

Abstract
Calcium influx into the presynaptic nerve terminal is well established as a trigger signal for transmitter release by exocytosis. By studying dissociated preoptic neurons with functional adhering nerve terminals, we here show that presynaptic Ca2+ influx plays dual and opposing roles in the control of spontaneous transmitter release. Thus, application of various Ca2+ channel blockers paradoxically increased the frequency of spontaneous (miniature) inhibitory GABA-mediated postsynaptic currents (mIPSCs). Similar effects on mIPSC frequency were recorded upon washout of Cd2+ or EGTA from the external solution. The results are explained by a model with parallel Ca2+ influx through channels coupled to the exocytotic machinery and through channels coupled to Ca2+-activated K+ channels at a distance from the release site.
The release of neurotransmitter from central nerve terminals is a process that has been extensively studied. Accordingly, the view is well established that action potential-evoked neurotransmitter release is triggered by the activation of voltage-gated Ca2+ channels and subsequent Ca2+ influx and local rise in intraterminal Ca2+ concentration (Katz & Miledi, 1968; Miledi, 1973). However, it has also long been known that quanta of transmitter are also released spontaneously in the absence of presynaptic impulse activity (Fatt & Katz, 1950, 1952; del Castillo & Katz, 1955). The physiological significance of the spontaneous neurotransmitter release is not yet well understood. However, evidence has recently been presented, suggesting that this type of release may carry synaptic information (Hirsch et al. 1999; Staley, 1999) and play important roles in the maintenance of synaptic structures (McKinney et al. 1999), synaptic plasticity (Jensen et al. 1999; Kombian et al. 2000), the regulation of neuronal impulse firing (Cohen & Miles, 2000) and the modulation of nociceptive transmission (Rhee et al. 2000). In addition, spontaneous synaptic transmission may be involved in the pathophysiology of amyotrophic lateral sclerosis (Andjus et al. 1997) and epilepsy (Hirsch et al. 1999; Cossart et al. 2001).
Like action potential-dependent release, spontaneous neurotransmitter release can be modulated by various means including the electrical activity of the neuron, neurotransmitters, neuropeptides, clinically used drugs and neurotoxins (for review see Bouron, 2001). In contrast to the evoked neurotransmitter release, the requirement of Ca2+ for triggering the action potential-independent neurotransmitter release is less clear. Thus, in several studies, spontaneous release was independent of Ca2+, or only weakly dependent on Ca2+ (Fatt & Katz, 1952; for example see also Capogna et al. 1993; Vaughan & Christie, 1997; Brussaard et al. 1999), but in other studies spontaneous release was influenced by Ca2+ influx through a number of different types of presynaptic Ca2+ channels (for example see Rhee et al. 2000). The rate of spontaneous release can also be changed by blocking low-threshold voltage-gated Ca2+ channels (Bao et al. 1998) or high-threshold voltage-gated Ca2+ channels (Koyama et al. 1999). However, despite intense efforts to clarify the mechanisms of regulation of spontaneous neurotransmitter release from central nerve terminals, the role of Ca2+ is still not clear.
In the present study, we investigate spontaneous GABA release from nerve terminals adhering to dissociated neurons from the rat medial preoptic nucleus (MPN). In previous studies of this preparation, we have shown that depolarization-evoked GABA release is mainly mediated by one predominant type of high-threshold Ca2+ channel that may be either N-, P- or Q-type, whereas L-type channels are not involved (Haage et al. 1998).
Here, we describe the seemingly paradoxical potentiating effects of Ca2+ channel blockers on the spontaneous GABA release. Our results lead us to the conclusion that Ca2+ influx into the presynaptic terminal may play dual and partly opposing roles. Although influx through some Ca2+ channels is crucial for a high spontaneous release frequency, this influx may be indirectly, negatively controlled via Ca2+ influx through other Ca2+ channels. The latter control is exerted by effects on the membrane potential via Ca2+-dependent K+ channels.
METHODS
Ethical approval of the procedures described was given by the local ethics committee for animal research.
Cell preparation
The methods used for cell preparation have been described elsewhere (Karlsson et al. 1997; Haage et al. 1998). In short, young (50-120 g) male Sprague-Dawley rats were killed by decapitation, the brain was removed and coronal slices containing the preoptic area were prepared. For recordings from neurons in intact slices, ∼150 μm thick slices were used. For further dissociation of single neurons with adhering synaptic terminals, 250-300 μm thick slices were used. In the latter case, the cells were isolated mechanically by application of a vibrating glass rod to the surface of the slice at the site of the medial preoptic nucleus (cf. Vorobjev, 1991). No enzymes were used. The dissociated cells had cell bodies measuring 10-15 μm at their longest axes, and often one or several neurites of length up to about 100 μm.
Electrophysiology
Whole-cell currents were measured under voltage-clamp conditions using the amphotericin B perforated-patch technique (Rae et al. 1991). Borosilicate glass pipettes with a resistance of 2-5 MΩ, when filled with intracellular solution and immersed in extracellular solution (see below), were used. The liquid-junction potential was measured as described by Neher (1992) and has been subtracted in all potential values given. An Axopatch 200A or Axopatch 200B amplifier, a Digidata 1200 interface, and the pCLAMP software (version 6.03-8; all from Axon Instruments) controlled via a 486 or Pentium processor-based personal computer were used to record electrical signals, which were low-pass filtered at 2-10 kHz (-3 dB). Series resistance compensation was not used, due to its introduction of extra noise and the small amplitude of the recorded signals under steady voltage conditions. Although the series resistance was not determined for all of the studied cells, previous estimation with the same preparation and recording techniques yielded values of 18 ± 3 MΩ (mean ± s.e.m.; n = 16; Johansson et al. 2001). Further, to avoid sudden large changes in series resistance, the time course of capacitative current in response to a -5 mV voltage step was repeatedly monitored during the experiments. Slow changes in series resistance less than about 20 % were accepted. Dissociated cells were studied using an inverted Zeiss Axiovert 25 microscope, whereas neurons in slices were studied using an upright Zeiss Axioskop FS microscope equipped with water-immersion objectives and differential interference contrast optics. In the latter case, the surfaces of visually identified neurons were cleaned by a stream of standard extracellular solution (for composition see below) applied from a glass pipette (see Edwards et al. 1989). A gravity-fed fast perfusion system, with the common outlet of a four- or eight-barrelled pipette positioned 100-200 μm from the studied cell, was used for continuous application of standard extracellular solution or of test solutions. Switching between solutions was controlled by solenoid valves, and the solution exchange time was, in good cases, less than 10 ms. All experiments were performed at room temperature (21-23 °C).
Solutions and chemicals
The standard extracellular solution used as control contained (mm): 137 NaCl, 5.0 KCl, 1.0 CaCl2, 1.2 MgCl2, 10 Hepes and 10 glucose. Glycine (3 μM) and tetrodotoxin (TTX; 2 μM, from Sigma or from Alomone Labs, Jerusalem, Israel) were routinely added, and pH was adjusted to 7.4 with NaOH. For recordings from neurons in slice preparations, the extracellular solution was oxygenated (95 % O2, 5 % CO2). The standard intracellular solution, used for filling the patch pipettes, contained (mm): 140 potassium gluconate, 3.0 NaCl, 1.2 MgCl2, 1.0 EGTA, 10 Hepes; pH was adjusted to 7.2 with KOH. As an alternative to K+, Cs+ was used to reduce the background noise. When Cs+ was used, pH was adjusted to 7.2 with CsOH. Amphotericin B (Sigma), prepared from a stock solution (6 mg amphotericin B dissolved in 100 μl dimethyl sulphoxide), was added to a final concentration of 120 μg amphotericin B per millilitre intracellular solution. The peptide toxins apamin, calciseptine, charybdotoxin and ω-conotoxins GVIA and MVIIC were purchased from Alomone Labs or alternatively from Latoxan (Valence, France), Bay K 8644; bicuculline methiodide, nifedipine, nimodipine, paxilline and tetraethylammonium (TEA) were from Sigma. The dihydropyridines nifedipine, nimodipine and Bay K 8644 were prepared from a stock solution (10 mm in dimethylsulphoxide). In all experiments dimethylsulphoxide was routinely added to the control solution to achieve the same concentration as in the dihydropyridine-containing test solutions. The peptide toxins were dissolved in extracellular solution. In experiments with charybdotoxin, bovine serum albumin (0.01 % w/v) was also added to both experimental and control solutions.
Analysis
All analysis and production of graphic presentations were performed using Clampfit (Axon Instruments), Mini Analysis (Synaptosoft, Inc.) and Origin (Microcal Software) software. The spontaneous currents were detected and counted semi-automatically using Clampfit and Mini Analysis software with consequent visual inspection. The amplitude threshold for detection of spontaneous postsynaptic currents was set above the noise level and kept constant in each experiment. The data are presented as means ± standard error of mean (s.e.m.), unless stated otherwise.
RESULTS
Organic Ca2+ channel blockers increase the frequency of spontaneous synaptic currents
As earlier reported (Haage et al. 1998), spontaneous ‘miniature’ GABA-mediated postsynaptic currents (mIPSCs) can be recorded from the dissociated MPN neurons, to which presynaptic nerve terminals are adhering. The frequency of detected mIPSCs depends on the background current noise, which varies somewhat with membrane potential and also depends on the ion composition of the pipette-filling solution. To better quantify the frequency of mIPSCs, we made the following experiments (unless stated otherwise) with Cs+ replacing K+ in the pipette filling solution, which resulted in significant reduction of the background noise, and with the postsynaptic cell voltage clamped at -4 mV. The voltage was chosen to give a relatively large driving force for Cl− without risking mistaking the mIPSCs for miniature excitatory postsynaptic currents, since the latter are commonly carried non-selectively by cations and thus also outwardly directed at positive voltages. Under these conditions, the detection limit for mIPSCs was a peak amplitude of ∼5 pA.
As earlier described, a burst of synaptic currents can be evoked by depolarization of the presynaptic terminals by transient perfusion with 140 mm KCl. Although the depolarization-induced synaptic current is not affected by the L-type Ca2+ channel blocker nifedipine (Haage et al. 1998), the situation is markedly different for the spontaneous mIPSCs described here. Upon switching from perfusion with control extracellular solution (using standard pipette filling solution; see Methods) to a solution containing 100 μM nimodipine, the frequency of mIPSCs unexpectedly increased markedly (Fig. 1A). On average, the mIPSC frequency transiently increased many-fold from 0.43 ± 0.13 Hz to a peak value of 3.8 ± 0.8 Hz (n = 16; Fig. 2A). The peak was reached within 10-20 s and thereafter the effect declined to a more steady level, reached after ∼1 min. The subsequent addition of 100 μM bicuculline methiodide reversibly abolished nearly all mIPSCs, suggesting that the current events triggered by nimodipine were due to GABAA receptor activation (Fig. 2A). Although the potentiating effect of nimodipine on the mIPSC frequency was not seen in all cells, it was clearly obtained in the majority of cells. In 73 % of the 350 neurons tested the mIPSC frequency increased in response to nimodipine, whereas in 19 % the frequency did not change significantly, and in 8 % a reduced frequency was recorded.
Figure 1. Ca2+ channel blockers increase the frequency of mIPSCs in dissociated neurons.
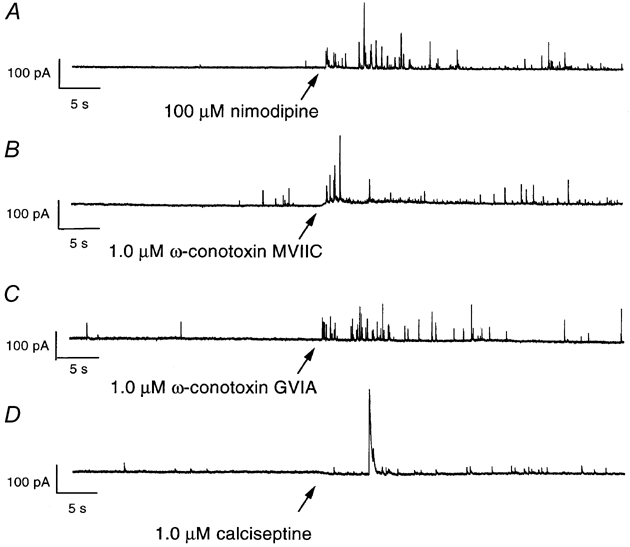
Nimodipine (100 μM, A), ω-conotoxin MVIIC (1.0 μM, B), ω-conotoxin GVIA (1.0 μM, C) and calciseptine (1.0 μM, D) were added to the external solution, starting at the time indicated by arrows. Note the resulting increase in mIPSC frequency. Current recorded with the postsynaptic membrane potential clamped at -4 mV. Currents presented in A and B were recorded from the same cell, C and D from two different cells.
Figure 2. Effect of Ca2+ channel blockers on the frequency of mIPSCs.
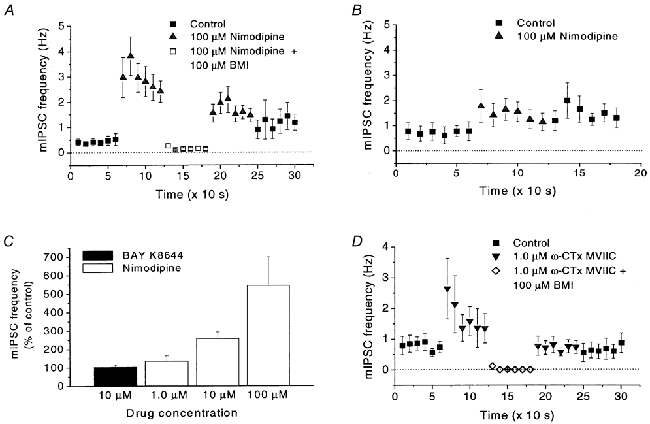
A, change in mIPSC frequency in dissociated neurons caused by nimodipine (100 μM). Note the increase caused by nimodipine and the block caused by addition of bicuculline methiodide (BMI; 100 μM). The number of mIPSCs was measured during 10 s intervals in 17 cells. B, in a slice preparation, nimodipine (100 μM) also affects the mIPSC frequency. Note the similar, although smaller, reaction to nimodipine as in dissociated neurons (A). The number of mIPSCs was measured as in A, in eight cells. C, effects of nimodipine (1.0, 10 and 100 μM) or Bay K 8644 (10 μM) on mIPSC frequency (measured for 1 min) in the same group of cells (n = 9). Note the lack of effect of Bay K 8644. D, effects of ω-conotoxin MVIIC (ω-CTx MVIIC; 1.0 μM) and of BMI (100 μM) on mIPSC frequency in 10 cells, measured as in A. For all recordings the postsynaptic membrane potential was clamped at -4 mV. Error bars indicate s.e.m.
To rule out the possibility that the observed effect was due to our preparation of dissociated neurons, we applied 100 μM nimodipine to MPN neurons in a slice preparation (see Methods). Although the increase in mIPSC frequency was less prominent and poorly reversible, the reaction to nimodipine was clear. The peak frequency (1.76 ± 0.65 Hz) was > twice the previous 60 s baseline frequency (0.73 ± 0.31 Hz, n = 8; Fig. 2B). Thus, the results suggest that the potentiating effect of nimodipine on mIPSC frequency may also be seen under physiological conditions. The smaller increase in mIPSC frequency and the slow time course of washout in the slice preparations are probably related to the more restricted diffusion of the experimental solutions, with a slow penetration into the slice and less steep concentration gradient reaching the recorded neurons.
Since Ca2+ is known to trigger transmitter release, the above effects of Ca2+ channel blockers were surprising. A number of earlier studies have demonstrated that dihydropyridine Ca2+ channel antagonists, e.g. nifedipine and nimodipine, as well as the agonist Bay K 8644, also may block K+ currents in some, but not all, preparations (A-type currents: Fagni et al. 1994; Mlinar & Enyeart, 1994; large-conductance Ca2+-activated currents: Avdonin et al. 1997). Although it may seem possible that our findings could be explained by dihydropyridine-mediated effects on K+ channels, several lines of evidence suggest that the potentiated mIPSC frequency was related to a block of Ca2+ channels. First, application of the agonist dihydropyridine Bay K 8644 (10 μM) did not affect the frequency of mIPSCs, whereas the antagonist nimodipine increased the mIPSC frequency in all three concentrations tested (1.0, 10 and 100 μM) (Fig. 2C). Another antagonist, nifedipine (100 μM), also potentiated the mIPSC frequency (in 4 of 7 cells tested; not further quantitatively analysed). Thus, the potentiating effect on mIPSC frequency is not general for all dihydropyridines, but only for those that are Ca2+ channel antagonists. This is, however, not completely ruling out a direct effect of dihydropyridines on K+ channels since in one report on delayed rectifier-like K+ currents of unusual pharmacology, the blocking effect on K+ channels was not shared by Bay K 8644 (Valmier et al. 1991). Stronger evidence for an effect on Ca2+ channels was provided by the use of other, non-dihydropyridine, Ca2+ channel blockers which all did potentiate the mIPSC frequency. Thus, application of 1.0 μM ω-conotoxin MVIIC, a blocker of N-, P- and Q-type Ca2+ channels, resulted in an increased frequency of mIPSCs (296 ± 54 % of previous baseline, n = 10), which were also sensitive to block by bicuculline methiodide (Fig. 1B and 2D). A similar, although smaller, effect (144 ± 21 %, n = 7) was seen at the application of the N-type Ca2+ channel blocker ω-conotoxin GVIA (1.0 μM; Fig. 1C). The L-type Ca2+ channel-blocking peptide calciseptine (1.0 μM) also caused an increase in mIPSC frequency (175 ± 11 %, n = 2; Fig. 1D).
The common known property of the above substances, which all were found to potentiate the mIPSC frequency, is a blocking effect on high-threshold Ca2+ channels. In previous studies of depolarization-evoked GABA release onto MPN neurons, mainly N-, P- and Q-type Ca2+ channels were found to mediate the Ca2+ influx triggering the release, whereas no evidence was found for a role of L-type channels (Haage et al. 1998). However, the potentiating effect of the L-type Ca2+ channel blockers found here implies a regulatory role also for presynaptic L-type channels in controlling spontaneous transmitter release.
Effects of Cd2+, Ni2+ and EGTA on mIPSC frequency
To use a more non-selective blocker of high-threshold Ca2+ channels, we proceeded to investigate the effects of Cd2+, which blocks L-, N-, P- and Q-type channels and to some extent also R-type channels. However, upon addition of 200 μM Cd2+ to the extracellular solution, the mIPSC frequency did not increase, but rather decreased to 63 ± 11 % (n = 11) of that in control (Fig. 3A). Further, although a potentiating effect of nimodipine (100 μM) on the mIPSC frequency was still present when nimodipine was added to the Cd2+ containing solution, this increase in mIPSC frequency was reduced to 38 ± 11 % (n = 11) of that obtained when nimodipine was added to control solution (Fig. 3A and D). In contrast, application of 200 μM Ni2+, which mainly blocks low-threshold Ca2+ channels, to the same series of cells caused only a small reduction of the mIPSC frequency (to 81 ± 11 %, n = 11) and did not significantly affect the response to nimodipine (96 ± 23 % remaining, n = 11; Fig. 3B and D). (The presence of a response, although significantly reduced, to nimodipine in 200 μM Cd2+ suggests that although Cd2+ blocks most high-threshold Ca2+ channels, some Ca2+ influx through L-type channels remains. This is consistent with earlier findings of a non-complete block of high-threshold Ca2+ currents by 100 μM Cd2+ (Lorenzon & Foehring, 1995; Sundgren-Andersson & Johansson, 1998). This was further supported by the experiments with EGTA below, showing a smaller response to nimodipine compared with that in Cd2+ as well as a larger reduction of baseline mIPSC frequency.)
Figure 3. Effects of Cd2+, Ni2+ and EGTA on mIPSC frequency and on response to nimodipine.
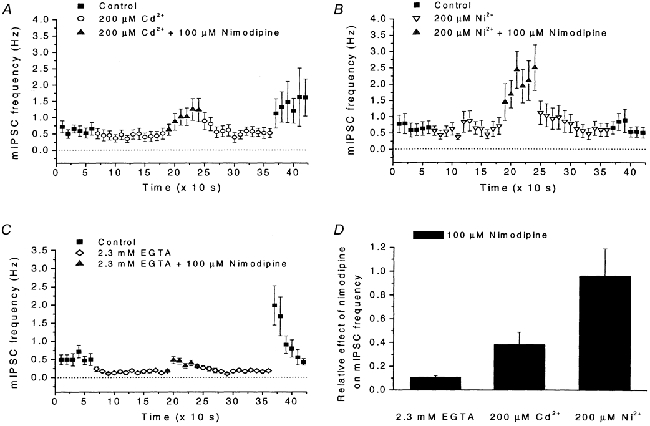
A, effects of Cd2+ (200 μM), and of nimodipine (100 μM) in the presence of Cd2+, on mIPSC frequency. Note the reduced mIPSC frequency, as well as the increase in mIPSC frequency after washout of Cd2+. Recording conditions and data presentation similar to those in Fig. 2A. Data from 11 cells. B, effects of Ni2+ (200 μM), and of nimodipine (100 μM) in the presence of Ni2+, on mIPSC frequency. Recording conditions and data presentation similar to those in A. Data from 11 cells. C, number of mIPSCs in control solution, in EGTA (2.3 mm) added to the control solution, and with nimodipine (100 μM) added to the EGTA-containing solution. Note the qualitatively similar effects compared with those seen with the application of Cd2+ in A. Recording conditions and data presentation similar to those in A. Data from 15 cells. D, summary of the effects of nimodipine (100 μM) on the mIPSC frequency (measured for 1 min) in the presence of EGTA (2.3 mm), Cd2+ (200 μM) or Ni2+ (200 μM). The effects are indicated relative to the response to nimodipine when added to control solution. Note that nimodipine evoked similar responses in Ni2+-containing solution and in control solution.
The above results showed that a rapid non-selective block, by Cd2+, of most high-threshold Ca2+ channel types does not cause potentiation of the mIPSC frequency although a more selective block of either of several Ca2+ channel types does. Thus, a likely interpretation is that although the potentiated mIPSC frequency is triggered by a reduced Ca2+ influx through some Ca2+ channels, influx through other Ca2+ channels is required for the effect. Additional observations on the effects of Cd2+ seem to support this idea of a ‘partial’ block of the Ca2+ channel population. During washout of Cd2+, when a partial block may be expected, the mIPSC frequency transiently increased to levels (1.6 ± 0.9 Hz, n = 11) well above the 60 s baseline frequency before addition of Cd2+ (0.61 ± 0.14 Hz, n = 11; Fig. 3A).
Effects similar to those seen with Cd2+ were recorded upon application of 2.3 mm EGTA, which was expected to give a free Ca2+ concentration of ∼100 nm. The mIPSC frequency was reduced in the presence of EGTA (to 52 ± 13 % of control, n = 15), the response to 100 μM nimodipine was markedly reduced in the presence of EGTA (to 12 ± 1 % of nimodipine-evoked effect in control, n = 15), but a dramatic increase of frequency was observed immediately after washout of EGTA (2.0 ± 0.5 Hz during the first 10 s after washout; compared with the baseline frequency in EGTA of 0.16 ± 0.03 Hz and in control of 0.53 ± 0.13 Hz, n = 15; Fig. 3C and D). Together, these observations suggest that a reduced influx of Ca2+ triggers some process leading to the dramatic increase in probability for spontaneous transmitter release when Ca2+ becomes available again.
We further made observations suggesting that the increase in mIPSC frequency after washout of Cd2+ and that caused by nimodipine depended on the same mechanism. This conclusion is based on the variability between cells in the response to nimodipine (in the presence of Cd2+). Using a one-way ANOVA procedure the cells were classified into two groups depending on whether they showed a significant (P < 0.05) response to nimodipine when added to control solution. After this classification, a clear correlation between the response to nimodipine and the effect of Cd2+ washout was evident. Cells that showed a significant response to nimodipine also showed a significant increase in mIPSC frequency after washout of Cd2+, whereas cells without a clear response to nimodipine did not (Fig. 4A and B). (The correlation coefficient obtained by a Spearman test was 0.75, P < 0.05, n = 11.) This correlation suggests the presence of a common causative factor underlying the response to nimodipine as well as the effect of washing out Cd2+.
Figure 4. Effects of Cd2+ and of nimodipine in the presence of Cd2+ on mIPSC frequency.
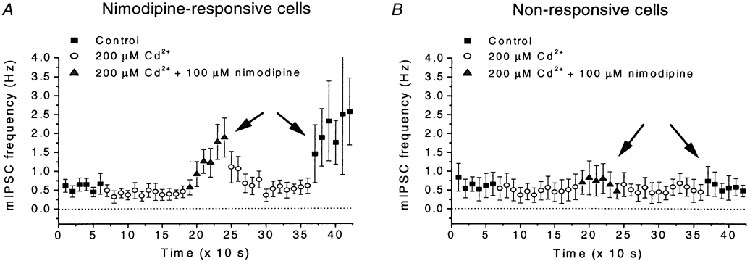
The cells were grouped on basis of a significant response to nimodipine (100 μM) when added to control solution. A, nimodipine-responsive cells (n = 6). B, non-responsive cells (n = 5). Note that nimodipine-sensitive cells also showed some response in Cd2+ (200 μM), and that the sensitivity to nimodipine was correlated with an increased mIPSC frequency after washout of Cd2+, which was not seen in the non-responsive cells. Correlation marked by arrows in A and B. Recording conditions and data presentation similar to those in Fig. 2A.
Relation between [Ca2+]o and mIPSC frequency
Due to the apparent role of Ca2+ influx in the above effects, we experimentally determined the relation between the external Ca2+ concentration, [Ca2+]o, and the mIPSC frequency in the absence of blocking agents as well as in response to 100 μM nimodipine. Whereas the frequency without blocker was only weakly dependent on [Ca2+]o, the effect of nimodipine showed a sublinear, roughly hyperbolic relation to [Ca2+]o (Fig. 5A). This relation was in reasonably good agreement with the previously established relation between [Ca2+]o and the postsynaptic current evoked by KCl-mediated depolarization of presynaptic terminals on MPN neurons (Fig. 5B; Haage et al. 1998). In this respect, the effect of nimodipine was similar to the effect of depolarization.
Figure 5. Relation between external Ca2+, mIPSC frequency and response to nimodipine.
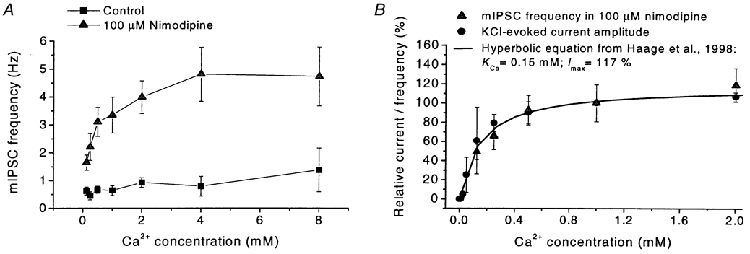
A, relation between external Ca2+ concentration and mIPSC frequency in standard extracellular solution (squares) and mIPSC frequency at the application of nimodipine (100 μM, triangles; measured during first 60 s after nimodipine application). Note the weak dependence of mIPSC frequency on external Ca2+ in control solution. Each point represents data from 15 cells, except for 1.0 mm Ca2+: 54 cells. Holding potential -4 mV. Error bars represent s.e.m.B, relation between external Ca2+ and mIPSC frequency at the application of nimodipine (100 μM, triangles) as in A, with superimposed relation between external Ca2+ and the peak amplitude of the GABAA receptor-mediated synaptic current evoked by the application of 140 mm K+ (filled circles; data from Haage et al. 1998). The smooth line is described by a hyperbolic equation with a half-saturating Ca2+ concentration of 0.15 mm and maximum current/mIPSC frequency 117 % of that at 1.0 mm Ca2+, as described by Haage et al. (1998).
A hypothetical model of presynaptic mechanisms controlling mIPSC frequency
One plausible model that could explain an increased mIPSC frequency caused by Ca2+ channel blockers involves Ca2+-activated K+ channels. The presence of Ca2+-activated K+ channels has been demonstrated in some nerve terminals (see reviews by Jackson, 1995; Meir et al. 1999), although their involvement in regulation of neurotransmission has been reported mainly for evoked release (Robitaille & Charlton, 1992; Robitaille et al. 1993). Our hypothesis depends on the functional/spatial separation between the Ca2+ channels that trigger exocytosis and the Ca2+ channels that couple to Ca2+-activated K+ channels (Fig. 6A). Such separation is clearly possible, and does not require a long distance. It has recently been demonstrated that even in individual membrane patches of hippocampal neurons, L-type Ca2+ channels may selectively couple to small-conductance Ca2+-activated K+ channels (SK channels) whereas N-type Ca2+ channels couple to large-conductance Ca2+-activated K+ channels (BK channels) (Marrion & Tavalin, 1998). We thus imagine that a block of Ca2+ channels that couple to K+ channels in the presynaptic membrane would lead to a reduced K+ conductance and consequent depolarization of the presynaptic terminal. If sufficient Ca2+ channels coupled to the exocytotic machinery were available (not blocked), they would be activated by the depolarization, and the resulting Ca2+ influx through those channels would increase the probability of transmitter release (Fig. 6B and C). It should be noted that the membrane potential of the presynaptic terminal, as a consequence of a high membrane resistance due to the small membrane area, is likely to fluctuate quite considerably in response to small currents (cf. the situation for very small neurons, Johansson & Århem, 1994). (For clarity, this dynamic situation is not illustrated in the simplified Fig. 6.) Therefore, it should be expected that high-threshold Ca2+ channels may be activated occasionally even in terminals that most of the time remain well polarized. This explains the findings that the basal mIPSC frequency depends on extracellular Ca2+ and on high-threshold Ca2+ channels. (Further arguments for the model are given in Discussion.)
Figure 6. Hypothetical model of nerve terminal on MPN neuron.
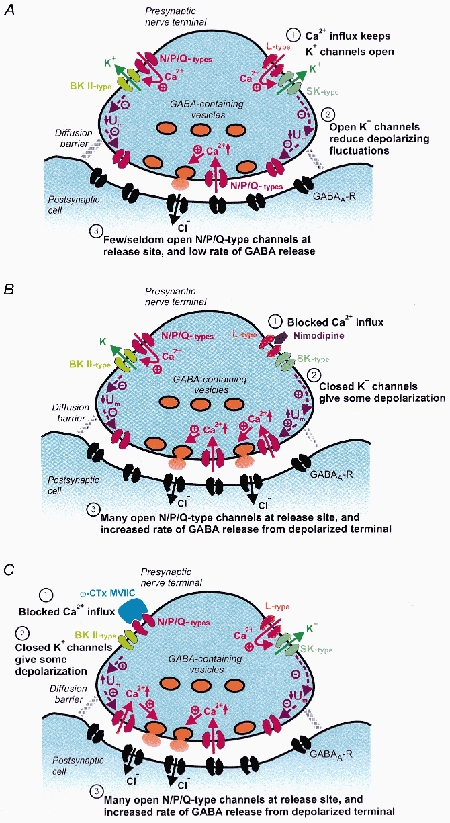
A, the presynaptic nerve terminal in control conditions. The terminal contains Ca2+ channels, of L- and N-, and P/Q-types, located outside the release site. Ca2+ influx through these channels leads to activation of Ca2+-gated K+ channels that exert major influence on the membrane potential. The membrane potential, in turn, controls Ca2+ channels, of N-, P- or Q-type, at the transmitter release site. Note that outside the release site, N- and P/Q-type Ca2+ channels are coupled to BK channels (left), whereas L-type Ca2+ channels are coupled to SK channels (right). B and C, blocking of the Ca2+ channels outside the release site by nimodipine (B) or ω-conotoxin MVIIC (C) is followed by closure of nearby Ca2+-gated K+ channels and depolarization. The depolarization causes activation of Ca2+ channels at the release site, and the Ca2+ influx triggers transmitter release. Note the presence of a diffusion barrier between the extrasynaptic part of the extracellular space and the synaptic cleft. This barrier is limiting rapid access of large blocking molecules to the Ca2+ channels at the release site. Note that the figure is a simplification and that the membrane potential of the terminal is expected to fluctuate around a mean value in A which differs from that in B and C.
Effects of K+ channel blockers on mIPSC frequency
The hypothetical model above requires the presence of presynaptic Ca2+-dependent K+ channels. Further, if the hypothesis is correct, blocking these channels should lead to presynaptic depolarization and increased mIPSC frequency. We tested both these requirements by applying K+ channel blockers to our preparation. Application of the bee venom apamin (1.0 μM), a well-known blocker of several SK channel types (Hugues et al. 1982), resulted in a dramatic increase in mIPSC frequency (694 ± 321 %, n = 17; Fig. 7A). We also applied charybdotoxin (ChTx; 200 nm) and paxilline (1.0 μM), which both block mainly BK channels (Miller et al. 1985; Knaus et al. 1994), although charybdotoxin may affect other types of K+ channels in addition (Schneider et al. 1989). Both drugs, when tested separately, induced a significant increase in mIPSC frequency (Fig. 7A), although the effect was smaller for paxilline (491 ± 245 %, n = 10, for charybdotoxin and 265 ± 89 %, n = 15, for paxilline). A similar increase in mIPSC frequency (217 ± 35 %, n = 13) was observed with the less selective K+ channel blocker tetraethylammonium (TEA, 10 mm; Fig. 7A). These results thus suggest that there are indeed Ca2+-activated K+ channels in the presynaptic terminals on the MPN neurons, and that blocking these channels results in an increased frequency of spontaneous transmitter release, in support of the above hypothesis.
Figure 7. Effects of K+ channel manipulation on mIPSC frequency.
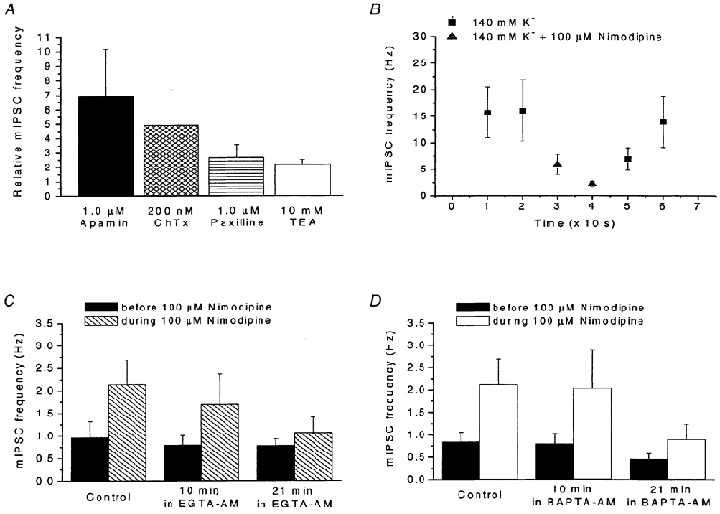
A, effects of the K+ channel blockers apamin (1.0 μM; n = 17), charybdotoxin (ChTx, 200 nm; n = 10), paxilline (1.0 μM; n = 15) and TEA (10 mm, n = 13) on mIPSC frequency. Data presented relative to control. The number of mIPSCs was measured during a 60 s period immediately before and just after the application of K+ channel blocker. B, effect of a high external K+ concentration (140 mm) on the response to nimodipine (100 μM). Frequency of mIPSCs in an external solution containing 140 mm K+ without Ca2+ channel blocker (squares) and after addition of 100 μM nimodipine (triangles). Note the reduced mIPSC frequency in the presence of nimodipine. Data from 7 cells. C and D, effects of the slow Ca2+ buffer EGTA-AM (C) and the fast Ca2+ buffer BAPTA-AM (D) on mIPSC frequency and on the response to nimodipine. The number of mIPSCs was measured during a 60 s period of nimodipine application to two groups of neurons (6 cells in each). Data presented relative to preceding baseline measured during a 60 s period immediately before addition of 100 μM nimodipine to the control solution and to Ca2+ buffer-containing solution after 10 and 21 min of preincubation with either 100 μM EGTA-AM or 100 μM BAPTA-AM. Note the block of response to nimodipine after 21 min of preincubation with EGTA-AM (C) and the reduced response caused by 21 min in BAPTA-AM (D). For all data, recording conditions similar to those in Fig. 2A. Error bars represent s.e.m.
Effects of nimodipine on mIPSC frequency in high external K+ concentration
Another prediction by the above-presented hypothesis is that application of Ca2+ channel blockers should not raise the mIPSC frequency when the presynaptic terminals are depolarized to near 0 mV, where Ca2+ influx through release-triggering Ca2+ channels is already at a maximum. We therefore depolarized the presynaptic terminals by a high external K+ concentration (cf. Haage et al. 1998) to test the validity of the prediction. For this, K+ was substituted for Na+ in the external solution, a procedure expected to depolarize the presynaptic terminals to near 0 mV. In the K+-rich external solution, the frequency of mIPSCs was very high (16 ± 5 Hz, n = 7). However, 10-20 s after addition of 100 μM nimodipine to the K+-rich solution, the mIPSC frequency was reduced to 27 ± 4 % (n = 7) of that before nimodipine application (Fig. 7B), in contrast with the potentiating effect of nimodipine when added to control solution. Again, the findings were thus consistent with the hypothesis above.
Intracellular Ca2+ buffers reduce the effect of nimodipine on mIPSC frequency
To test whether the nimodipine-induced increase of mIPSC frequency is due to increased Ca2+ influx, the presynaptic intracellular Ca2+ concentration was manipulated. For that we applied the cell-permeable Ca2+ buffers that do not alter extracellular Ca2+, BAPTA-AM and EGTA-AM. These forms of BAPTA and EGTA enter the terminals by uptake and accumulate intracellularly following cleavage by unspecific esterases (Cummings et al. 1996). After 21 min of perfusion with 100 μM EGTA-AM the effect of nimodipine on mIPSC frequency was only 29 ± 14 % of that observed in control (n = 6; Fig. 7C). In similarity to EGTA-AM, 100 μM BAPTA-AM significantly reduced the effect of nimodipine, to 27 ± 8 % (n = 6; Fig. 7D). The common reducing effect of both BAPTA-AM and EGTA-AM on the nimodipine-evoked increase of mIPSC frequency suggests that the effect of nimodipine depends on a rise in intraterminal Ca2+ concentrations. Thus, these results were in agreement with our general hypothesis.
Does closure of the presynaptic Ca2+-activated K+ channels explain the potentiating effects of the Ca2+ channel blockers on the mIPSC frequency?
If the Ca2+ channel blockers increase the mIPSC frequency because of closure of K+ channels and resulting depolarization, as suggested above, then blockage of these K+ channels by other agents may be expected to mask the effect of the Ca2+ channel blockers. Such masking effects could then also be used to identify the specific K+ channels involved. We therefore attempted to identify the K+ channels by using the K+ channel blockers apamin, charybdotoxin, TEA and paxilline. As noted above, application of each of these blockers alone caused an increase in mIPSC frequency, and therefore their target Ca2+-dependent K+ channels seemed possible as candidate mediators of effects due to reduced Ca2+ influx.
Also in the presence of 1.0 μM apamin (2 min pre-application), nimodipine was effective in increasing the mIPSC frequency (to a peak of 6.4 ± 1.4 Hz; Fig. 8A). Similar results were obtained in the presence of 200 nm charybdotoxin (peak 5.6 ± 1.1 Hz; Fig. 8B). In the presence of 10 mm TEA, nimodipine was even more effective than when applied to control solution. A peak frequency of 11.9 ± 2.5 Hz was obtained in TEA, whereas the corresponding peak frequency in control solution was 8.7 ± 1.8 Hz (n = 15; Fig. 8C). Similarly, in 1.0 μM paxilline, the effect of nimodipine was larger (peak at 8.0 ± 1.7 Hz, n = 15) than when applied to control solution (peak at 4.5 ± 0.9 Hz, n = 15; Fig. 8D). Therefore, it seems unlikely that the effects of nimodipine were mediated by K+ channels sensitive to any of these four K+ channel blockers. However, as indicated below, we expect that other K+ channels, not sensitive to these toxins, are also present in the nerve terminals.
Figure 8. Effects of K+ channel blockers on response to nimodipine.
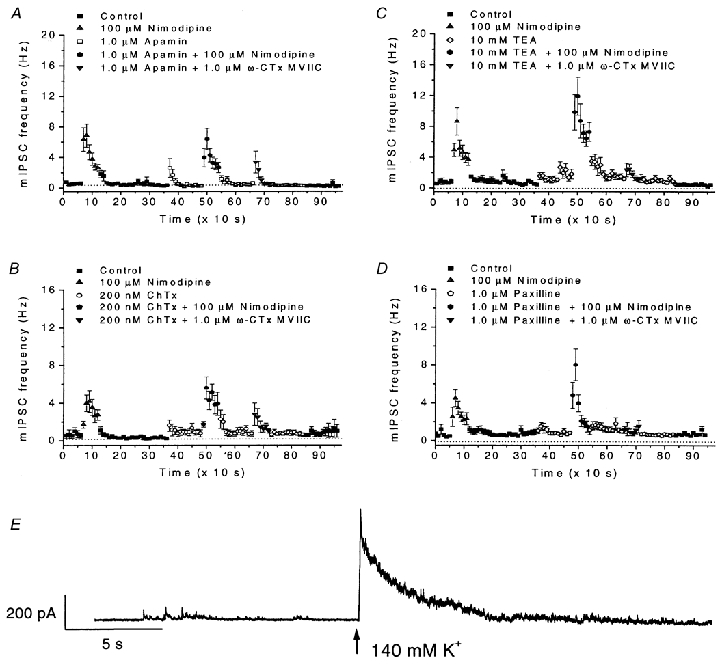
A-D, effects of the K+ channel blockers apamin (1.0 μM; A), charybdotoxin (ChTx, 200 nm; B), TEA (10 mm; C) and paxilline (1.0 μM; D) on mIPSC frequency and on the responses to nimodipine (100 μM) and to ω-conotoxin MVIIC (ω-CTx MVIIC; 1.0 μM). Data from 17 (A), 10 (B), 13 (C) and 15 (D) cells. Note the scale in A and B differs from that in C and D. Note also the large effect of nimodipine, but small effect of ω-conotoxin MVIIC, in the presence of the K+-channel blockers in C and D. Data presentation similar to those in Fig. 2A. E. Effect of raising the external K+ concentration (to 140 mm at the time indicated by an arrow) in the presence of a mixture of apamin (1.0 μM), charybdotoxin (ChTx; 200 nm) and TEA (10 mm). The cells were pre-treated with the mixture of K+-channel blockers for 5 min. Note the dramatic increase of mIPSC frequency upon application of K+-rich solution. For all data, recording conditions similar to those in Fig. 2A. Error bars represent s.e.m.
The situation for ω-conotoxin MVIIC differed from that of nimodipine. Although application of ω-conotoxin MVIIC (down-pointing triangles in Fig. 8A-D) within 10 s dramatically increased the mIPSC frequency in the presence of 1.0 μM apamin (from 0.6 ± 0.2 to 3.4 ± 1.4 Hz, n = 17; Fig. 8A) or 200 nm charybdotoxin (from 0.8 ± 0.3 to 2.8 ± 1.2 Hz, n = 10; Fig. 8B), the effect ω-conotoxin MVIIC was reduced or masked in the presence of TEA (increased from 1.2 ± 0.2 to 2.4 ± 0.9 Hz, n = 13; Fig. 8C) and nearly abolished in the presence of 1.0 μM paxilline (increased from 1.0 ± 0.2 to 1. 2 ± 0. 4 Hz, n = 15; Fig. 8D). (The effects of ω-conotoxin MVIIC under different conditions were, due to the poor reversibility of this substance, compared between different groups of cells.)
The reduced effect of ω-conotoxin MVIIC in the presence of TEA or paxilline is consistent with the hypothesis that the Ca2+ influx through ω-conotoxin MVIIC-sensitive channels activates K+ channels sensitive to TEA and paxilline. This pharmacological profile, with sensitivity to TEA but not charybdotoxin or apamin, matches that of large-conductance Ca2+-activated K+ channels found in the neurohypophysial terminals of another type of hypothalamic neuron (Wang et al. 1992) and may be due to the presence of a β4 subunit (Behrens et al. 2000; Meera et al. 2000).
The potentiated effect of nimodipine in the presence of paxilline and TEA suggests that the nimodipine-sensitive L-type channels are not coupled to the same K+ channels as ω-conotoxin MVIIC-sensitive channels. The potentiated effect, however, provides additional support that the action of nimodipine is depolarizing. A larger depolarization is expected when K+ channels, which counteract depolarization, are blocked. If our general hypothesis (Fig. 6) is correct, the findings imply that the L-type channels are coupled to as yet unidentified K+ channels that are not blocked by apamin, charybdotoxin, TEA or paxilline. In this case, we should expect a depolarization of the presynaptic terminals and an increased mIPSC frequency as a consequence of raising the external K+ concentration, even in the presence of the K+ channel blockers. Indeed, application of K+-rich (140 mm) solution caused a dramatic increase in mIPSC frequency in cells that were pretreated with a mixture of TEA, charybdotoxin and apamin for 5 min (Fig. 8E). (The effect was not quantified due to the extremely high frequency where individual mIPSCs could not be resolved.) These results thus suggest that there are remaining K+ channels insensitive to the blockers used. Among these channels may be Ca2+-activated K+ channels that interact with L-type Ca2+ channels.
Effects of long-duration application of ω-conotoxin MVIIC on mIPSC frequency and on response to nimodipine
Our findings above show that block of Ca2+ channels can give a paradoxical increase in spontaneous transmitter release, although our hypothetical model suggests that Ca2+ channels are also required to trigger release. Why then, are the release-triggering Ca2+ channels at the active zones only affected by the small blocking ion Cd2+ and not by the large-molecular blockers nimodipine and ω-conotoxin MVIIC? In the case of nimodipine, we expect no L-type channels at the release site, but in the case of ω-conotoxin MVIIC, we do expect that N-, P- or Q-type Ca2+ channels are contributing to the release in this preparation (Haage et al. 1998) as well as in many other preparations (for review see Dunlap et al. 1995). A likely explanation involves different time courses of toxin effects on the Ca2+-mediated control of the release-triggering machinery and on the control of the Ca2+-activated K+ channels. A slow time course (time constant > 1.5 min) of ω-conotoxin MVIIC-mediated block of K+-evoked GABA release has previously been noted for this preparation (Haage et al. 1998). Also in other preparations the blocking effect on synaptic transmission is slow (for example see Wheeler et al. 1994), in contrast to the fast potentiating effect on mIPSC frequency reported here (maximum within 10 s; Fig. 2D). Differences in time course could be due, for example, to separate locations of the Ca2+ channels coupled to the two different systems, as suggested in Fig. 6, with different access and consequently different concentration of the toxin at the two locations. In contrast to ω-conotoxin MVIIC, small ions of high mobility, such as Cd2+, may quickly reach the release site in the synaptic cleft and thereby block the Ca2+ influx necessary for release.
We tested the proposed requirement of functional ω-conotoxin MVIIC-sensitive Ca2+ channels (at the release site) for the nimodipine-evoked increase in mIPSC frequency. We thus applied ω-conotoxin MVIIC (1.0 μM) for 28 min and investigated the effect on basal mIPSC frequency as well as the effect on nimodipine-evoked potentiation of mIPSC frequency. After an initially increased mIPSC frequency, the toxin induced a slow gradual decline of basal mIPSC frequency to 53 ± 5 % (n = 5) of control after 27-28 min (Fig. 9A). Further, also the response to nimodipine (measured relative to the basal mIPSC frequency in ω-conotoxin MVIIC-containing solution during the 60 s interval immediately preceding each application of nimodipine) declined gradually with time in the presence of ω-conotoxin MVIIC (Fig. 9B). After 20 min in ω-conotoxin MVIIC, the response to nimodipine was 44 ± 12 % of that in control solution (n = 5).
Figure 9. Effects of long-duration application of ω-conotoxin MVIIC on basal mIPSC frequency and on response to nimodipine.
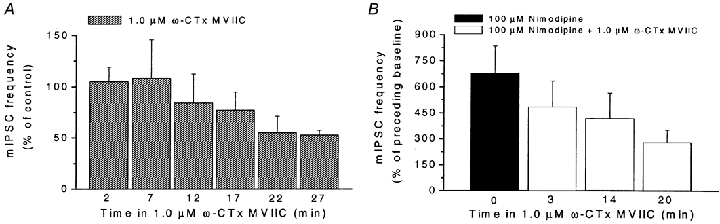
A, change in mIPSC frequency caused by long duration application of ω-conotoxin MVIIC (ω-CTx MVIIC; 1.0 μM) in 5 cells. The number of mIPSCs was measured during 60-s periods with 4-min intervals. Note the slow decline (after the initial increase) in mIPSC frequency in the presence of ω-conotoxin MVIIC. Data presented relative to control. B, reduced response to nimodipine (100 μM) caused by long-duration application of ω-conotoxin MVIIC (ω-CTx MVIIC; 1.0 μM) in 5 cells. The number of mIPSCs was measured during a 60-s period of nimodipine application. Data presented relative to preceding baseline. The baseline was measured during a 60-s period immediately before addition of nimodipine (in the presence of ω-CTx MVIIC, for 2nd to 4th bar). Note the considerable decrease of response to nimodipine after prolonged preincubation with ω-conotoxin MVIIC. For all data, recording conditions similar to those in Fig. 2A. Error bars represent s.e.m.
The findings thus showed that ω-conotoxin MVIIC-sensitive Ca2+ channels contributed to the basal mIPSC frequency, and that the blocking effect showed a considerably slower time course than the potentiating effect on the mIPSC frequency. The findings further showed that the effect of nimodipine also depended on functional ω-conotoxin MVIIC-sensitive Ca2+ channels, in agreement with our proposed hypothesis.
DISCUSSION
In the present work, evidence for the existence of several Ca2+ and K+ channel types in the presynaptic nerve terminals of rat MPN neurons was presented. Further, a contribution of several of these channel types in controlling spontaneous transmitter release was demonstrated. The main emphasis was put in efforts to explain the apparent dual and opposing roles of Ca2+ influx in controlling the spontaneous release.
The study was performed using dissociated neurons with functional adhering nerve terminals (cf. Haage et al. 1998). This preparation has the advantage that the major parts of the presynaptic neuron, including cell body, dendrites and major parts of the axon have been removed. Thus, although MPN neurons can generate several types of Ca2+-mediated impulses when fast Na+-dependent spikes are blocked (Sundgren-Andersson & Johansson, 1998), in the present study, propagation of such impulses from the soma to the nerve terminals can be excluded. Therefore, the observed effects on mIPSC frequency are likely to be mediated by direct actions on the nerve terminal.
Ca2+ and K+ channel types controlling spontaneous transmitter release
The pharmacological evidence suggested that a number of ion channel types are present in MPN nerve terminals. The effects on mIPSC frequency show that some channels of these types are open under ‘resting’ conditions without Na+-dependent spikes and control the frequency of spontaneous transmitter release. The presence of the previously demonstrated N- and P/Q-type Ca2+ channels (Haage et al. 1998) was here supported by the effect of ω-conotoxin GVIA and the even larger effect of ω-conotoxin MVIIC on the mIPSC frequency. In addition, there was evidence for apamin-sensitive SK channels and paxilline-sensitive BK channels. Interestingly, evidence for BK channels in hippocampal presynaptic terminals was recently presented by Hu et al. (2001), although they were unable to find any drug-free conditions where these channels affected impulse-evoked transmission. Further, also dihydropyridine- and calciseptine-sensitive L-type Ca2+ channels appear to play a prominent role in the spontaneous neurotransmitter release in the present study, although they do not contribute to transient K+-evoked transmitter release from these nerve terminals (Haage et al. 1998). (The difference is easily explained by the postulated effect via the membrane potential, which is ‘overruled’ in the case of K+-evoked release where the membrane potential is set by the external K+ concentration.) In contrast to the findings of Parfitt & Madison (1993) and of Bao et al. (1998), we found no evidence for a contribution of Ni2+-sensitive low-threshold T-type Ca2+ channels to the spontaneous transmitter release, in our system.
Mechanisms for increased mIPSC frequency at blocked Ca2+ influx
The major unexpected finding in the present study was that application of Ca2+ channel blockers potentiated the frequency of ‘miniature’ synaptic currents. This was unexpected, since it is well established that Ca2+ influx mediates transmitter release (for review see Bennett, 1999), and since Ca2+ channel blockers reduce the GABA release evoked by depolarization of presynaptic terminals in the MPN (Haage et al. 1998). While the effects of nimodipine and nifedipine could possibly be due to direct block of K+ channels, the similar effect of other Ca2+ channel blockers (calciseptine, ω-conotoxin MVIIC and ω-conotoxin GVIA), the lack of effect of Bay K 8644 (L-type Ca2+ channel agonist that may block K+ channels in some preparations), the effects of washing out Cd2+ or EGTA, and the correlation of the effect of washing out EGTA with the effect of nimodipine, all suggest that the effects were caused by a reduced Ca2+ influx.
A simple model that could account for the ‘paradoxical’ increase in mIPSC frequency caused by the Ca2+ channel blockers was presented in Fig. 6. A main question when interpreting our results in terms of the proposed model is how a reduced Ca2+ influx could cause an increased Ca2+ influx. The time course of the observed effects suggests an explanation. The increase of mIPSC frequency upon application of the Ca2+ channel blockers was rapid, with peak responses usually obtained within 10-20 s. This is considerably quicker than the blocking effects of some of these drugs on the GABA release evoked by K+-mediated depolarization, where the effects of ω-conotoxin MVIIC and ω-conotoxin GVIA, as well as of ω-agatoxin IVA, developed over several minutes (Haage et al. 1998; Wheeler et al. 1994). The slow blocking effects of synaptic transmission contrast with the findings that ω-conotoxin GVIA, ω-agatoxin IVA and ω-conotoxin MVIIC (in the case of N-type channels) may block their target Ca2+ channels rapidly, in some cases within a few seconds (Bargas et al. 1994; Randall & Tsien, 1995; McDonough et al. 1996). The most likely explanation for the difference in the present preparation is that to block the release, the toxin has to interact with Ca2+ channels at the synaptic release site, whereas Ca2+ channels coupled to K+ channels may be expressed also in the terminal membrane outside the synaptic cleft. The latter channels should be more directly exposed to the toxin-containing perfusion, whereas access to the release site in the synaptic cleft should be slower for large blocking molecules, such as ω-conotoxin MVIIC (although the access may be only marginally reduced for small ions such as Cd2+). The structures that provide the mechanically and chemically strong attachment of presynaptic terminals to the postsynaptic cells (see Pfenninger, 1971; Cotman & Taylor, 1972), the electron-dense material (Gray, 1959), the dense or intermediate plaque (Peters et al. 1991), the filaments and fibrils (Ichimura & Hashimoto, 1988) in the synaptic cleft and/or the adherens junctions seen at many synapses (Uchida et al. 1996) are likely to contribute to the barrier for diffusion into the synaptic cleft (see Südhof, 2001, for a review of the different cell adhesion molecules contributing to these structures). It has earlier been suggested that the structures in the synaptic cleft may interfere with the diffusion of substances from the synaptic cleft to the surrounding extracellular space (Peters et al. 1991). Supporting the idea of a diffusion barrier, the present study showed that the potentiating effect of ω-conotoxin MVIIC on mIPSC frequency (due to Ca2+ channels coupled to K+ channels) was much quicker than the inhibiting effect (due to Ca2+ channels coupled to the release machinery). When Ca2+ channels coupled to K+ channels are blocked, the depolarization induced by K+ channel closure is expected to nearly instantaneously spread to the membrane of the synaptic cleft, where the Ca2+ channels (not yet blocked), in turn, are quickly activated and trigger release in accordance with the proposed model (Fig. 6B and C).
Recent findings support the idea that the localization of the Ca2+ channels within the presynaptic terminal may affect their coupling to transmitter release. Different spatial localization of Ca2+ channel subtypes within a presynaptic terminal was clearly demonstrated by Wu et al. (1999) and also correlated with the effectiveness of coupling to transmitter release (see also Chuhma et al. 2001).
Types of Ca2+-activated K+ channels
Although evidence for the presence of presynaptic Ca2+-dependent K+ channels, as well as a depolarizing action of the Ca2+ channel blockers were presented above, the K+ channels involved have not been unambiguously identified. In the case of ω-conotoxin MVIIC, the effect was reduced or masked when TEA or paxilline were present. Thus, the effect of ω-conotoxin MVIIC was at least partly mediated by Ca2+ channels coupled to TEA- and paxilline-sensitive K+ channels. A likely candidate K+ channel type is the type II BK channel that is insensitive to charybdotoxin (Reinhart et al. 1989) and is present in other (neurohypophysial) nerve terminals (Wang et al. 1992; Dopico et al. 1999). Recently, it has been shown that the presence of a β4 subunit in BK channels introduces insensitivity to charybdotoxin as well as to iberiotoxin (Behrens et al. 2000; Meera et al. 2000) although the sensitivity to paxilline is preserved (Hu et al. 2001). Thus, it seems likely that some of the presynaptic BK channels in the MPN contain β4 subunits.
The effect of nimodipine on mIPSC frequency in MPN neurons observed in the present study was not blocked or masked by apamin, charybdotoxin, TEA or paxilline. Although we were unable to block the proposed K+ channels interacting with nimodipine-sensitive Ca2+ channels, we showed that there are remaining K+ channels, which can mediate the effect of nimodipine. The SK- channel subtype (SK1) insensitive to all K+ channel blockers used is the most likely candidate.
Control of impulse-triggered versus spontaneous neurotransmission
Although the probability of spontaneous transmitter release can be regulated in parallel with the probability of impulse-triggered release (Prange & Murphy, 1999), separate control is suggested by differential effects of Ca2+ channel blockers on these two types of release (for example see Bao et al. 1998). Recent findings also demonstrate that spontaneous synaptic currents may selectively be subject to short-term potentiation in the absence of potentiation of impulse-evoked transmission (Kombian et al. 2000). Further, findings by Hirsch et al. (1999) demonstrate that selective dysfunction of spontaneous release may be associated with seizure activity although impulse-evoked release is intact, thus providing evidence for a physiological role of spontaneous release (see also Staley, 1999). Our present findings suggest a novel mechanism for the control of such spontaneous release.
In conclusion, the present study shows that Ca2+ channel blockers, frequently used to inhibit impulse-mediated neurotransmission, paradoxically can increase the frequency of spontaneous ‘miniature’ synaptic currents. Therefore, Ca2+ influx is likely to play dual and opposing roles in controlling neurotransmission. The described evidence is consistent with a mechanism where Ca2+ channels together with Ca2+-dependent K+ channels control the frequency of spontaneous neurotransmitter release.
Acknowledgments
This work was supported by the Swedish Research Council (Project No. 11202), the Royal Swedish Academy of Sciences, Magn. Bergvalls Stiftelse, Åke Wibergs Stiftelse, Umeå University and a ‘Spjutspets’ grant from Umeå Sjukvård. M. D. and E. M. were supported by scholarships from the Swedish Institute.
REFERENCES
- Andjus PR, Stevic-Marinkovic Z, Cherubini E. Immunoglobulins from motoneurone disease patients enhance glutamate release from rat hippocampal neurones in culture. Journal of Physiology. 1997;504:103–112. doi: 10.1111/j.1469-7793.1997.103bf.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avdonin V, Shibata EF, Hoshi T. Dihydropyridine action on voltage-dependent potassium channels expressed in Xenopus oocytes. Journal of General Physiology. 1997;109:169–180. doi: 10.1085/jgp.109.2.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao J, Li JJ, Perl ER. Differences in Ca2+ channels governing generation of miniature and evoked excitatory synaptic currents in spinal laminae I and II. Journal of Neuroscience. 1998;18:8740–8750. doi: 10.1523/JNEUROSCI.18-21-08740.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bargas J, Howe A, Eberwine J, Cao Y, Surmeier DJ. Cellular and molecular chracterization of Ca2+ currents in acutely isolated, adult rat neostriatal neurons. Journal of Neuroscience. 1994;14:6667–6686. doi: 10.1523/JNEUROSCI.14-11-06667.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrens R, Nolting A, Reimann F, Schwarz M, Waldschütz R, Pongs O. hKCNMB3 and hKCNMB4, cloning and characterization of two members of the large-conductance calcium-activated potassium channel β subunit family. FEBS Letters. 2000;474:99–106. doi: 10.1016/s0014-5793(00)01584-2. [DOI] [PubMed] [Google Scholar]
- Bennett MR. The concept of a calcium sensor in transmitter release. Progress in Neurobiology. 1999;59:243–277. doi: 10.1016/s0301-0082(99)00004-0. [DOI] [PubMed] [Google Scholar]
- Bouron A. Modulation of spontaneous quantal release of neurotransmitters in the hippocampus. Progress in Neurobiology. 2001;63:613–635. doi: 10.1016/s0301-0082(00)00053-8. [DOI] [PubMed] [Google Scholar]
- Brussaard AB, Devay P, Leyting-Vermeulen JL, Kits KS. Changes in properties and neurosteroid regulation of GABAergic synapses in the supraoptic nucleus during the mammalian female reproductive cycle. Journal of Physiology. 1999;516:513–524. doi: 10.1111/j.1469-7793.1999.0513v.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capogna M, Gähwiler BH, Thompson SM. Mechanism of mu-opioid receptor-mediated presynaptic inhibition in the rat hippocampus in vitro. Journal of Physiology. 1993;470:539–558. doi: 10.1113/jphysiol.1993.sp019874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuhma N, Koyano K, Ohmori H. Synchronisation of neurotransmitter release during postnatal development in a calyceal presynaptic terminal of rat. Journal of Physiology. 2001;530:93–104. doi: 10.1111/j.1469-7793.2001.0093m.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen I, Miles R. Contributions of intrinsic and synaptic activities to the generation of neuronal discharges in in vitro hippocampus. Journal of Physiology. 2000;524:485–502. doi: 10.1111/j.1469-7793.2000.00485.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cossart R, Dinocourt C, Hirsch JC, Merchan-Perez A, De Felipe J, Ben-Ari Y, Esclapez M, Bernard C. Dendritic but not somatic GABAergic inhibition is decreased in experimental epilepsy. Nature Neuroscience. 2001;4:52–62. doi: 10.1038/82900. [DOI] [PubMed] [Google Scholar]
- Cotman CW, Taylor DJ. Isolation and structural studies on synaptic complexes from rat brain. Journal of Cell Biology. 1972;55:696–711. doi: 10.1083/jcb.55.3.696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings DD, Wilcox KS, Dichter MA. Calcium dependent paired-pulse facilitation of miniature EPSC frequency accompanies depression of EPSCs at hippocampal synapses in culture. Journal of Neuroscience. 1996;16:5312–5323. doi: 10.1523/JNEUROSCI.16-17-05312.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DelCastillo J, Katz B. Local activity at a depolarized nerve-muscle junction. Journal of Physiology. 1955;128:396–411. doi: 10.1113/jphysiol.1955.sp005315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dopico AM, Widmer H, Wang G, Lemos JR, Treistman SN. Rat supraoptic magnocellular neurones show distinct large conductance, Ca2+-activated K+ channel subtypes in cell bodies versus nerve endings. Journal of Physiology. 1999;519:101–114. doi: 10.1111/j.1469-7793.1999.0101o.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunlap K, Luebke JI, Turner TJ. Exocytotic Ca2+ channels in mammalian central neurons. Trends in Neurosciences. 1995;18:89–98. [PubMed] [Google Scholar]
- Edwards FA, Konnerth A, Sakmann B, Takahashi T. A thin slice preparation for patch clamp recordings from neurones of the mammalian central nervous system. Pflügers Archiv. 1989;414:600–612. doi: 10.1007/BF00580998. [DOI] [PubMed] [Google Scholar]
- Fagni L, Bossu JL, Bockaert J. Inhibitory effects of dihydropyridines on macroscopic K+ currents and on the large-conductance Ca2+-activated K+ channel in cultured cerebellar granule cells. Pflügers Archiv. 1994;429:176–182. doi: 10.1007/BF00374310. [DOI] [PubMed] [Google Scholar]
- Fatt P, Katz B. Some observations on biological noise. Nature. 1950;166:597–598. doi: 10.1038/166597a0. [DOI] [PubMed] [Google Scholar]
- Fatt P, Katz B. Spontaneous subthreshold activity at motor nerve endings. Journal of Physiology. 1952;117:109–128. [PMC free article] [PubMed] [Google Scholar]
- Gray EG. Axo-somatic and axo-dendritic synapses of the cerebral cortex: an electron microscope study. Journal of Anatomy. 1959;95:101–106. [PMC free article] [PubMed] [Google Scholar]
- Haage D, Karlsson U, Johansson S. Heterogeneous presynaptic Ca2+ channel types triggering GABA release onto medial preoptic neurons from rat. Journal of Physiology. 1998;507:77–91. doi: 10.1111/j.1469-7793.1998.077bu.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch JC, Agassandian C, Merchan-Perez A, Ben-Ari Y, Defelipe J, Esclapez M, Bernard C. Deficit of quantal release of GABA in experimental models of temporal lobe epilepsy. Nature Neuroscience. 1999;2:499–500. doi: 10.1038/9142. [DOI] [PubMed] [Google Scholar]
- Hu H, Shao L-R, Chavoshy S, Gu N, Trieb M, Behrens R, Laake P, Pongs O, Knaus HG, Ottersen OP, Storm JF. Presynaptic Ca2+-activated K+ channels in glutamatergic hippocampal terminals and their role in spike repolarization and regulation of transmitter release. Journal of Neuroscience. 2001;21:9585–9597. doi: 10.1523/JNEUROSCI.21-24-09585.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hugues M, Romey G, Duval D, Vincent JP, Lazdunski M. Apamin as a selective blocker of the calcium-dependent potassium channel in neuroblastoma cells: voltage-clamp and biochemical characterization of the toxin receptor. Proceedings of the National Academy of Sciences of the USA. 1982;79:1308–1312. doi: 10.1073/pnas.79.4.1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichimura T, Hashimoto PH. Structural components in the synaptic cleft captured by freeze-substitution and deep etching of directly frozen cerebellar cortex. Journal of Neurocytology. 1988;17:3–12. doi: 10.1007/BF01735373. [DOI] [PubMed] [Google Scholar]
- Jackson MB. Presynaptic excitability. International Review of Neurobiology. 1995;38:201–251. doi: 10.1016/s0074-7742(08)60527-9. [DOI] [PubMed] [Google Scholar]
- Jensen K, Jensen MS, Lambert JDC. Post-tetanic potentiation of GABAergic IPSCs in cultured rat hippocampal neurones. Journal of Physiology. 1999;519:71–84. doi: 10.1111/j.1469-7793.1999.0071o.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson S, Århem P. Single-channel currents trigger action potentials in small cultured hippocampal neurons. Proceedings of the National Academy of Sciences of the USA. 1994;91:1761–1765. doi: 10.1073/pnas.91.5.1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson S, Druzin M, Haage D, Wang M-D. The functional role of a bicuculline-sensitive Ca2+-activated K+ current in rat medial preoptic neurons. Journal of Physiology. 2001;532:625–635. doi: 10.1111/j.1469-7793.2001.0625e.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson U, Sundgren AK, Nasstrom J, Johansson S. Glutamate-evoked currents in acutely dissociated neurons from the rat medial preoptic nucleus. Brain Research. 1997;759:270–276. doi: 10.1016/s0006-8993(97)00262-x. [DOI] [PubMed] [Google Scholar]
- Katz B, Miledi R. The role of calcium in neuromuscular facilitation. Journal of Physiology. 1968;195:481–492. doi: 10.1113/jphysiol.1968.sp008469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knaus HG, McManus OB, Lee SH, Schmalhofer WA, Garcia-Calvo M, Helms LM, Sanchez M, Giangiacomo K, Reuben JP, Smith AB, Kaczorowski GJ, Garcia ML. Tremorgenic indole alkaloids potently inhibit smooth muscle high-conductance calcium-activated potassium channels. Biochemistry. 1994;33:5819–5828. doi: 10.1021/bi00185a021. [DOI] [PubMed] [Google Scholar]
- Kombian SB, Hirasawa M, Mouginot D, Chen X, Pittman QJ. Short-term potentiation of miniature excitatory synaptic currents causes excitation of supraoptic neurons. Journal of Neurophysiology. 2000;83:2542–2553. doi: 10.1152/jn.2000.83.5.2542. [DOI] [PubMed] [Google Scholar]
- Koyama S, Kubo C, Rhee JS, Akaike N. Presynaptic serotonergic inhibition of GABAergic synaptic transmission in mechanically dissociated rat basolateral amygdala neurons. Journal of Physiology. 1999;518:525–538. doi: 10.1111/j.1469-7793.1999.0525p.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenzon NM, Foehring RC. Characterization of pharmacologically identified voltage-gated calcium channel currents in acutely isolated rat neocortical neurons. I. Adult neurons. Journal of Neurophysiology. 1995;73:1430–1442. doi: 10.1152/jn.1995.73.4.1430. [DOI] [PubMed] [Google Scholar]
- McDonough SI, Swartz KJ, Mintz IM, Boland LM, Bean BP. Inhibition of calcium channels in rat central and peripheral neurons by ω-conotoxin MVIIC. Journal of Neuroscience. 1996;16:2612–2623. doi: 10.1523/JNEUROSCI.16-08-02612.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinney RA, Capogna M, Dürr R, Gähwiler BH, Thompson SC. Miniature synaptic events maintain dendritic spines via AMPA receptor activation. Nature Neuroscience. 1999;2:44–49. doi: 10.1038/4548. [DOI] [PubMed] [Google Scholar]
- Marrion NV, Tavalin SJ. Selective activation of Ca2+-activated K+ channels by co-localized Ca2+ channels in hippocampal neurons. Nature. 1998;395:900–905. doi: 10.1038/27674. [DOI] [PubMed] [Google Scholar]
- Meera P, Wallner M, Toro L. A neuronal β subunit (KCNMB4). makes the large conductance, voltage- and Ca2+-activated K+ channel resistant to charybdotoxin and iberiotoxin. Proceedings of the National Academy of Sciences of the USA. 2000;97:5562–5567. doi: 10.1073/pnas.100118597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meir A, Ginsburg S, Butkevich A, Kachalsky SG, Kaiserman I, Ahdut R, Demirgoren S, Rahamimoff R. Ion channels in presynaptic nerve terminals and control of transmitter release. Physiological Reviews. 1999;79:1019–1088. doi: 10.1152/physrev.1999.79.3.1019. [DOI] [PubMed] [Google Scholar]
- Miledi R. Transmitter release induced by injection of calcium ions into nerve terminals. Proceedings of the Royal Society of London B. 1973;183:421–425. doi: 10.1098/rspb.1973.0026. [DOI] [PubMed] [Google Scholar]
- Miller C, Moczydlowski E, Latorre R, Phillips M. Charybdotoxin, a protein inhibitor of single Ca2+-activated K+ channels from mammalian skeletal muscle. Nature. 1985;313:316–318. doi: 10.1038/313316a0. [DOI] [PubMed] [Google Scholar]
- Mlinar B, Enyeart JJ. Identical inhibitory modulation of A-type potassium currents by dihydropyridine calcium channel agonists and antagonists. Molecular Pharmacology. 1994;46:743–749. [PubMed] [Google Scholar]
- Neher E. Correction for liquid junction potentials in patch clamp experiments. Methods in Enzymology. 1992;207:123–131. doi: 10.1016/0076-6879(92)07008-c. [DOI] [PubMed] [Google Scholar]
- Parfitt KD, Madison DV. Phorbol esters enhance synaptic transmission by a presynaptic, calcium-dependent mechanism in rat hippocampus. Journal of Physiology. 1993;471:245–268. doi: 10.1113/jphysiol.1993.sp019900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters A, Palay SL, Webster HdeF. The Fine Structure of the Nervous System. 3. New York: Oxford University Press; 1991. [Google Scholar]
- Pfenninger KH. The cytochemistry of synaptic densities. II. Proteinaceous components and mechanisms of synaptic connectivity. Journal of Ultrastructure Research. 1971;35:451–475. doi: 10.1016/s0022-5320(71)80005-9. [DOI] [PubMed] [Google Scholar]
- Prange O, Murphy TH. Correlation of miniature synaptic activity and evoked release probability in cultures of cortical neurons. Journal of Neuroscience. 1999;19:6427–6438. doi: 10.1523/JNEUROSCI.19-15-06427.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rae J, Cooper K, Gates P, Watsky M. Low access resistance perforated patch recordings using amphotericin B. Journal of Neuroscience Methods. 1991;37:15–26. doi: 10.1016/0165-0270(91)90017-t. [DOI] [PubMed] [Google Scholar]
- Randall A, Tsien RW. Pharmacological dissection of multiple types of Ca2+ channel currents in rat cerebellar granule neurons. Journal of Neuroscience. 1995;15:2995–3012. doi: 10.1523/JNEUROSCI.15-04-02995.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhart PH, Chung S, Levitan IB. A family of calcium-dependent potassium channels from rat brain. Neuron. 1989;2:1031–1041. doi: 10.1016/0896-6273(89)90227-4. [DOI] [PubMed] [Google Scholar]
- Rhee JS, Wang ZM, Nabekura J, Inoue K, Akaike N. ATP facilitates spontaneous glycinergic IPSC frequency at dissociated rat dorsal horn interneuron synapses. Journal of Physiology. 2000;524:471–483. doi: 10.1111/j.1469-7793.2000.t01-1-00471.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robitaille R, Charlton MP. Presynaptic calcium signals and transmitter release are modulated by calcium-activated potassium channels. Journal of Neuroscience. 1992;12:297–305. doi: 10.1523/JNEUROSCI.12-01-00297.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robitaille R, Garcia ML, Kaczorowski GJ, Charlton MP. Functional colocalization of calcium and calcium-gated potassium channels in control of transmitter release. Neuron. 1993;11:645–655. doi: 10.1016/0896-6273(93)90076-4. [DOI] [PubMed] [Google Scholar]
- Schneider MJ, Rogowski RS, Krueger BK, Blaustein MP. Charybdotoxin blocks both Ca2+-activated K+ channels and Ca2+-independent voltage-gated K+ channels in rat brain synaptosomes. FEBS Letters. 1989;250:433–436. doi: 10.1016/0014-5793(89)80771-9. [DOI] [PubMed] [Google Scholar]
- Staley KJ. Quantal GABA release: noise or not? Nature Neuroscience. 1999;2:494–495. doi: 10.1038/9139. [DOI] [PubMed] [Google Scholar]
- Sundgren-andersson AK, Johansson S. Calcium spikes and calcium currents in neurons from the medial preoptic nucleus of rat. Brain Research. 1998;783:194–209. doi: 10.1016/s0006-8993(97)01342-5. [DOI] [PubMed] [Google Scholar]
- Uchida N, Honjo Y, Johnson KR, Wheelock MJ, Takeichi M. The catenin/cadherin adhesion system is localized in synaptic junctions bordering transmitter release zones. Journal of Cell Biology. 1996;135:767–779. doi: 10.1083/jcb.135.3.767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valmier J, Richard S, Devic E, Nargeot J, Simmoneau M, Baldy-Moulinier M. Dihydropyridines interact with calcium-independent potassium currents in embryonic mammalian sensory neurons. Pflügers Archiv. 1991;419:281–287. doi: 10.1007/BF00371108. [DOI] [PubMed] [Google Scholar]
- Vaughan CW, Christie MJ. Presynaptic inhibitory action of opioids on synaptic transmission in the rat periaqueductal gray in vitro. Journal of Physiology. 1997;498:463–472. doi: 10.1113/jphysiol.1997.sp021872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vorobjev VS. Vibrodissociation of sliced mammalian nervous tissue. Journal of Neuroscience Methods. 1991;38:145–150. doi: 10.1016/0165-0270(91)90164-u. [DOI] [PubMed] [Google Scholar]
- Wang G, Thorn P, Lemos JR. A novel large-conductance Ca2+-activated potassium channel and current in nerve terminals of the rat neurohypophysis. Journal of Physiology. 1992;457:47–74. doi: 10.1113/jphysiol.1992.sp019364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler DB, Randall A, Tsien RW. Roles of N-type and Q-type Ca2+ channels in supporting hippocampal synaptic transmission. Science. 1994;264:107–111. doi: 10.1126/science.7832825. [DOI] [PubMed] [Google Scholar]
- Wu L-G, Westenbroek RE, Borst JGG, Catterall WA, Sakmann B. Calcium channel types with distinct presynaptic localization couple differentially to transmitter release in single calyx-type synapses. Journal of Neuroscience. 1999;19:726–736. doi: 10.1523/JNEUROSCI.19-02-00726.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]