

Abstract
Exposure of PC12 cells to chronic hypoxia (CH; 10 % O2, 24 h) augments catecholamine secretion via formation of a Cd2+-resistant Ca2+ influx pathway, and up-regulates native L-type Ca2+ channels. These effects are mimicked by exposure of cells to Alzheimer's disease-associated amyloid β peptides (AβPs). Since pathological effects of AβPs have been associated with increased levels of reactive oxygen species (ROS), the involvement of ROS in hypoxia-mediated up-regulation of exocytosis and Ca2+ channel activity was examined. Both melatonin and ascorbic acid (two structurally unrelated antioxidants) fully blocked the enhancement of catecholamine secretion caused by CH (as determined amperometrically). Enhanced immunofluorescence, observed in chronically hypoxic cells using a primary monoclonal antibody raised against the N-terminus of AβP, was also suppressed by melatonin. Ascorbic acid, melatonin and ebselen (an additional antioxidant) also fully prevented augmentation of whole-cell Ca2+ currents caused by CH (as monitored using whole-cell patch-clamp recordings). Exposure of normoxic cells to H2O2 (40 μM, 24 h), like hypoxia, caused Ca2+ channel up-regulation. Importantly, AβP formation appeared to be an absolute requirement for the effects of hypoxia, since the ability of CH to augment exocytosis and Ca2+ channel activity was blocked by two novel inhibitors of γ secretase, an enzyme complex required for AβP formation. Our results indicate that the effects of hypoxia require ROS generation from AβPs, and suggest that elevated levels of ROS mediate hypoxic and AβP-mediated pathological remodelling of Ca2+ homeostasis.
The incidence of dementias such as Alzheimer's disease (AD) is significantly increased in patients who have previously suffered prolonged hypoxic or ischaemic episodes arising, for example, as a consequence of cardiovascular dysfunction such as stroke or arrhythmia (Tatemichi et al. 1994; Kokmen et al. 1996; Moroney et al. 1996). Ischaemia involves alteration of a number of parameters, including lack of substrates, accumulation of metabolic products, acidosis and reduction of ATP and O2 levels, each of which is essential to cellular homeostasis, yet our understanding of the influences of each of these parameters to cell injury or destruction is far from complete. However, the clear link between hypoxic/ischaemic episodes and increased incidence of AD strongly suggests that one or more of these parameters are capable of precipitating this increasingly widespread disease.
A defining feature of AD is the appearance of fibrillar deposits consisting of amyloid β peptides (AβPs; reviewed by Mattson, 1997; Selkoe, 2001). AβPs are 39-43 amino acid peptide cleavage products derived from amyloid precursor protein (APP; Glenner & Wong, 1984; Masters et al. 1985). APP is one of only a few gene products whose expression is increased following a period of cerebral ischaemia (Kogure & Kato, 1993; Koistinaho et al. 1996). The major (non-amyloidogenic) cleavage product of APP, sAPPα, is neuroprotective (Mattson, 1997; Selkoe, 2001), and so increased expression of APP may be considered a defence mechanism against ischaemia. However, increased APP levels would also provide increased substrate for formation of toxic AβPs (Mattson, 1997; Selkoe, 2001) and, indeed, AβP production is increased following ischaemia (Yokoto et al. 1996; Jendroska et al. 1997).
The mechanisms underlying the neuronal toxicity of AβPs appear complex and remain to be fully resolved. Toxicity involves disruption of Ca2+i homeostasis (Fraser et al. 1997; Mattson, 1997) which may be oxidative and involve free radical damage (Behl et al. 1994; Schubert et al. 1995). In addition, other studies have shown that AβPs disrupt Ca2+i homeostasis by forming Ca2+-permeable pores or channels (Arispe et al. 1996; Kawahara et al. 1997; Rhee et al. 1998) which may account for increased central synaptic activity. Indeed, increased activity such as enhancement of long-term potentiation and elevated glutamate release has been demonstrated in hippocampal neurones exposed to AβPs in vitro (Arias et al. 1995; Wu et al. 1995).
Our recent studies have provided clear evidence that Ca2+ homeostasis and neurosecretion are altered when cells are exposed to hypoxic conditions, and that these effects are mimicked by AβPs. Using the catecholamine secreting cell line PC12, we have shown that chronic hypoxia (10 % O2, 24 h) leads to excessive stimulus-evoked neurosecretion due to the emergence of a Cd2+-resistant Ca2+ influx pathway tightly coupled to the exocytotic machinery (Taylor et al. 1999). In addition, a selective up-regulation of L-type Ca2+ channels was observed (Green & Peers, 2001). These effects were mimicked by direct application of AβPs to the cells (Taylor et al. 1999). In the present study, we have examined whether AβP formation is a necessary step in these effects of hypoxia, and also report the involvement of reactive oxygen species (ROS) in mediating these effects of chronic hypoxia. Such studies were prompted not only by the well-recognized involvement of oxidative stress associated with neurotoxic effects of AβPs (Mattson, 1997; Miranda et al. 2000; Varadarajan et al. 2000; Selkoe, 2001), but also by recent, contested reports which suggest that ROS levels increase during prolonged hypoxia (Chandel et al. 1998; Hohler et al. 1999; Chandel & Schumacker, 2000). In addition to this it has recently been shown that oxidative stress increases AβPs, either by direct addition of H2O2 (Misonou et al. 2000), or indirectly through addition of mercury (Olivieri et al. 2000). Our results indicate that ROS are likely important mediators in the effects of both chronic hypoxia and AβPs and, importantly, that these effects of hypoxia require AβP production.
METHODS
Cell culture
PC12 cells were cultured in RPMI 1640 culture medium (containing l-glutamine) supplemented with 20 % fetal calf serum and 1 % penicillin-streptomycin (Gibco, Paisley, Strathclyde, UK) as previously described (Taylor et al. 1999; Taylor & Peers, 1999). Cells were incubated at 37 °C in a humidified atmosphere of 5 % CO2-95 % air, passaged every 7 days and used for up to 20 passages. Cells used for experiments were transferred to smaller flasks in 10 ml of medium, to which was added 1 μM dexamethasone (Sigma, Poole, UK, from a stock solution of 1 mm in Ultrapure water), and were cultured for a further 72-96 h to enrich catecholamine stores (Tischler et al. 1983). Cells exposed to chronic hypoxia were treated identically, except that for 24 h prior to experiments they were transferred to a humidified incubator equilibrated with 10 % O2, 5 % CO2 and 85 % N2. Following this period in chronic hypoxia, cells were exposed to room air for no longer than 1 h before experimentation. Peptides used in this study were dissolved in Ultrapure water and stored frozen in aliquots until required, so that they only underwent one freeze-thaw cycle before being applied directly to the cells. Gel electrophoresis of peptide samples revealed that they were applied to cells in the unaggregated form. When used, antioxidants were included for the culture period in normoxia, hypoxia or during exposure to AβP(1-40), as were the dipeptide-aldehyde γ secretase inhibitors, 2-napthyl-Val-Phe-CHO (NVP) and Boc-Gly-Val-valinal (GVV).
Amperometry
Each experimental day, PC12 cells were plated onto poly-l-lysine-coated coverslips and allowed to adhere for 1 h under either normoxic or hypoxic (10 % O2) conditions, as required. Fragments of coverslip were then transferred to a recording chamber (volume 80 μl) which was continually perfused under gravity (flow rate 1-2 ml min−1) with a solution of composition (in mm): NaCl 135, KCl 5, MgSO4 1.2, CaCl2 2.5, Hepes 5 and glucose 10 (pH 7.4, osmolarity adjusted to 300 mosmol l−1 with sucrose, 21-24 °C). High [K+] solutions contained 50 mm K+ and the [Na+] was reduced accordingly to maintain iso-osmolarity.
Carbon fibre microelectrodes (proCFE, Dagan Instruments, MN, USA) with a diameter of 5 μm were positioned adjacent to individual PC12 cells using a micromanipulator and were polarized to +800 mV to allow the oxidation of released catecholamine. Resulting currents were recorded using an Axopatch 200A amplifier (with extended voltage range), filtered at 1 kHz and digitized at 2 kHz before storage on computer. All acquisition was performed using a Digidata 1200 interface and Fetchex software from the pCLAMP 6.0.3 suite (Axon Instruments). Unless otherwise stated, each experiment consisted of current recordings of a control period during which cells were perfused only with normoxic external medium. This was then exchanged for a depolarizing test solution (containing 50 mm K+) and amperometric signals were recorded for a further period of 1-4 min. Catecholamine secretion was apparent as discrete spike-like events, each corresponding to the released contents of a single vesicle of catecholamine (Wightman et al. 1991; Chow & Von Ruden, 1995). The perfusate was then exchanged for one containing 50 mm K+ but also 200 μM Cd2+ to block native, voltage-gated Ca2+ channels (see Taylor et al. 1999). Secretory events were never seen unless the electrode was polarized and adjacent to a cell. Quantification of release was achieved by determining spike frequency using Mini Analysis Program (Synaptosoft Inc., Leonia, NJ, USA). This allowed visual inspection of each event so that artefacts (due, for example, to solution switches) could be rejected from the analysis. Results are presented as individual examples or means ± standard error of the mean and statistical comparisons were made using Student's unpaired t test.
Electrophysiology
Cells were plated onto coverslips, fragments of which were subsequently placed in a perfusion chamber exactly as for amperometric recordings, except that perfusing solution was of composition (in mm): NaCl 110, CsCl 5, MgCl2 0.6, BaCl2 20, Hepes 5, glucose 10 and tetraethylammonium chloride 20 (pH 7.4). The osmolarity of the perfusate was adjusted to 300 mosmol l−1 by addition of sucrose. Patch pipettes (5-7 MΩ resistance) were filled with a solution of (in mm): CsCl 130, EGTA 1.1, MgCl2 2, CaCl2 0.1, NaCl 10, Hepes 10 and Na2ATP 2 (pH 7.2). After establishing the whole-cell configuration, cells were voltage-clamped at −80 mV and whole cell capacitance determined from analog compensation. To evoke whole-cell Ca2+ channel currents, 200 ms voltage ramps were applied from −100 to +100 mV at a frequency of 0.2 Hz (Green & Peers, 2001). Evoked currents were filtered at 1 kHz, digitized at 2 kHz and stored on computer for off-line analysis. All results are presented as mean ± s.e.m. current densities spanning the voltage range −60 to +60 mV, which covers their full activation range, and statistical analysis performed using Student's unpaired t tests. All data tested were taken from current densities measured at +20 mV (where I-V relationships were maximal).
Immunocytochemistry
Immunofluorescent labelling with a monoclonal antibody raised against the extracellular N-terminal five residues of AβP (3D6 antibody; Johnson-Wood et al. 1997) was performed as previously described (Taylor et al. 1999) with cells plated onto coverslips and subjected to normoxic or other conditions as described above. Cells were fixed by immersion in 4 % paraformaldehyde in 0.1 m phosphate buffer (pH 7.4) for 15 min, and then rinsed thoroughly in several changes of 0.1 m phosphate-buffered saline (PBS). The cells were then incubated for 20 min at room temperature in PBS containing 10 % normal goat serum (NGS). At this time cells which were to be permeabilized (insets of Fig. 7) also had Triton X-100 (0.2 %) added. Cells were then thoroughly washed again with PBS for several changes. The coverslips were then incubated with the 3D6 antibody (diluted to 0.5 μg ml−1 in PBS) in 24-well microtitration plates on a shaker for 18 h at 4 °C. After two 10 min rinses in PBS, the cells were incubated for 2 h in a 1/200 dilution of Cy2 conjugated anti-mouse IgG (Jackson ImmunoResearch, PA, USA). Control experiments (Fig. 2E and F) varied as detailed in the legend of Fig. 2. After two further 10 min rinses in PBS, the coverslips were mounted onto glass microscope slides with glycerol-PBS and the edges of the coverslips were sealed with clear nail polish. The cells were examined using a Zeiss Axioskop epifluorescence microscope using a No. 10 (fluorescein) filter set. Photographs were taken using a Kodak MDS120 digital camera system.
Figure 7. Immunofluorescent labelling of chronically hypoxic cells is suppressed by γ secretase inhibitors.
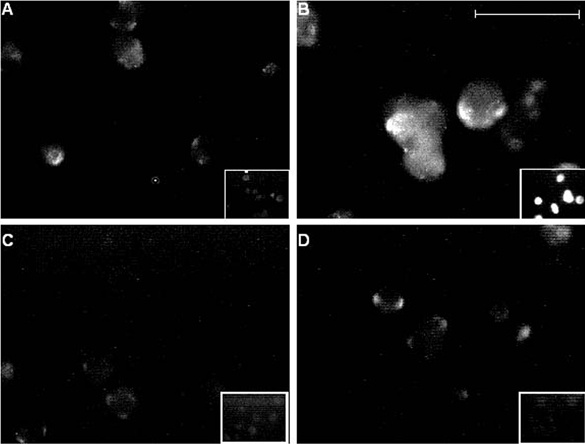
Fluorescence images of PC12 cells cultured normoxically (A) or under chronically hypoxic conditions (B-D). Hypoxic cells shown in C were also exposed to 10 μM GVV during the hypoxic period, and in D were exposed to 10 μM NVP. Fluorescence was detected using the 3D6 monoclonal antibody raised against the extracellular N-terminus of AβP as the primary antibody, and was applied to intact (non-permeabilized; main images) and permeabilized cells (inset images) in each case. Scale bar represents 40 μm (200 μm for insets) and is applicable to all panels.
Figure 2. Immunofluorescent labelling of chronically hypoxic cells is suppressed by melatonin.
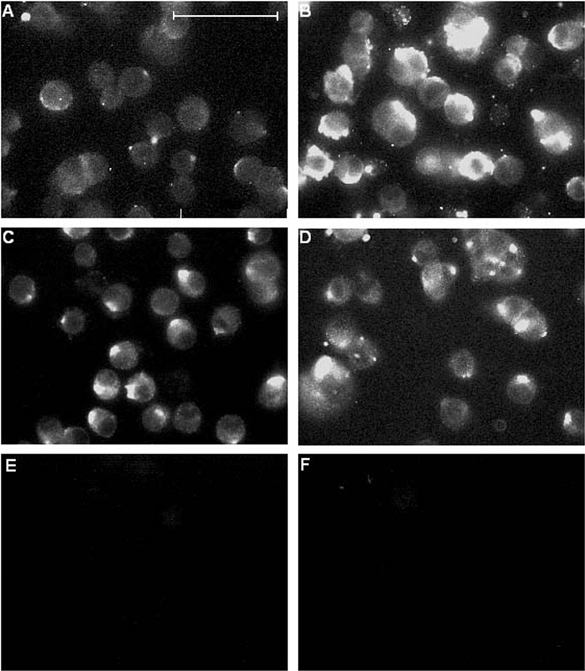
Fluorescence images of PC12 cells cultured in the presence or absence of 150 μM melatonin either normoxically (A and C, respectively), or under chronically hypoxic conditions (B and D, respectively). Fluorescence was detected using the 3D6 monoclonal antibody raised against the extracellular N-terminus of AβP as the primary antibody. E, background fluorescence (obtained by repeating staining procedure but omitting application of primary (3D6) antibody). F, immunofluorescence from cells conducted as in A and B, except that the primary antibody (3D6) was pre-incubated in excess (3 μM) AβP(1-40) before being applied to cells. Scale bar in A represents 40 μm and is applicable to all panels.
RESULTS
Exposure of control (normoxically cultured) PC12 cells to perfusate containing 50 mm K+ evokes exocytotic release of catecholamine which is dependent on Ca2+ influx through voltage-gated Ca2+ channels, since it is almost completely abolished in the presence of 200 μM Cd2+ (Fig. 1A and E; see also Taylor & Peers, 1999). Following a 24 h period of chronic hypoxia (10 % O2), secretory responses were significantly enhanced as compared with controls (P < 0.03), in agreement with our earlier studies (Taylor et al. 1999; see Fig. 1B and E), and approximately 35 % of the K+-evoked secretion remained in the presence of Cd2+ (significantly greater secretion than seen in controls; P < 0.001), indicating that chronic hypoxia induces a Ca2+ influx pathway coupled to exocytosis which is resistant to Cd2+ blockade. However, when the antioxidants melatonin (150 μM; Fig. 1C and E) or ascorbic acid (200 μM; Fig. 1D and E) were added to the culture medium during the 24 h period of chronic hypoxia, no such increases in exocytosis were observed (P > 0.5 for each antioxidant vs. controls), and Cd2+ almost completely prevented secretion (P < 0.01 for each antioxidant vs. Cd2+-resistant secretion observed in the absence of antioxidant).
Figure 1. Antioxidants prevent Cd2+-resistant exocytosis in chronically hypoxic PC12 cells.
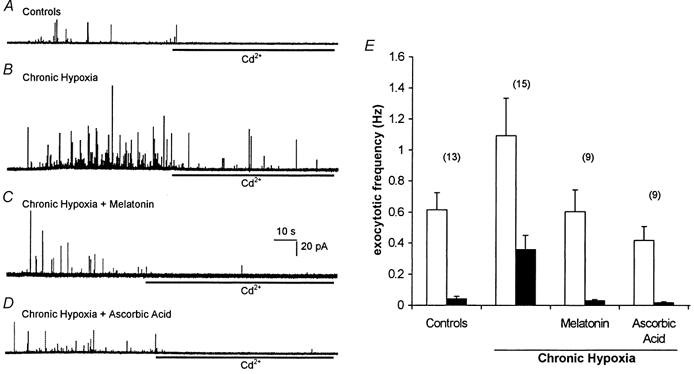
A, amperometric recording of exocytosis from a representative control PC12 cell. Secretion was evoked by exposure to a perfusate containing 50 mm K+ (application period commencing at the beginning of the trace). In this trace and those of B-D, cells were exposed to 200 μM Cd2+ in the continued presence of 50 mm K+ for the period indicated by the horizontal bars. B, as A, except that the recording was made from a cell previously cultured for 24 h in 10 % O2. Note that Cd2+ does not prevent fully the secretory response. C and D, as B, except that during the exposure to hypoxia, the cells were also exposed to 150 μM melatonin (C) or 200 μM ascorbic acid (D). Scale bars apply to all traces. E, bar graph showing mean (with vertical s.e.m. bars, taken from the number of cells indicated above each bar) exocytotic frequency in the four cell groups indicated in A-D before (□) and during (▪) exposure to Cd2+.
We have previously shown that chronic hypoxia induces the appearance of AβPs in the plasma membrane of PC12 cells (Taylor et al. 1999). Figure 2 shows fluorescence images of intact (i.e. non-permeabilized) PC12 cells following exposure to a monoclonal antibody, 3D6 (Johnson-Wood et al. 1997), raised against the N-terminus of AβP. Clearly, immunofluorescence was enhanced in cells exposed to chronic hypoxia for 24 h (Fig. 2B) as compared with controls (Fig. 2A). The enhanced fluorescence was greatly attenuated, however, when cells were exposed to 150 μM melatonin (Fig. 2D), despite the fact that melatonin exposure caused a slight increase in autofluorescence itself (Fig. 2C).
We have recently reported that native L-type voltage-gated Ca2+ channels, whilst contributing approximately 40 % to the total whole-cell Ca2+ current, are not coupled to depolarization-evoked catecholamine secretion (Taylor & Peers, 1999; Green & Peers, 2001). However, exposure of cells to chronic hypoxia or to amyloid peptides AβP(1-40), AβP(1-42) or AβP(25-35) (but not the reverse sequence peptide AβP(40-1)) causes a selective up-regulation of L-type Ca2+ channel activity or expression (Green & Peers, 2001), an effect which is clearly distinct from potentiation of exocytosis. Figure 3A illustrates this enhancement of whole-cell Ca2+ current in response to chronic hypoxia. Importantly, hypoxic augmentation of currents was fully reversed by 150 μM melatonin (Fig. 3B; a statistically significant effect; P < 0.02). This action of melatonin was most likely due to its antioxidant properties, since hypoxic augmentation of currents was also prevented by ascorbic acid (200 μM; Fig. 3C) and ebselen (10 μM; Fig. 3D). Interestingly, ascorbic acid treatment in itself caused a small but significant increase in Ca2+ channel current density (P < 0.02vs. controls; Fig. 3C), an effect for which we cannot presently account, but no further augmentation was observed following CH treatment. Thus ROS production appears to mediate both augmentation of exocytosis and enhancement of Ca2+ current density.
Figure 3. Antioxidants prevent augmentation of Ca2+ currents by chronic hypoxia.
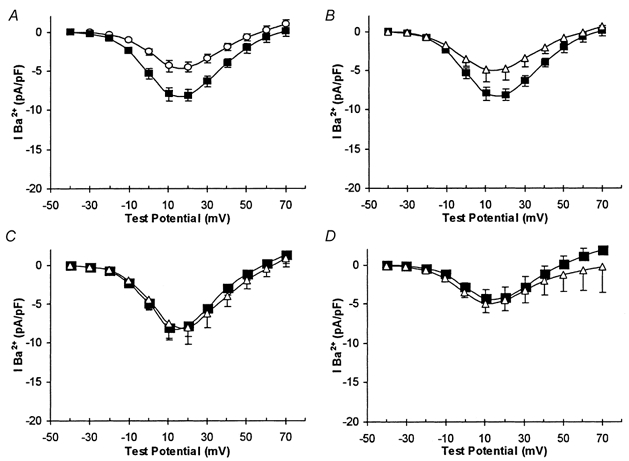
All plots are of mean current density vs. voltage plots (with vertical s.e.m. bars). A, recordings obtained from control cells (○, n = 9) and cells cultured under CH conditions (▪, n = 8). B, recordings obtained from CH cells in the absence (▪, n = 8) and presence (▵, n = 6) of 150 μM melatonin. C, recordings obtained from control (▵, n = 11) and CH (▪, n = 6) which were also incubated in the presence of 200 μM ascorbic acid. D, control (▵, n = 8) and CH (▪, n = 10) currents recorded in cells exposed to 10 μM ebselen during the 24 h period of CH or normoxia.
To test this idea further, we attempted to mimic the effects of chronic hypoxia by exposing cells for 24 h to 40 μM H2O2. As illustrated in Fig. 4, Ca2+ current densities were significantly enhanced (P < 0.005) following exposure to H2O2. Furthermore, this enhancement was selectively due to up-regulation of L-type Ca2+ channels, since exposure of H2O2-treated cells to 2 μM nifedipine (a selective blocker of L-type channels) reduced current densities by ca 77 %. Such a degree of inhibition is almost identical to the effects of nifedipine on chronically hypoxic and amyloid peptide treated cells (Green & Peers, 2001), and is significantly greater (P < 0.01) than the inhibitory effects of nifedipine on control cells (Green & Peers, 2001), indicating a selective enhancement of L-type channels caused by H2O2. In contrast to this effect on L-type Ca2+ channels, incubation of cells with 40 μM H2O2 failed to induce Cd2+-resistant, K+-evoked exocytosis. As exemplified in Fig. 4B (and the inset bar graph which shows mean data), application of 200 μM Cd2+ almost completely abolished ongoing secretory responses when cells previously incubated with 40 μM H2O2 were stimulated with 50 mm K+. Thus, whilst H2O2 could mimic the effects of chronic hypoxia and exposure to AβP on up-regulation of L-type Ca2+ channels, it was unable to induce Cd2+-resistant Ca2+ influx.
Figure 4. H2O2 mimics hypoxic and amyloid peptide augmentation of current density, but does not induce Cd2+-resistant evoked exocytosis.
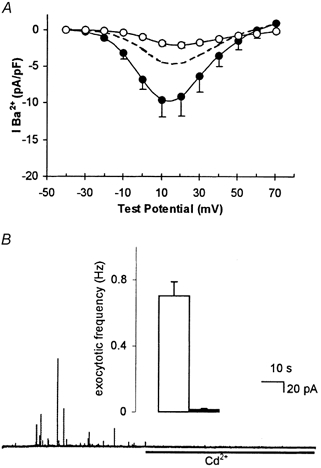
A, mean current density vs. voltage plots (with vertical s.e.m. bars, where visible behind symbols) obtained from seven cells exposed to 40 μM H2O2 for 24 h. Recordings were made in the absence (•) or presence (○) of 2 μM nifedipine. Dashed line indicates, for ease of comparison, control current density (taken from Fig. 3A). B, amperometric recording of exocytosis from a representative PC12 cell exposed to 40 μM H2O2 for 24 h. Secretion was evoked by exposure to a perfusate containing 50 mm K+ (application period commencing at the beginning of the trace), and 200 μM Cd2+ was applied for the period indicated by the horizontal bar. Inset shows mean ± s.e.m. exocytotic frequency in H2O2-treated cells before (□) and during (▪) exposure to Cd2+.
The findings reported thus far, together with our previous studies (Taylor et al. 1999; Green & Peers, 2000) indicated that the effects of CH and exposure to AβPs were qualitatively indistinguishable, raising the distinct possibility that the effects of chronic hypoxia were, in fact, mediated by AβP production. To investigate this possibility, we examined the actions of two novel γ secretase inhibitors to interfere with the actions of CH (γ secretase is an enzyme complex required to cleave AβP from amyloid precursor protein; see Discussion). These dipeptide-aldehyde inhibitors were 2-napthyl-Val-Phe-CHO (NVP) and Boc-Gly-Val-valinal (GVV), and when cells were incubated with either compound (each at a concentration of 10 μM, added for the 24 h hypoxic period), the effects of hypoxia were fully prevented. Thus, the Cd2+-resistant component of catecholamine secretion, and indeed the augmentation of total secretion, evoked by exposure of cells to 50 mm K+ was fully reversed, being not significantly different from control (normoxically cultured) cells (P > 0.4 for both compounds; Fig. 5A-C). In addition, whilst neither inhibitor exerted significant effects on control Ca2+ channel currents (Fig. 6A), hypoxic enhancement of whole cell Ca2+ channel currents was fully prevented (Fig. 6B), whilst currents recorded from AβP-treated cells remained significantly greater than controls in the presence of either inhibitor (e.g. P < 0.01 at +20 mV, Fig. 6C). The ability of these inhibitors to prevent hypoxic augmentation of secretion and Ca2+ current density were associated with prevention of increased immunofluorescence detected as described above using the monoclonal antibody 3D6 (Fig. 7A-D). The simultaneous addition of 10 μM NVP to cells treated with 40 μM H2O2 had no effect on the current enhancement caused by the H2O2 (Fig. 4); current densities still had a 2.4-fold increase compared to control cells (n = 6; data not shown). These results strongly suggest that AβP formation is an absolute requirement for hypoxia to exert its effects on exocytosis and Ca2+ current augmentation.
Figure 5. Inhibitors of γ secretase prevent hypoxic augmentation of exocytosis.
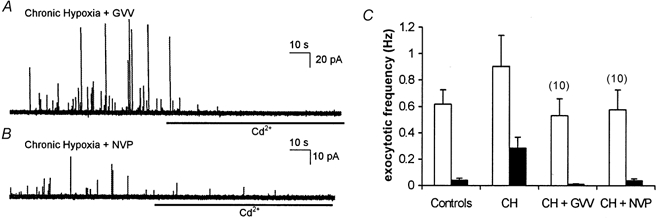
A, amperometric recording of exocytosis from a representative PC12 cell which had been cultured under chronically hypoxic conditions but in the additional presence of the γ secretase inhibitor, GVV (10 μM). Secretion was evoked by exposure to a perfusate containing 50 mm K+ (application period commencing at the beginning of the trace). B, as A, but the cell was incubated with another γ secretase inhibitor, NVP (10 μM). In both traces cells were exposed to 200 μM Cd2+ in the continued presence of 50 mm K+ for the period indicated by the horizontal bars. C, bar graph showing mean (with vertical s.e.m. bars, taken from the number of cells indicated above each bar) exocytotic frequency in the two cell groups indicated in A and B before (□) and during (▪) exposure to Cd2+. Control and CH data taken from Fig. 1 for ease of comparison.
Figure 6. Inhibitors of γ secretase suppress enhancement of currents by hypoxia, but not by AβP.
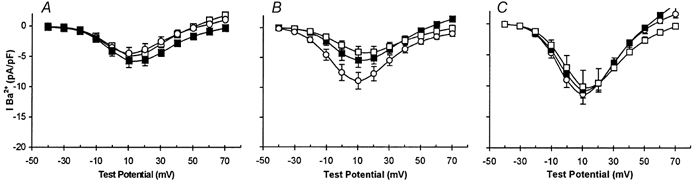
A-C, plots of mean current density vs. voltage (with vertical s.e.m. bars). A, data obtained from control cells in the absence (○, n = 8) and in the presence of the γ secretase inhibitors GVV and NVP (▪, n = 7; □, n = 8). B, data obtained from cells cultured under CH conditions in the absence (○, n = 9) and presence of the γ secretase inhibitors GVV and NVP (▪, n = 10; □, n = 8). C, recordings obtained from normoxically cultured cells exposed to 100 nm AβP(1-40) cells in the absence (○, n = 8) and presence of the γ secretase inhibitors GVV and NVP (▪, n = 8; □, n = 11).
DISCUSSION
Prolonged periods of hypoxia induce a wide variety of cellular responses which, physiologically, form part of the adaptive response to environmental changes such as acclimatization to high altitude (Bunn & Poyton, 1997; Lopez-Barneo et al. 2001). However, such induced changes may also contribute to pathological remodelling of cellular processes, as part of the reaction to reduced availability of O2 caused by disease or accident. Importantly, hypoxic/ischaemic episodes can precipitate the onset of dementias including AD (Tatemichi et al. 1994; Kokmen et al. 1996; Moroney et al. 1996), and so an understanding of the mechanisms underlying pathological remodelling of cell function following prolonged hypoxia is of great potential importance in the future design of therapeutic strategies aimed at preventing the onset of AD following O2 deprivation.
Our previous studies demonstrated two distinct alterations of cell function induced by hypoxia which are and mimicked by AβPs. Firstly, CH induced a Cd2+-resistant Ca2+ influx pathway which, whilst difficult to detect electrophysiologically due to its small amplitude (Green & Peers, 2001), was tightly coupled to exocytosis (Taylor et al. 1999; see also Fig. 1 and Fig. 2). Secondly, chronic hypoxia dramatically increased the component of whole-cell Ca2+ current attributable to L-type channels, whilst Ca2+ influx through other, non-L-type channel types (N-type and P-/Q-type; Liu et al. 1996; Taylor & Peers, 1999) was unaffected (Green & Peers, 2001). Disruption of Ca2+ homeostasis is an important factor leading to neurodegeneration in AD, and numerous studies have indicated that AβPs form Ca2+-permeable channels in lipid bilayers and vesicles (Arispe et al. 1996; Kawahara et al. 1997; Rhee et al. 1998). Thus, the Ca2+ influx pathway coupled to secretion, together with the increase in L-type Ca2+ channel activity or expression (Taylor et al. 1999; Green & Peers, 2001) is likely to contribute to the excessive Ca2+ influx associated with amyloid neurodegeneration.
Reactive oxygen species (ROS) have long been known to be involved in cell damage and death induced by amyloid peptides, and the ability of these peptides to increase ROS levels is closely associated with disruption of Ca2+ homeostasis and toxicity (e.g. Behl et al. 1994; Harris et al. 1995; Schubert et al. 1995; Guo et al. 1999; Miranda et al. 2000; Pratico & Delanty, 2000; Varadarajan et al. 2000; Selkoe, 2001)). Indeed, antioxidant therapy has shown improvements in AD patients (reviewed by Pratico & Delanty, 2000).
Given (i) the strong association of ROS-mediated cell damage and disruption of Ca2+ homeostasis with amyloid peptides, (ii) the association of hypoxic/ischaemic episodes with increased subsequent development of dementia and (iii) our previous evidence suggesting that CH alters Ca2+ influx via amyloid peptide formation, the present study was directed firstly at investigating the potential involvement of ROS in mediating Cd2+-resistant exocytosis and up-regulation of L-type Ca2+ channels caused by CH. To this end we examined the ability of various antioxidants to interfere with these effects of hypoxia, and particularly focused on the actions of melatonin, an endogenous antioxidant whose levels decline with age in humans, as late onset dementias become more prevalent (Reiter, 1995). Clearly, all antioxidants examined abolished the effects of CH, indicating that induction of a Cd2+-resistant Ca2+ influx pathway coupled to exocytosis, and the up-regulation of native voltage-gated Ca2+ channels, was mediated by increased ROS levels. In further support of this, exposure of cells to H2O2 (Fig. 4) mimicked the actions of CH in enhancing L-type current density. Such a finding is in accordance with previous studies described above which indicate that many cellular effects of this and related peptides are mediated by ROS. Indeed, Thomas et al. (1998) have shown that H2O2 can specifically stimulate current through L-type Ca2+ channels. Furthermore, we have recently shown that L-type Ca2+ channel up-regulation by AβP(1-40) is fully prevented by a variety of antioxidants (Giles et al. 2001).
In addition to the augmentation of L-type Ca2+ channels, we also show that the Cd2+-resistant Ca2+ influx pathway tightly coupled to secretion, apparent after a period of CH, is also inhibited by antioxidants (Fig. 1). It is possible that antioxidants merely prevent the coupling of this novel channel to exocytosis or, more likely, that they act by preventing peptide aggregation and insertion into the membrane. Presumably, channel formation requires soluble peptides to aggregate and be inserted into the membrane, as previously suggested (Arispe et al. 1996; Kawahara et al. 1997; Rhee et al. 1998). It has been established in PC12 cells that aggregation can be prevented by antioxidants (Tomiyama et al. 1996). Our immunohistochemical studies (Fig. 2) also suggest that this is the case, since hypoxia-induced 3D6 immunoreactivity was suppressed by melatonin, indicating a reduced presence of AβPs in the membrane. Importantly, although others (e.g. Reeve et al. 2001) have suggested ROS decrease in chronic hypoxia, the findings presented here indicate that ROS levels increase during prolonged hypoxia, as demonstrated previously in these cells (Hohler et al. 1999), and that increased ROS levels mediate both the induction of the Cd2+-resistant Ca2+ influx pathway and the up-regulation of native Ca2+ channels, two distinct effects. An important observation, however, was that H2O2 could mimic only the effect of hypoxia to up-regulate Ca2+ channels and not the ability of hypoxia to induce Cd2+-resistant evoked exocytosis (Fig. 4B). This is because the latter effect (induction of a Cd2+-resistant Ca2+ influx pathway coupled to evoked exocytosis) requires increased levels of amyloid peptides to aggregate and form channels, whereas the former (L-type channel up-regulation) requires ROS derived from amyloid peptides, and this effect could be mimicked by H2O2 application. It is noteworthy, however, that we cannot at present discount other ROS species in these effects.
Whether prolonged hypoxia was indeed increasing ROS levels independently of the AβP and that these ROS were leading to increased production of AβP as suggested (Misonou et al. 2000; Olivieri et al. 2000), or CH was inducing formation of the AβP through other pathways and that the produced AβP was then increasing ROS levels (Huang et al. 1999; Varadarajan et al. 2000) which were mediating the effects shown, needed to be determined. The antioxidants would prevent the effects shown in either case. To that end we then looked at these CH-induced ROS-mediated effects in the presence of AβP production inhibitors.
Both AβP(1-40) and AβP(1-42) are cleaved from amyloid precursor protein by the sequential actions of β and γ secretase (e.g. Mattson, 1997). Recent evidence suggests that γ secretase is an enzyme complex, consisting of presenilin 1 fragments coupled to additional proteins. Our understanding of the role of this complex in both physiological and pathophysiological cellular events is incomplete, yet is likely to benefit from recently developed γ secretase inhibitors. We examined the actions of two such compounds to interfere with the effects of chronic hypoxia (Figs 6, 7 and 8). Production of AβP(1-40) and AβP(1-42) by γ secretase are both prevented by NPV (Sinha & Lieberberg, 1999), whereas GVV preferentially inhibits production of AβP(1-40) (Murphy et al. 2000). We found that both agents were effective in inhibiting the actions of hypoxia, indicating that AβP production is a necessary step in hypoxic augmentation of currents and exocytosis in PC12 cells. Furthermore, given the preferential ability of GVV to block AβP(1-40) formation, our results suggest that AβP(1-40) may be the form of peptide required to mediate this action of hypoxia. Clearly ROS are therefore mediating the effects of AβP rather than its production during prolonged hypoxia; in the absence of AβPs due to γ secretase inhibition, CH no longer caused current augmentation. If CH did increase ROS levels independently from AβP production then we would expect this ROS to cause current enhancement since addition of H2O2 augmented Ca2+ currents.
The question of whether or not ROS levels increase during hypoxia is not generally established and requires further study. Indeed, in our study, hypoxia seems to induce formation of AβPs without increasing ROS levels and ROS production may only arise due to the increased presence of AβPs. A generally accepted view is that ROS levels decline as available O2 declines (e.g. Reeve et al. 2001). However, Schumacker and colleagues have provided evidence which points to mitochondria as a source of increased ROS production during hypoxia (Chandel et al. 1998; Chandel & Schumacker, 2000), and indeed Hohler et al. (1999) have demonstrated such an effect in PC12 cells. Such contrasting findings most likely arise from the difficulties associated with directly measuring ROS levels (Semenza, 2000). This is a crucial point to clarify in the context of the present study, especially since oxidative stress (i.e. increased ROS levels) can precipitate AβP accumulation (Misonou et al. 2000). Our data support the idea proposed earlier (Hohler et al. 1999) that ROS levels do indeed increase in these cells during prolonged hypoxia although through increased production of AβPs.
In summary, our results indicate that chronic hypoxia can precipitate increased formation of AβP, and lead to the induction of Cd2+-resistant exocytosis and up-regulation of native Ca2+ channels via increased ROS production. Importantly, these actions of hypoxia are absolutely dependent on AβP formation, and these peptides are likely to be the site of ROS generation. We suggest, therefore, that pathological remodelling of cell function in response to prolonged hypoxia - as manifested here by excessive neurosecretion and Ca2+ channel up-regulation - involves elevated ROS levels generated by increased levels of AβPs. These findings are important as they demonstrate a clear biochemical link between O2 deprivation and AβP formation which mirror clinical findings of a link between ischaemic episodes and Alzheimer's disease.
Acknowledgments
This work was supported by The Alzheimer's Society, the Wellcome Trust and the Medical Research Council through a PhD studentship to K.N.G.
REFERENCES
- Arias C, Arrieta I, Tapia R. Beta-amyloid peptide fragment 25–35 potentiates the calcium-dependent release of excitatory amino acids from depolarized hippocampal slices. Journal of Neuroscience Research. 1995;41:561–566. doi: 10.1002/jnr.490410416. [DOI] [PubMed] [Google Scholar]
- Arispe N, Pollard HB, Rojas E. Zn2+ interaction with Alzheimer amyloid beta protein calcium channels. Proceedings of the National Academy of Sciences of the USA. 1996;93:1710–1715. doi: 10.1073/pnas.93.4.1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behl C, Davis JB, Lesley R, Schubert D. Hydrogen peroxide mediates amyloid beta protein toxicity. Cell. 1994;77:817–827. doi: 10.1016/0092-8674(94)90131-7. [DOI] [PubMed] [Google Scholar]
- Bunn HF, Poyton RO. Oxygen sensing and molecular adaptation to hypoxia. Physiological Reviews. 1997;76:839–885. doi: 10.1152/physrev.1996.76.3.839. [DOI] [PubMed] [Google Scholar]
- Chandel NS, Maltepe E, Goldwasser E, Mathieu CE, Simon MC, Schumacker PT. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proceedings of the National Academy of Sciences of the USA. 1998;95:11715–11720. doi: 10.1073/pnas.95.20.11715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandel NS, Schumacker PT. Cellular oxygen sensing by mitochondria: old questions, new insight. Journal of Applied Physiology. 2000;88:1880–1889. doi: 10.1152/jappl.2000.88.5.1880. [DOI] [PubMed] [Google Scholar]
- Chow RH, Von Ruden L. Electrochemical detection of secretion from single cells. In: Sakmann B, Neher E, editors. Single Channel Recording. 2. New York: Plenum Press; 1995. pp. 245–275. [Google Scholar]
- Fraser SP, Suh Y-H, Djamgoz MBA. Ionic effects of the Alzheimer's disease beta-amyloid precursor protein and its metabolic fragments. Trends in Neurosciences. 1997;20:67–72. doi: 10.1016/s0166-2236(96)10079-5. [DOI] [PubMed] [Google Scholar]
- Giles GI, Tasker KM, Johnson RK, Jacob C, Peers C, Green KN. Electrochemistry of chalcogen compounds: prediction of antioxidant activity. Chemical Communications. 2001;23:2490–2492. doi: 10.1039/b107972g. [DOI] [PubMed] [Google Scholar]
- Glenner GG, Wong CW. Alzheimer's disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochemical and Biophysical Research Communications. 1984;120:885–890. doi: 10.1016/s0006-291x(84)80190-4. [DOI] [PubMed] [Google Scholar]
- Green KN, Peers C. Amyloid β peptides mediate hypoxic augmentation of Ca2+ channels. Journal of Neurochemistry. 2001;77:953–956. doi: 10.1046/j.1471-4159.2001.00338.x. [DOI] [PubMed] [Google Scholar]
- Guo Q, Sebastian L, Sopher BL, Miller MW, Ware CB, Martin GM, Mattson MP. Increased vulnerability of hippocampal neurons from presenilin-1 mutant knock-in mice to amyloid beta-peptide toxicity: central roles of superoxide production and caspase activation. Journal of Neurochemistry. 1999;72:1019–1029. doi: 10.1046/j.1471-4159.1999.0721019.x. [DOI] [PubMed] [Google Scholar]
- Harris ME, Hensley K, Butterfield DA, Leedle RA, Carney JM. Direct evidence of oxidative injury produced by the Alzheimer's beta-amyloid peptide (1–40) in cultured hippocampal neurons. Experimental Neurology. 1995;131:193–202. doi: 10.1016/0014-4886(95)90041-1. [DOI] [PubMed] [Google Scholar]
- Hohler B, Lange B, Holzapfel B, Goldenberg A, Hanze J, Sell A, Testan H, Moller W, Kummer W. Hypoxic upregulation of tyrosine hydroxylase gene expression is paralleled, but not induced, by increased generation of reactive oxygen species in PC12 cells. FEBS Letters. 1999;457:53–56. doi: 10.1016/s0014-5793(99)00999-0. [DOI] [PubMed] [Google Scholar]
- Huang X, Atwood CS, Hartshorn MA, Multhaup G, Goldstein LE, Scarpa RC, Cuajungco MP, Gray DN, Lim J, Moir RD, Tanzi RE, Bush AI. The Aβ peptide of Alzheimer's disease directly produces hydrogen peroxide through metal ion reduction. Biochemistry. 1999;38:7609–7616. doi: 10.1021/bi990438f. [DOI] [PubMed] [Google Scholar]
- Jendroska K, Hoffmann OM, Patt S. Amyloid beta peptide and precursor protein (APP). in mild and severe brain ischemia. Annals of the New York Academy of Sciences. 1997;826:401–405. doi: 10.1111/j.1749-6632.1997.tb48492.x. [DOI] [PubMed] [Google Scholar]
- Johnson-Wood K, Lee M, Motter R, Hu K, Gordon G, Barbour R, Khan K, Gordon M, Tan H, Games D, Leiberburg I, Schenk D, Seubert P, McConlogue L. Amyloid precursor protein processing and Aβ42 deposition in a transgenic mouse model of Alzheimer's disease. Proceedings of the National Academy of Sciences of the USA. 1997;94:1550–1556. doi: 10.1073/pnas.94.4.1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawahara M, Arispe N, Kuroda Y, Rojas E. Alzheimer's disease amyloid beta-protein forms Zn2+-sensitive, cation selective channels across excised membrane patches from hypothalamic neurons. Biophysical Journal. 1997;73:67–75. doi: 10.1016/S0006-3495(97)78048-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kogure K, Kato H. Altered gene expression in cerebral ischemia. Stroke. 1993;24:2121–2127. doi: 10.1161/01.str.24.12.2121. [DOI] [PubMed] [Google Scholar]
- Koistinaho J, Pyykonen I, Keinanen R, Hokfelt T. Expression of beta-amyloid precursor protein mRNAs following transient focal ischemia. NeuroReport. 1996;7:2727–2731. doi: 10.1097/00001756-199611040-00064. [DOI] [PubMed] [Google Scholar]
- Kokmen E, Whisnant JP, O'Fallon WM, Chu CP, Beard CM. Dementia after ischemic stroke: a population-based study in Rochester, Minnesota (1960–1984) Neurology. 1996;46:154–159. doi: 10.1212/wnl.46.1.154. [DOI] [PubMed] [Google Scholar]
- Liu H, Felix R, Gurnett CA, De Waard M, Witcher DR, Campbell KP. Expression and subunit interaction of voltage-dependent Ca2+ channels in PC12 cells. Journal of Neuroscience. 1996;16:7557–7565. doi: 10.1523/JNEUROSCI.16-23-07557.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Barneo J, Pardal R, Ortega-Saenz P. Cellular mechanisms of oxygen sensing. Annual Review of Physiology. 2001;63:259–287. doi: 10.1146/annurev.physiol.63.1.259. [DOI] [PubMed] [Google Scholar]
- Masters CL, Multhaup G, Simms G, Pottgiesser J, Martins RN, Beyreuther K. Neuronal origin of a cerebral amyloid: neurofibrillary tangles of Alzheimer's disease contain the same protein as the amyloid of plaque cores and blood vessels. EMBO Journal. 1985;4:2757–2763. doi: 10.1002/j.1460-2075.1985.tb04000.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP. Cellular actions of α-amyloid precursor protein and its soluble and fibrillogenic derivatives. Physiological Reviews. 1997;77:1081–1132. doi: 10.1152/physrev.1997.77.4.1081. [DOI] [PubMed] [Google Scholar]
- Miranda S, Opazo C, Larrondo LF, Munoz FJ, Ruiz F, Leighton F, Inestrosa NC. The role of oxidative stress in the toxicity induced by amyloid beta-peptide in Alzheimer's disease. Progress in Neurobiology. 2000;62:633–648. doi: 10.1016/s0301-0082(00)00015-0. [DOI] [PubMed] [Google Scholar]
- Misonou H, Morishima-Kawashima M, Ihara Y. Oxidative stress induces intracellular accumulation of amyloid beta-protein (Abeta) in human neuroblastoma cells. Biochemistry. 2000;39:6951–6959. doi: 10.1021/bi000169p. [DOI] [PubMed] [Google Scholar]
- Moroney JT, Bagiella E, Desmond DW, Paik MC, Stern Y, Tatemichi TK. Risk factors for incident dementia after stroke. Role of hypoxic and ischemic disorders. Stroke. 1996;27:1283–1289. doi: 10.1161/01.str.27.8.1283. [DOI] [PubMed] [Google Scholar]
- Murphy MP, Uljon SN, Fraser PE, Fauq A, Lookingbill HA, Findlay KA, Smith TA, Lewis PA, McLendon DC, Wang R, Golde TE. Presenilin 1 regulates pharmacologically distinct secretase activities. Implications for the role of presenilin in γ-secretase cleavage. Journal of Biological Chemistry. 2000;275:26277–26284. doi: 10.1074/jbc.M002812200. [DOI] [PubMed] [Google Scholar]
- Olivieri G, Brack CH, Muller-Spahn F, Stahelin HB, Herrmann M, Renard P, Brockhaus M, Hock C. Mercury induces cell cytotoxicity and oxidative stress and increases β-amyloid secretion and tau phosphorylation in SHSY5Y neuroblastoma cells. Journal of Neurochemistry. 2000;74:231–236. doi: 10.1046/j.1471-4159.2000.0740231.x. [DOI] [PubMed] [Google Scholar]
- Pratico D, Delanty N. Oxidative injury in diseases of the central nervous system: focus on Alzheimer's disease. American Journal of Medicine. 2000;109:577–585. doi: 10.1016/s0002-9343(00)00547-7. [DOI] [PubMed] [Google Scholar]
- Reeve HL, Michelakis E, Nelson DP, Weir EK, Archer SL. Alterations in a redox oxygen sensing mechanism in chronic hypoxia. Journal of Applied Physiology. 2001;90:2249–2256. doi: 10.1152/jappl.2001.90.6.2249. [DOI] [PubMed] [Google Scholar]
- Reiter RJ. Melatonin reduces kainate-induced lipid peroxidation in homogenates of different brain regions. FASEB Journal. 1995;9:526–533. doi: 10.1096/fasebj.9.12.7672513. [DOI] [PubMed] [Google Scholar]
- Rhee SK, Quist AP, Lal R. Amyloid α protein (1–42) forms calcium-permeable, Zn2+-sensitive channel. Journal of Biological Chemistry. 1998;273:13379–13382. doi: 10.1074/jbc.273.22.13379. [DOI] [PubMed] [Google Scholar]
- Schubert D, Behl C, Lesley R, Brack A, Dargusch R, Sagara Y, Kimura H. Amyloid peptides are toxic via a common oxidative mechanism. Proceedings of the National Academy of Sciences of the USA. 1995;92:1989–1993. doi: 10.1073/pnas.92.6.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer's disease: genes, proteins and therapy. Physiological Reviews. 2001;81:741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- Semenza GL. Mechanisms of oxygen homeostasis, circa 1999. Advances in Experimental Medicine and Biology. 2000;475:303–310. doi: 10.1007/0-306-46825-5_29. [DOI] [PubMed] [Google Scholar]
- Sinha S, Lieberberg I. Cellular mechanisms of β-amyloid production and secretion. Proceedings of the National Academy of Sciences of the USA. 1999;96:11049–11053. doi: 10.1073/pnas.96.20.11049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatemichi TK, Paik M, Bagiella E, Desmond DW, Stern Y, Sano M, Hauser WA, Mayeux R. Risk of dementia after stroke in a hospitalized cohort: results of a longitudinal study. Neurology. 1994;44:1885–1891. doi: 10.1212/wnl.44.10.1885. [DOI] [PubMed] [Google Scholar]
- Taylor SC, Batten TFC, Peers C. Hypoxic enhancement of quantal catecholamine secretion: evidence for the involvement of amyloid α-peptides. Journal of Biological Chemistry. 1999;274:31217–31223. doi: 10.1074/jbc.274.44.31217. [DOI] [PubMed] [Google Scholar]
- Taylor SC, Peers C. Store-operated Ca2+ influx and voltage-gated Ca2+ channels coupled to exocytosis in pheochromocytoma (PC12) cells. Journal of Neurochemistry. 1999;73:874–880. doi: 10.1046/j.1471-4159.1999.0730874.x. [DOI] [PubMed] [Google Scholar]
- Thomas GP, Sims SM, Cook MA, Karmazyn M. Hydrogen peroxide-induced stimulation of L-type calcium current in guinea pig ventricular myocytes and its inhibition by adenosine A1 receptor activation. Journal of Pharmacological and Experimental Therapy. 1998;286:1208–1214. [PubMed] [Google Scholar]
- Tischler AS, Perlman RL, Morse GM, Sheard BE. Glucocorticoids increase catecholamine synthesis and storage in PC12 pheochromocytoma cell cultures. Journal of Neurochemistry. 1983;40:364–370. doi: 10.1111/j.1471-4159.1983.tb11291.x. [DOI] [PubMed] [Google Scholar]
- Tomiyama T, Shoji A, Kataoka K, Suwa Y, Asano S, Kaneko H, Endo N. Inhibition of amyloid beta protein aggregation and neurotoxicity by rifampicin. Its possible function as a hydroxyl radical scavenger. Journal of Biological Chemistry. 1996;271:6839–6844. doi: 10.1074/jbc.271.12.6839. [DOI] [PubMed] [Google Scholar]
- Varadarajan S, Yatin S, Aksenova M, Butterfield DA. Alzheimer's amyloid beta-peptide-associated free radical oxidative stress and neurotoxicity. Journal of Structural Biology. 2000;130:184–208. doi: 10.1006/jsbi.2000.4274. [DOI] [PubMed] [Google Scholar]
- Wightman RM, Jankowski JA, Kennedy RT, Kwagoe KT, Schroeder TJ, Leszczyszyn DJ, Near JA, Diliberto EJ, Jr, Viveros OH. Temporally resolved catecholamine spikes correspond to single vesicle release from individual chromaffin cells. Proceedings of the National Academy of Sciences of the USA. 1991;88:10754–10758. doi: 10.1073/pnas.88.23.10754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Anwyl R, Rowan MJ. Beta-amyloid-(1–40) increases long-term potentiation in rat hippocampus in vitro. European Journal of Pharmacology. 1995;284:R1–3. doi: 10.1016/0014-2999(95)00539-w. [DOI] [PubMed] [Google Scholar]
- Yokota M, Saido TC, Tani E, Yamaura I, Minami N. Cytotoxic fragment of amyloid precursor protein accumulates in hippocampus after global forebrain ischemia. Journal of Cerebral Blood Flow and Metabolism. 1996;16:1219–1223. doi: 10.1097/00004647-199611000-00016. [DOI] [PubMed] [Google Scholar]