquired under high-caloric diets, some control of food intake may persist, reflecting redundancy of biological control in the central regulatory system controlling body weight (Proietto et al. 2000). In particular amylin, a peptide cosecreted with insulin in humans (Juhl et al. 2000) and in laboratory rodents (Stridsberg et al. 1993; Leckström et al. 1999), has been recently categorised as an ‘adiposity signal’, along with leptin and insulin (Woods & Seeley, 2000). The central target for amylin, a circulating peptide that decreases meal size in addition to its neurally mediated slowing effect on intestinal motility (Lutz et al. 1995), was identified as the area postrema (AP) (Lutz et al. 1997), where the blood- brain barrier is absent and amylin has access to and specifically excites neurones (Riediger et al. 2001).
The present study was designed to investigate the efficiency of salmon calcitonin (sCT) compared to leptin. sCT is an easily available amylin analogue which has been shown to act effectively as an anorectic compound via amylin receptors of the AP (Lutz et al. 2000). Experimental animals were C57Bl/6J mice fed chow or chow plus a palatable high-caloric supplement diet, two obesity mouse models, knockout mice lacking the melanocortin-4 receptor (MC4-r-KO) and the leptin-deficient ob/ob mouse. In addition, sCT responsiveness was studied in fatty (fa/fa) rats carrying a leptin receptor defect (Chua et al. 1996) and in wild-type (+/+) controls.
METHODS
Animals and food intake measurements
Food intake studies were carried out in adult C57Bl/6J (n = 61), ob/ob (C57Bl/6J background, n = 17) and homozygous MC4-r-KO (n = 16) mice and in wild-type and fatty (fa/fa) Zucker rats (n = 4/4). All animals in each experimental group were of the same sex and age. C57Bl/6J and ob/ob mice were either received from Harlan Winkelmann (Borchen, FRG) or born in our own colony. MC4-r-KO mice were reared in our breeding colony derived from breeding pairs kindly provided by Dennis Huszar (Millenium Pharmaceuticals, Inc., Cambridge, MA, USA). For studies on wild-type and fa/fa rats, a strain reared in our own colony was used. Animals were maintained in single cages at 22 °C and a 12 :12 light : dark cycle with water and pelleted chow (Altromin, Lage, FRG) ad libitum. Pellets were fixed in a device that allowed us to correct measurements for lost crumbs. Two experimental groups of C57Bl/6J mice (both n = 10) additionally received white chocolate (Wissoll, Muehlheim, FRG), a highly palatable, high-caloric (PHC) dietary supplement (24 kJ g−1), with caloric contents consisting of 39 % carbohydrates, 56 % fat and 5 % proteins compared to 62 % carbohydrates, 10 % fat and 28 % proteins for the standard chow (17 kJ g−1). Body mass and food intake were determined daily at the end of the light phase. To supplement body weight determination with data for fat content, body composition was determined for some of the animal samples as previously described (Markewicz et al. 1993). These animals were exposed to CO2 for 30 s and then decapitated. For plasma insulin measurement blood was collected after decapitation or by retroorbital puncturing after short anaesthesia (< 20 s) with a CO2 : O2 (3 : 1) mixture. All procedures were carried out according to the German guidelines for animal experimentation and approved by the local veterinary control institution for animal care and use.
Treatment protocol
On the day before treatment, average food intake during the preceding 2 days and the current body masses were matched as closely as possible between one half of the experimental group of mice receiving sCT and/or leptin and the other half serving as controls and receiving phosphate-buffered saline (PBS). sCT (Calcitonin-ratiopharm 50, Mr = 3432 Da, 1 i.u. = 0.167 μg) was injected subcutaneously (s.c.) or in a few cases intraperitoneally, in various doses 1 h before lights-off. In some experiments, sCT was given in two equal doses at 5 and 1 h before lights-off, but as no significant difference between different application protocols was observed, data were pooled for evaluation. Rats were treated with sCT according to the schedule established in mice. Following a previous study (Doering et al. 1998), leptin was injected s.c. in two equal doses at the beginning and the end of the light phase at an overall dose of 200 pmol g−1 day−1 (R&D Systems GmbH, Wiesbaden, FRG, Mr = 16 kDa). Dose calculations are based on the body masses determined on the treatment day. Repeated treatments within the same experimental group were carried out at intervals of at least 4 days and with experimental and control groups reversed.
Evaluation
Energy intake was calculated assuming a gross caloric content of 17 kJ g−1 for the chow and 24 kJ g−1 for the chocolate. The treatment effects were evaluated during the 24 h period starting on the evening after the injections (the effect period). For each treated animal, the individual deviation of its energy intake from the average intake of the control animals was calculated (a) for the effect period and (b) for the 2 following days and the 2 days preceding the treatment day, the latter time periods serving as control days to account for the mostly minor chance differences existing between the individual and its control mean. By calculating the difference between the average deviation during the control days and the deviation in the effect period the individual net treatment effect was obtained.
Apart from determining the effects in absolute terms, they were also calculated as percentage changes relative to the energy intake during the two pre-treatment days. Least square means ± s.e.m. were calculated by 2-way ANOVA (SigmaStat) for C57Bl/6J mice, with pharmacological treatment and experimental days as factors, and separate calculations were made for the two diet components. For comparison of the effects on energy intake under the two diets (chow and chow + PHC diet) the factors were diet and treatment.
To compare the responsiveness to sCT of the different strains investigated, dose-response relationships were analysed both per animal and per unit body mass by calculating log-linear regressions. For the data obtained from mice in the dose range of 50-500 pmol day−1 (≈1-9 pmol g−1 day−1), in which differences were most distinct, comparisons between strains were made by applying 2-way ANOVA to the sCT-treated animals with strain and dose as factors. Within each strain the effects seen in the sCT treated vs. control animals were determined by 2-way ANOVA with dose and treatment as factors. The same analysis was carried out on the rat data obtained in the dose range of 1-9 pmol g−1 day−1 (≈750-5000 pmol day−1 for fa/fa and ≈400-2500 pmol day−1 for +/+ rats).
RESULTS
Effects of leptin and sCT on chow-intake in normal mice are additive
In chow-fed C57Bl/6J mice, leptin significantly decreased 24-h post-treatment food intake by 0.6 g or 13 % (P < 0.05, Fig. 1). In comparison, an approximately 100-fold lower dose of sCT (2.1 pmol g−1 day−1 sCT vs. 200 pmol g−1 day−1 leptin) had a more potent effect, reducing chow intake by twice as much (1.2 g or 26 %, P < 0.001). Combining both peptides reduced food intake by 2 g or 43 % (P < 0.001), suggesting an additive effect of leptin and sCT on food intake in normal, chow-fed mice.
Figure 1. Effects of 1-day treatments with leptin, sCT or a combination of both on energy intake of chow-fed C57Bl/6 mice.
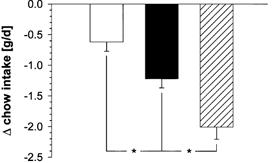
Shown are the average differences in 24-h post-treatment energy intake (least square means + s.e.m.) between the experimental and the control animals (n = 14). Leptin (□, n = 13): 200 pmol g−1 day−1; sCT (▪, n = 14): 2.1 pmol g−1 d−1; combined treatment ( , n = 8). *P < 0.05.
, n = 8). *P < 0.05.
sCT acts in C57Bl/6J mice made leptin-resistant by a supplementary PHC diet
When normal C57Bl/6J mice were offered white chocolate as a supplementary PHC diet in addition to standard chow (Fig. 2), their chow intake decreased to less than 20 % of normal (not shown), approximately 85 % of the total gross caloric intake being now derived from chocolate. After more than 40 days on this supplementary diet, C57Bl/6J mice no longer responded to leptin treatment with decreases in intake, neither for chow nor chocolate (Fig. 2A-C, left panel). In contrast, a more than 100-fold lower sCT dose (1.6 pmol g−1 day−1), administered first to half of the animal sample and, after a 6 day interval, to the other half, potently inhibited chocolate intake. The inhibition was associated with a slight but significant increase in chow intake (Fig. 2A-C, middle panels), but this did not compensate energetically for the reduced chocolate intake (Fig. 2C). Repetition of the leptin injection after the sCT treatment confirmed the complete lack of leptin effects on food intake under this dietary regimen (Fig. 2A-C, right panel).
Figure 2. Effects of 1-day treatments with leptin or sCT on chow and chocolate intake of C57Bl/6 mice which had received both foods simultaneously for > 40 days.
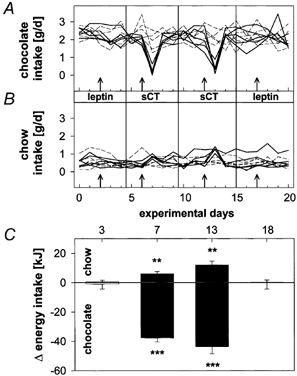
Shown are time courses of daily chocolate (A) and chow intakes (B). Arrows indicate treatment with leptin (200 pmol g−1 day−1) or sCT (1.6 pmol g−1 day−1). Continuous lines: experimental animals (n = 5); dashed lines: control animals (n = 5). After the first sCT treatment, control animals were used as experimental animals, and vice versa. C, average differences in 24-h post-treatment energy intake (least square means + s.e.m.) between the experimental and the control animals, shown separately for chocolate and chow after treatment with leptin (outer columns) or sCT (inner columns). **P < 0.01, ***P < 0.001.
Figure 3 compares the changes in energy intake induced by leptin and sCT, depending on the diet, expressed as a percentage of the pre-treatment energy intake. The distinct anorectic effect of leptin seen in chow-fed animals had completely disappeared after 40 days of supplementary chocolate feeding and was not re-established by the intervening sCT treatments. In contrast to the loss of leptin responsiveness, the anorectic sCT effect was doubled in chocolate-fed animals compared to the effect in the chow-fed animals. When calculating cumulative energy intakes for 10-day periods, the average intake of mice on combined PHC + chow (n = 10) was 613 ± 16 kJ which was significantly higher (P < 0.02 than 532 ± 26 kJ determined for the same mice receiving only chow for 10 subsequent days. This difference corresponded to the observation that the average body weight of 31 ± 1 g attained by the mice at the end of the combined PHC + chow feeding periods (n = 10) was significantly higher (P < 0.001) than the weight of 24 ± 1 g attained by mice of the same age after chow feeding (n = 41), and body composition analysis showed a body fat content of 32 ± 2 % for the PHC-fed mice, i.e. about twice the normal body fat content of chow-fed mice.
Figure 3. Comparison between the effects of 1-day treatments with leptin or sCT on energy intake of C57Bl/6J mice maintained either on chow only or on chow plus chocolate.
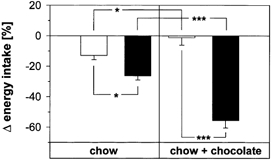
Shown are average differences in 24-h post-treatment energy intake (least square means + s.e.m. between the experimental and the control animals corresponding to Figs 1 and 2, expressed as a percentage of the average intake during the two pre-treatment days. Leptin (□): 200 pmol g−1 day−1; sCT (▪): 1.6 pmol g−1 day−1. Chow-fed mice (left panel): n = 13 leptin-treated mice, n = 14 sCT-treated mice, n = 14 controls; chow + chocolate-fed mice (right panel): n = 10 leptin-treated mice, n = 10 sCT-treated mice, n = 10 controls. *P < 0.05, ***P < 0.001. Chow-fed mice were 3 and 7 months old with no detectable difference between groups; animals fed the supplementary PHD diet were 7 months old.
Mice that received the PHC-diet for a longer period (3 months) and had become leptin resistant responded to a sCT treatment of 1.5 pmol g−1 day−1 with a reduction of energy intake of −38 ± 7 kJ, corresponding to −50 ± 7 % (n = 5/5), i.e. their responsiveness was at least as pronounced as that of the PHC-fed animals shown in Fig. 2. Thus, more prolonged PHC feeding did not attenuate the sCT effect, and Fig. 4 rather suggests that sCT responsiveness may even have increased with the degree of obesity in PHC-fed animals since the sCT treatment reduced energy intake progressively with increasing body mass.
Figure 4. Correlation between body mass and sCT responsiveness in PHC-fed C57Bl/6J mice.
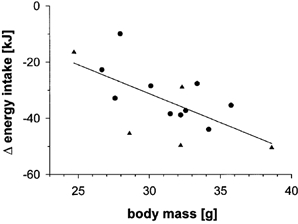
▴, mice having received PHC diet for > 80 days (sCT dose 1.5 pmol g−1 day−1, n = 5); •, mice on PHC diet (sCT dose 1.6 pmol g−1 day−1, n = 2× 5) from which the results of Fig. 2 were obtained. r =×0.60, P < 0.05.
sCT effects are attenuated in chow-fed, genetically obese animal models
In contrast to the C57Bl/6J mice, ob/ob and MC4-r-KO mice are genetically prone to becoming obese because leptin signalling is impaired. The ob/ob defect precludes leptin synthesis, and in homozygous MC4-r-KO mice one out of the two main central pathways emerging from the ARC as a central leptin target is affected: the inhibitory melanocyte stimulating hormone (αMSH) pathway is blocked, while the stimulatory neuropeptide Y (NPY) pathway is preserved. In order to take into account the basal differences in body mass between the strains investigated (C57Bl/6J: 24 ± 1 g; MC4-r-KO: 56 ± 2 g; ob/ob: 60 ± 2 g), the percentage changes in energy intake were related to the sCT dose both per animal and per unit body mass. Figure 5 compares the relative responsiveness to various sCT doses in the three strains investigated by presenting log-linear dose-effect curves with 95 % confidence ranges. Figure 5A shows the log-linear regressions for doses per animal. Figure 5B presents the same relationships for the range of doses per unit body mass up to 10 pmol g−1 day−1. Application of 2-way ANOVA to the data revealed that sCT did act in both C57Bl/6J and MC4-r-KO mice (P < 0.001), but the average response of the MC4-r-KO mice was significantly reduced in comparison to that of the C57Bl/6J mice (P < 0.001). In the ob/ob mice, however, energy intake did not differ between sCT- and PBS-treated animals up to a dose of 9 pmol g−1 day−1 (P =0.65). Not shown in Fig. 5B are findings that the maximum sCT dose of 18 pmol g−1 day−1 applied to lean mice decreased their food intake by nearly 50 %. The same dose per unit body mass applied to ob/ob mice also induced a significant (P < 0.01) but much smaller reduction in food intake by 17 ± 3 % in comparison to control animals (n = 6/6) indicating that obesity due to lack of leptin was associated with a distinctly reduced though not fully abolished sCT responsiveness.
Figure 5. Effects of 1-day treatments with sCT at various doses on energy intake of chow-fed C57Bl/6J mice, two genetically obese mouse strains and of wild-type and fatty (fa/fa) rats.
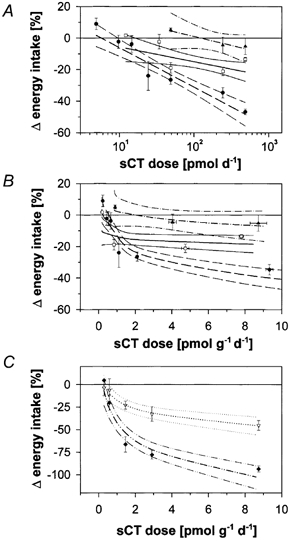
Shown are, for mice, mean values ± s.e.m. and linear regressions with their 95 % confidence ranges of effects vs. log dose per animal (A) and the corresponding relationship for dose per unit body mass with linearized dose scale (B). sCT effects are measured as average differences in 24-h post-treatment energy intake between the experimental and the control animals expressed as a percentage of the average intake during the two pre-treatment days. C57Bl/6J mice (individual treatments: n = 61): - - -, •; MC4-r-KO mice (n = 33):–, □; ob/ob mice (n = 12): - ·- ·, ▴. For rats, mean values ± s.e.m. and regressions were determined in the same way as for the mouse models and are shown for wild-type (·· - ··-, ♦) and fatty (fa/fa) (········, ▿) animals (C).
For chow-fed, wild-type rats weighing 278 ± 10 g, Fig. 5C demonstrates that for a given sCT dose the percentage reduction in energy intake was more pronounced than that of chow-fed C57Bl/6J mice, both for equal doses per unit body mass, as well as for equal doses per animal (not shown). The fa/fa rats weighing 530 ± 11 g and being grossly obese because of their leptin receptor defect, were again less sCT responsive than the wild-type rats, but the degree of response attenuation was much smaller than in the leptin-deficient ob/ob mice irrespective of whether the dose was standardized per animal (not shown) or per unit body mass.
Plasma insulin levels differ between the investigated animal models
To obtain information about the occurrence of hyperinsulinaemia, which generally tends to develop in states of obesity, plasma insulin measurements were carried out. In order to minimize influences of food intake on plasma insulin concentrations the time chosen for blood sampling, by decapitation or retroorbital puncturing, was about 3-1 h before lights-off. Blood was collected in heparinized tubes on ice and centrifuged and plasma aliquots were stored at −80 °C until being assayed. Plasma insulin was determined using a commercial radioimmunoassay (RIA) kit (Serono Diagnostics, Freiburg, FRG) with rat insulin (Novo Industries Laboratories, Bagsvaerde, DK) as standard as previously described (Schmidt et al. 2001). The RIA values were corrected by considering the heparin and buffer dilution. To decrease variability due to interassay variability pooled plasma samples were measured in each assay and the deviations from the long-term mean insulin level of the pool were use for appropriate correction. The average plasma insulin concentrations obtained in this way for the investigated animal models are listed in Table 1. Compared with chow-fed C57Bl/6J mice, the PHC-diet induced a 3- to 4-fold increase. This level was, however, greatly exceeded by the plasma insulin levels occurring in the genetically obese mice among which plasma insulin in the ob/ob mice was almost 5-fold higher than in the MC4-r-KO mice. The insulin levels of the leptin receptor-deficient fa/fa rats were more than 10-fold higher than in wild-type rats, but the degree of hyperinsulinaemia caused by the genetic leptin receptor defect was much less than in the leptin-deficient ob/ob mice, although the fa/fa and ob/ob animals were similarly obese and of the same age.
Table 1.
Insulin plasma concentrations representative for the animal models studied for sCT responsiveness
| Species | Strain/ genotype | Dietary regimen | Plasma insulin (mu;U ml−1) | n |
|---|---|---|---|---|
| Mice | C57Bl/6J | Chow | 20 ± 2 | 15 |
| C57Bl/6J | Chow + PHC-diet | 70 ± 7 | 25 | |
| MC4-r-KO | Chow | 489 ± 43 | 13 | |
| ob/ob | Chow | 2348 ± 270 | 26 | |
| Rats | Zucker +/+ | Chow | 36 ± 5 | 22 |
| Zucker fa/fa | Chow | 453 ± 108 | 6 |
Shown are average means ± s.e.m., n = number of animals.
DISCUSSION
Leptin and amylin are categorised as adiposity signals based on their actions on energy balance (Woods & Seeley, 2000). For each of these peptides circulating in the blood separate signalling systems exist which encompass the signal molecule itself, its specific neuronal receptor, and the brain pathways downstream to the receptor. For functional leptin resistance, which is the most frequent disturbance of the leptin-signalling system causing impaired control of energy balance in obese individuals, there is no reason to assume, a priori, that the amylin-signalling system is impaired too. Thus, when considering pharmacological approaches to obesity treatment it appears that the redundancy of energy balance control should be taken into account. Proceeding from this idea sCT as an amylin analogue (Lutz et al. 2000) was used in the present study on the premise that a major problem in treating obese humans is their preference for obesity-promoting high-sugar-high-fat diets over healthy low-caloric, high-fibre diets. This was taken into account by using comparable feeding conditions in exploring this pharmacological approach. In contrast to previous studies in rodents on high-caloric diets, the diet regimen used here for the first time offered the choice between feeding on chow and on the PHC diet. With 85 % of the gross caloric intake of untreated animals being derived from the PHC diet and only 15 % from chow, the role of diet preference for ingestion is documented, and information about the effect of anorectic substances on this choice can be obtained. The main result of this study confirmed the basic premise that sCT remained effective under conditions of leptin resistance induced by palatable high-caloric food. It was further shown that the anorectic action of sCT may be limited, though not fully abolished, in several genetic obesity models in which leptin responsiveness is disturbed.
Potential modes of sCT action
The observation that sCT did not measurably reduce food intake in ob/ob mice at doses effective in other animal models indicates that this peptide does not reduce food intake as a consequence of general malaise mediated by non-specifically responsive ‘emetic chemoreceptors’ (Carpenter et al. 1983) in the AP, which have been categorised in the rat as different from other receptors such as those involved in salt and fluid balance and especially those which are glucose-responsive and postulated to be mediators of satiety (Adachi et al. 1991). More than 90 % of the AP neurones excited by amylin were also found to be excited by glucose (Riediger et al. 1999a) and, consequently, these neurones are well-characterized as potential targets for satiety signals and probably also mediate the specific anorectic action of sCT as an amylin analogue, as indicated by two independent approaches (Lutz et al. 2000; Riediger et al. 2001). That C57Bl/6J mice on the combined chow and PHC diet reduced PHC intake but simultaneously increased chow intake in response to sCT also does not indicate malaise. Both observations are in line with the notion that, for amylin, taste aversion and malaise may be excluded as mechanisms underlying its anorectic action (Lutz et al. 1995; Morley et al. 1997; Rowland & Richmond, 1999). The set of related receptor subtypes to which sCT binds in the rat brain includes amylin receptors as a subset (Christopoulos et al. 1995). The excitatory action of sCT on neurones accessible from the blood side of the brain, i.e. in circumventricular organs (CVOs) devoid of the blood- brain barrier, was characterized for two CVOs. In the subfornical organ, where calcitonin and amylin probably exert their actions as dipsogens, neuronal excitation by sCT was longer lasting than excitation by calcitonin and amylin (Riediger et al. 1999b). Excitation of AP neurones by sCT was also shown to be longer lasting than by amylin and, thus, corresponded to the observation that the anorectic action of sCT is prolonged in comparison to that of amylin (Riediger et al. 2000).
Differential actions of leptin and sCT
The present study demonstrates for the first time that conventionally fed laboratory mice of the C57Bl/6J strain responded to sCT and leptin in an additive manner. Since sCT may be considered as an amylin analogue with respect to its anorectic action (Lutz et al. 2000), these data suggest that amylin and leptin as endogenous adiposity signals may also act additively. Most important among the observations made in this study is the result that nutritional influences do not abolish the anorectic action of sCT even if they abolish the anorectic action of leptin completely. In a state of functional leptin resistance induced by long-term feeding of a combination of chow and the PHC diet, the leptin effect was absent, as expected, while the sCT effect was even more pronounced, although, interestingly, it was exerted specifically by reducing the intake of the palatable high-caloric component of the diet. This differential sCT action was also seen at an early stage with the combined PHC and chow diet, when C57Bl/6J mice still responded to leptin with parallel reductions of both chow and PHC intake, while sCT strongly reduced PHC intake but slightly enhanced chow intake (I. Schmidt, unpublished observation).
In contrast to functional leptin resistance, however, sCT responsiveness was attenuated to different degrees in the animal models used in this study in which disturbances of the leptin-signalling system resulted from diverse genetic defects. MC4-r-KO mice with their non-functional αMSH system displayed quantitatively reduced responsiveness to sCT at a dose range which was effective in chow-fed C57Bl/6J mice. Mice with the ob/ob defect do not produce leptin and did not respond measurably to sCT in the low to middle dose range effective in the MC4-r-KO mice, but when the dose per unit body mass was elevated to the maximum dose applied to C57Bl/6J mice, the ob/ob mice, too, reduced their food intake slightly. Mediation of this sCT effect by amylin receptors is a likely assumption. Studies on the influence of different amylin doses on short-term food intake of ob/ob mice and heterozygotes (ob/c) after 15-16 h food deprivation showed that 30 min after access to food only the ob/ob mice had significantly reduced their food intake and only in response to the highest amylin dose, but after 60 min the lower amylin doses significantly reduced food intake in the ob/c mice but not in the ob/ob mice which only responded to the highest amylin dose (Morley et al. 1994). The observed less distinct genotype influence on amylin responsiveness suggests that the hunger signals generated during starvation prior to feeding might have initially overridden the more subtle genotype differences in responsiveness to amylin. Moreover, the ob/c heterozygotes differ from wild-type animals and, thus, the data are not directly comparable to the observations of this study in which ob/ob and wild-type mice were compared. Although the sCT responsiveness of the leptin receptor-deficient fa/fa rats was reduced too in comparison to wild-type rats, they were more responsive to sCT than each of the mouse strains investigated in this study, indicating that even massive hyperleptinaemia as the consequence of the fa/fa defect (Wu-Peng et al. 1997) does not impair the sCT action. Thus, taken together, genetic impairment of the leptin-signalling system does not per se inactivate the amylin signalling system by which the anorectic sCT action is mediated.
Are sCT actions altered by obesity-related systemic hormonal disturbances?
In this study diverse graded and directional changes in sCT responsiveness were observed in functionally leptin-resistant mice and in the investigated models of genetic obesity. These changes do not seem to correlate with the degree of disturbance of the leptin system. These inconsistencies suggests that secondary disturbances, rather than the defective leptin system itself, might have become effective in altering sCT responsiveness. Further, the degree of obesity or the high triglyceride content, respectively, as well as the underlying hyperphagia, also do not seem to correlate with the degree to which sCT responsiveness may be attenuated. The chow-fed genetic obesity models used in this study are generally hyperinsulinaemic (Pieber et al. 1994; Huszar et al. 1997; Leckström et al. 1999; Fan et al. 2000), suggesting a role for hyperinsulinaemia as a relevant secondary disturbance. Our own measurements allow quantitative comparisons, because they were obtained using the same assay procedure. For the genetically obese animals used in this study it is tempting to speculate that the higher their plasma insulin level, the lower might be their sCT responsiveness.
Perspectives: potential roles of hyperinsulinaemia and hyperamylinaemia in altering sCT responsiveness
Since insulin secretion is, in principle, associated with cosecretion of amylin (Young, 1994; Castillo et al. 1995) and since sCT exerts its anorectic effects by acting on amylin receptors of the AP (Lutz et al. 2000), we can speculate that hyperamylinaemia, too, is a potential modulatory factor accounting for reduced sCT responsiveness in genetic obesity models. The endogenous amylin levels of ob/ob mice exceed those of wild-type mice by one order of magnitude (Leckström et al. 1999) and tend to increase further during the animal's lifetime, similar to insulin (Höppener et al. 1999). Hyperamylinaemia was shown to accompany hyperinsulinaemia in rats as the consequence of acquired obesity, as well as of genetic obesity due to the fa/fa defect (Pieber et al. 1994). Currently there is no direct evidence for hyperamylinaemia in MC4-r-KO mice, but it is likely to exist, considering the link established by cosecretion between insulin and amylin. Although the relationship between insulin and amylin was reported to vary between the animal models investigated, the large differences in plasma insulin levels between the models used in this study raise the question of whether the differences in plasma amylin levels might have been similarly pronounced.
Moderate elevations of plasma amylin may be expected for the chocolate-fed C57Bl/6J mice of this study on the basis of the observed moderate increase in plasma insulin and of measurements in the same strain after long-term feeding of a high-fat diet (Mulder et al. 2000). However, their sCT responsiveness was enhanced rather than reduced, suggesting that the quality of the diet might counteract to some extent the attenuation of the sCT action by hyperinsulinaemia and/or hyperamylinaemia. This assumption is supported by the higher sCT responsiveness of chocolate-fed in comparison to chow-fed MC4-r-KO mice (I. Schmidt, unpublished observation).
In conclusion, the elucidation of the hitherto unknown factors accounting for unchanged or even enhanced sCT responsiveness in the medically prevailing conditions of diet-induced obesity and functional leptin resistance, respectively, appears to be the most interesting aspect of this study. Future studies aimed at understanding whether and how reduced responsiveness to sCT, as observed in models of genetic obesity, might result from excessive hyperinsulinaemia and/or hyperamylinaemia would promote insights into reciprocal control of the redundant system of adiposity signals.
Acknowledgments
We are very grateful to Dennis Huszar (Millenium Pharmaceuticals, Inc., Cambridge, MA, USA) for providing us with breeding pairs of MC4-r-KO-mice for this study, and to Anke Hinney and Johannes Hebebrand (Marburg, FRG) for getting us started on this interesting topic and for all their of encouragement along the way. We would also like to thank Christina Schubert for help with the animal experiments. We gratefully acknowledge financial support from the Deutsche Forschungsgemeinschaft (Schm 680/4-1) and the Bundesministerium fuer Bildung und Forschung as part of the German Human Genome Project (01 KW 0009).
REFERENCES
- Adachi A, Kobashi M, Miyoshi N, Tsukamoto G. Chemosensitive neurons in the area postrema of the rat and their possible functions. Brain Research Bulletin. 1991;26:137–140. doi: 10.1016/0361-9230(91)90198-s. [DOI] [PubMed] [Google Scholar]
- Carpenter DO, Briggs DB, Strominger N. Responses of neurons of canine area postrema to neurotransmitters and peptides. Cellular and Molecular Neurobiology. 1983;3:113–126. doi: 10.1007/BF00735276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castillo MJ, Scheen AJ, Lefebvre PJ. Amylin/islet amyloid polypeptide: biochemistry, physiology, pathophysiology. Diabete et Metabolisme. 1995;21:3–25. [PubMed] [Google Scholar]
- Christopoulos G, Paxinos G, Huang XF, Beaumont K, Toga AW, Sexton PM. Comparative distribution of receptors for amylin and the related peptides calcitonin gene related peptide and calcitonin in rat and monkey. Canadian Journal of Physiology and Pharmacology. 1995;73:1037–1041. doi: 10.1139/y95-146. [DOI] [PubMed] [Google Scholar]
- Chua SC, Chung WK, Wu-Peng XS, Zhang Y, Liu SM, Tartaglia L, Leibel RL. Phenotypes of mouse diabetes and rat fatty due to mutations in the OB (leptin) receptor. Science. 1996;271:994–996. doi: 10.1126/science.271.5251.994. [DOI] [PubMed] [Google Scholar]
- Doering H, Schwarzer K, Nuesslein-Hildesheim B, Schmidt I. Leptin selectively increases energy expenditure of food-restricted lean mice. International Journal of Obesity. 1998;22:83–88. doi: 10.1038/sj.ijo.0800547. [DOI] [PubMed] [Google Scholar]
- Fan W, Dinulescu DM, Butler AA, Zhou J, Marks DL, Cone RD. The central melanocortin system can directly regulate serum insulin levels. Endocrinology. 2000;141:30772–3079. doi: 10.1210/endo.141.9.7665. [DOI] [PubMed] [Google Scholar]
- Heymsfield SB, Greenberg AS, Fujioka K, Kushner R, Hunt T, Lubina JA, Patane J, Self B, Hunt P, McCanish M. Recombinant leptin for weight loss in obese and lean adults. Journal of the American Medical Association. 1998;282:1568–1575. doi: 10.1001/jama.282.16.1568. [DOI] [PubMed] [Google Scholar]
- Höppener JWM, Oosterwijk C, Nieuwenhuis MG, Posthuma G, Thijssen JHH, Vroom TM, Ahrén B, Lips CJM. Extensive islet amyloid formation is induced by development of type II diabetes mellitus and contributes to its progression; pathogenesis of diabetes in a mouse model. Diabetologia. 1999;42:427–434. doi: 10.1007/s001250051175. [DOI] [PubMed] [Google Scholar]
- Hukshorn CJ, Saris WHM, Westerterp MS, Farid AR, Smith FJ, Campfield LA. Weekly subcutaneous pegylated recombinant native human leptin (PEG-OB). administration in obese men. Journal of Clinical Endocrinology and Metabolism. 2000;85:4003–4009. doi: 10.1210/jcem.85.11.6955. [DOI] [PubMed] [Google Scholar]
- Huszar D, Lynch CA, Fairchild-Huntress V, Dunmore JH, Fang Q, Berkemeier LR, Gu W, Kesterson RA, Boston BA, Cone RD, Smith FJ, Campfield LA, Burn P, Lee F. Targeted disruption of the melanocortin-4 receptor results in obesity in mice. Cell. 1997;88:131–141. doi: 10.1016/s0092-8674(00)81865-6. [DOI] [PubMed] [Google Scholar]
- Juhl CB, Porksen N, Sturis J, Hansen AP, Veldhuis JD, Pincus S, Fineman M, Schmidtz O. High-frequency oscillations in circulating amylin concentrations in healthy humans. American Journal of Physiology - Endocrinology and Metabolism. 2000;278:E481–490. doi: 10.1152/ajpendo.2000.278.3.E484. [DOI] [PubMed] [Google Scholar]
- Leckström A, Lundquist I, Ma Z, Westermark P. Islet amyloid polypeptide and insulin relationship in a longitudinal study of the genetically obese (ob/ob). mouse. Pancreas. 1999;18:266–273. doi: 10.1097/00006676-199904000-00008. [DOI] [PubMed] [Google Scholar]
- Lin S, Thomas TC, Storlien LH, Huang XF. Development of high fat diet-induced obesity and leptin resistance in C57Bl/6J mice. International Journal of Obesity. 2000;24:639–646. doi: 10.1038/sj.ijo.0801209. [DOI] [PubMed] [Google Scholar]
- Lu H, Duanmu Z, Houck C, Jen KLC, Buison A, Dunbar JC. Obesity due to high fat diet decreases the sympathetic nervous and cardiovascular responses to intracerebroventricular leptin in rats. Brain Research Bulletin. 1998;47:331–335. doi: 10.1016/s0361-9230(98)00086-0. [DOI] [PubMed] [Google Scholar]
- Lutz TA, Geary N, Szabady MM, Del Prete E, Scharrer E. Amylin decreases meal size in rats. Physiology and Behavior. 1995;58:1197–1202. doi: 10.1016/0031-9384(95)02067-5. [DOI] [PubMed] [Google Scholar]
- Lutz TA, Senn M, Althaus J, Del Prete E, Ehrensperger F, Scharrer E. Lesion of the area postrema nucleus of the solitary tract (AP/NTS) attenuates the anorectic effects of amylin and calcitonin gene-related peptide (CGRP) in rats. Peptides. 1997;19:309–317. doi: 10.1016/s0196-9781(97)00292-1. [DOI] [PubMed] [Google Scholar]
- Lutz TA, Tschudy S, Rushing PA, Scharrer E. Amylin receptors mediate the anorectic action of salmon calcitonin (sCT) Peptides. 2000;21:233–238. doi: 10.1016/s0196-9781(99)00208-9. [DOI] [PubMed] [Google Scholar]
- Markewicz B, Kuhmichel G, Schmidt I. Onset of excess fat deposition in Zucker rats with and without decreased thermogenesis. American Journal of Physiology. 1993;265:E478–486. doi: 10.1152/ajpendo.1993.265.3.E478. [DOI] [PubMed] [Google Scholar]
- Morley JE, Flood JF, Horowitz M, Morley PMK, Walter MJ. Modulation of food intake by peripherally administered amylin. American Journal of Physiology. 1994;267:R178–184. doi: 10.1152/ajpregu.1994.267.1.R178. [DOI] [PubMed] [Google Scholar]
- Morley JE, Suarez MD, Mattamal M, Flood JF. Amylin and food intake in mice: effects on motivation to eat and mechanism of action. Pharmacology, Biochemistry and Behavior. 1997;56:123–129. doi: 10.1016/S0091-3057(96)00168-2. [DOI] [PubMed] [Google Scholar]
- Mulder H, Martensson H, Sundler F, Ahren B. Differential changes in islet amyloid polypeptide (Amylin) and insulin mRNA expression after high-fat diet induced insulin resistance in C57Bl/6J mice. Metabolisms Clinical and Experimental. 2000;49:1518–1522. doi: 10.1053/meta.2000.18511. [DOI] [PubMed] [Google Scholar]
- Pelleymounter MA, Cullen MU, Healy D, Hecht R, Winters D, McCaleb M. Efficacy of exogenous recombinant murine leptin in lean and obese 10- to 20-mo-old female CD! mice. Amercian Journal of Physiology. 1998;275:R950–959. doi: 10.1152/ajpregu.1998.275.4.R950. [DOI] [PubMed] [Google Scholar]
- Pieber TR, Roitelman J, Lee Y, Luskey KL, Stein DT. Direct plasma radioimmunoassay for rat amylin (1–37): Concentrations with acquired and genetic obesity. American Journal of Physiology. 1994;267:E156–164. doi: 10.1152/ajpendo.1994.267.1.E156. [DOI] [PubMed] [Google Scholar]
- Proietto J, Fam BC, Ainslie DA, Thorburn AW. Novel anti-obesity drugs. Expert Opinion on Investigational Drugs. 2000;9:1317–1326. doi: 10.1517/13543784.9.6.1317. [DOI] [PubMed] [Google Scholar]
- Riediger T, Lutz TA, Schmid HA, Scharrer E. The area postrema as a major target for peptides controlling food intake. Society for Neuroscience Abstracts. 2000;26:369.10. [Google Scholar]
- Riediger T, Rauch M, Jurat G, Schmid HA. Central nervous targets for pancreatic amylin. Pflügers Archiv. 1999a;437:R142. abstract. [Google Scholar]
- Riediger T, Schmid HA, Lutz TA, Simon E, Scharrer E. Amylin potently activates area postrema neurons: possible involvement of cGMP-signaling. American Journal of Physiology - Regulatory, Integrative and Comparative Physiology. 2001;281:R1833–1843. doi: 10.1152/ajpregu.2001.281.6.R1833. [DOI] [PubMed] [Google Scholar]
- Riediger T, Schmid HA, Young AA, Simon E. Pharmacological characterization of amylin-related peptides activating subfornical organ neurons. Brain Research. 1999b;837:161–168. doi: 10.1016/s0006-8993(99)01697-2. [DOI] [PubMed] [Google Scholar]
- Rowland NE, Richmond RM. Area postrema and the anorectic actions of dexfenfluramine and amylin. Brain Research. 1999;820:86–91. doi: 10.1016/s0006-8993(98)01348-1. [DOI] [PubMed] [Google Scholar]
- Schmidt I, Fritz A, Schölch C, Schneider D, Simon E, Plagemann A. The effect of leptin treatment on the development of obesity in overfed suckling Wistar rats. International Journal of Obesity. 2001;25:1168–1174. doi: 10.1038/sj.ijo.0801669. [DOI] [PubMed] [Google Scholar]
- Sinha MK, Caro JF. Clinical aspects of leptin. Vitamins and Hormones. 1998;54:1–30. doi: 10.1016/s0083-6729(08)60919-x. [DOI] [PubMed] [Google Scholar]
- Stridsberg M, Sandler S, Wilander E. Cosecretion of islet amyloid polypeptide (IAPP) and insulin from isolated rat pancreatic islets following stimulation or inhibition of β-cell function. Regulatory Peptides. 1993;45:363–370. doi: 10.1016/0167-0115(93)90362-c. [DOI] [PubMed] [Google Scholar]
- Widdowson PS, Upton R, Buckingham R, Arch J, Williams G. Inhibition of food response to intracerebroventricular injection of leptin is attenuated in rats with diet-induced obesity. Diabetes. 1997;46:1782–1785. doi: 10.2337/diab.46.11.1782. [DOI] [PubMed] [Google Scholar]
- Woods SC, Seeley RJ. Adiposity signals and the control of energy homeostasis. Nutrition. 2000;16:894–902. doi: 10.1016/s0899-9007(00)00454-8. [DOI] [PubMed] [Google Scholar]
- Wu-Peng X, Chua SC, Jr, Okada N, Liu SM, Nicolson M, Leibel RL. Phenotype of the obese Koletsky (f) rat due to Tyr763Stop mutation in the extracellular domain of the leptin receptor (Lepr) Diabetes. 1997;46:513–518. doi: 10.2337/diab.46.3.513. [DOI] [PubMed] [Google Scholar]
- Young AA. Amylin regulation of fuel metabolism. Journal of Cellular Biochemistry. 1994;55(suppl.):12–18. doi: 10.1002/jcb.240550003. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Procenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372:425–432. doi: 10.1038/372425a0. [DOI] [PubMed] [Google Scholar]